Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(2):595-610. doi:10.7150/jca.50785 This issue Cite
Research Paper
Immunological Role and Prognostic Value of APBB1IP in Pan-Cancer Analysis
Qianyun Ge1,2, Ganxun Li1,2, Jin Chen1,2, Jia Song1,2, Guangzhen Cai1,2, Yi he1,2, Xuewu Zhang1,2, Huifang Liang1,2, Zeyang Ding1,2 ![]() , Bixiang Zhang1,2
, Bixiang Zhang1,2 ![]()
1. Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
2. Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Received 2020-7-17; Accepted 2020-11-2; Published 2021-1-1
Abstract

Objective: APBB1IP is a Rap1-binding protein that mainly acts as a regulator of leukocyte recruitment and pathogen clearance through complement-mediated phagocytosis. However, the role of APBB1IP in tumor immunity remains unclear. This study was carried out to evaluate the prognostic landscape of APBB1IP in pan-cancer analysis and investigate the relationship between APBB1IP expression and immune infiltration.
Methods: We explored the expression pattern and prognostic value of APBB1IP in pan-cancer analysis through Kaplan-Meier Plotter and multiple databases, including TCGA, Oncomine. We then assessed the correlation between APBB1IP expression and immune cell infiltration using the TIMER database. Furthermore, we identified the proteins that interact with APBB1IP and performed epigenetic and transcriptional analyses. Multivariate Cox regression analyses were applied to construct a prognostic model, which consisted of APBB1IP and its interacting proteins, based on the lung cancer cohorts from the Gene Expression Omnibus (GEO) database.
Results: The expression of APBB1IP was correlated with the prognosis of several types of cancer. APBB1IP upregulation was found to be associated with increased immune cell infiltration, especially for CD8+ T cells, natural killer (NK) cells, and immune regulators. A link was found between APBB1IP and immune-related proteins including RAP1A/B, TLN1/2 and VCL in the interaction network.
Conclusion: APBB1IP can serve as a prognostic biomarker in pan-cancer analysis. APBB1IP upregulation was correlated with increased immune-cell infiltration, and the expression APBB1IP in different tumors might be related to the tumor immune microenvironment.
Keywords: APBB1IP, pan-cancer analysis, immune infiltration, prognosis, tumor immune microenvironment
Introduction
The amyloid β (A4) precursor protein-binding, family B, member 1 interacting protein (APBB1IP) was identified as a binding partner of the amyloid β (A4) precursor protein-binding, family B, member 1 (APBB1) [1]. Subsequently, APBB1IP was found to interact with the small guanosine triphosphatase (GTPase) Rap1 in a yeast two-hybrid screen [2]. APBB1IP belongs to the MRL (Mig-10/RIAM/Lamellipodin) family of adaptor proteins, which have a proline-rich region at the C terminus and a highly conserved pattern of 27 amino acids in a predicted coiled-coil region immediately N-terminal to the RA domain [2].
APBB1IP is an intrinsic element of the integrin activation machinery, and is required for Rap1-induced affinity changes in β1 and β2 integrins in T cells [2]. Moreover, APBB1IP was demonstrated to also be involved in Rap1-mediated activation of αIIbβ3 integrin in platelets [3]. Based on these molecular mechanisms, the APBB1IP mainly is thought to mainly function in the activation and modulation of innate immune responses, as a regulator of leukocyte recruitment and pathogen clearance through complement-mediated phagocytosis [4-6]. However, APBB1IP also plays a central role in cancer cell migration and invasion, and APBB1IP-depleted melanoma cells displayed decreased persistent cell migration directionality, thereby reducing cancer invasion [7]. However, there are no systemic studies on the role of APBB1IP in different human cancers, and it remains unclear whether the effects of APBB1IP might be context-dependent, varying in different cancers.
The tumor microenvironment (TME) contains various cells, with infiltrating immune cells accounting for a large proportion [8]. The roles and prognostic value of infiltrating immune cells have been extensively studied in various malignant tumors [9, 10]. Immunotherapy, as an alternative approach to anticancer treatment, has been developed in recent years to reactivate the adaptive and innate immune systems and create a robust antitumoral immune response. For instance, mall-molecule inhibitors of cytotoxic T lymphocyte associated antigen 4 (CTLA4), programmed death-1 (PD-1), and programmed death ligand-1 (PD-L1) were found to have promising antitumor effects on non-small-cell lung carcinoma and colorectal cancer [11, 12]. Unfortunately, only a small proportion of patients with certain cancer types respond well to current immunotherapies [12]. Thus, it is necessary to explore additional potential targets.
In this study, we evaluated the expression signature and prognostic value of the APBB1IP gene in pan-cancer analysis using the TCGA pan-cancer database and Kaplan-Meier Plotter. We then explored the potential relationships between APBB1IP expression and immune infiltration levels. Finally, the potential mechanisms were also explored by bioinformatic analysis.
Methods
Data Acquisition
Mutation data, RNA sequencing data, and clinical data for 33 cancer types were obtained from TCGA using UCSC Xena [13]. GSE13213 [14] and GSE29016 [15] gene expression profiles were retrieved from the gene expression omnibus database. The inclusion and exclusion criteria adopted for the samples were (1) the loss of expression of APBB1IP; (2) the loss of follow-up information; (3) uncertain TNM stage; and (4) accompanied by other diseases. In GSE13213 and GSE29016, 117 and 68 lung cancer samples were retrieved respectively for subsequent analysis. Other data were obtained from public databases as described in the corresponding parts.
APBB1IP gene expression analysis
The mRNA expression of APBB1IP in different cancer types was analyzed in the ONCOMINE database. [16] The threshold was set to a P-value of 0.01 and fold-change of 2. APBB1IP expression in normal tissue and tumor tissue was compared across cancers using the Wilcoxon test, and the results were visualized using the R packages “ggpubr”.
Survival analysis
The prognosis of patients with high- tumors and low-APBB1IP tumors was compared using univariate Cox analysis for each cancer subtype, and genes with a P value < 0.05 were considered as prognostic genes. Overall survival analysis was performed via Kaplan-Meier survival analysis using the “survival” and “survminer” R packages.
Kaplan-Meier Plotter Database Analysis
Kaplan-Meier plotter was used to assess the effects of 54,675 genes on survival using 10,461 cancer samples. These samples include 5,143 breast, 1,816 ovarian, 2,437 lung, and 1,065 gastric cancer samples on the HGU133 Plus 2.0 array with a mean follow-up of 69, 40, 49, and 33 months, respectively. The correlation between APBB1IP expression and survival in 21 different cancers was analyzed using Kaplan-Meier plotter [17]. The hazard ratio (HR), 95% confidence interval and log-rank P-value were also computed.
TIMER analysis
The TIMER database was used to systematically analyze the tumor-infiltrating immune cells (TIICs) in 32 cancer types in more than 10,000 samples from The Cancer Genome Atlas (TCGA) database [18]. TIMER applies a previously published statistical deconvolution method to infer the abundance of tumor-infiltrating immune cells (TIICs) from gene expression profiles [19]. Gene modules were used to analyze APBB1IP expression in several types of cancer and the correlation of APBB1IP expression with the abundance of infiltrating immune cells, after which Spearman's rho value and statistical significance were obtained. The correlation between APBB1IP expression and several immune cell markers was also analyzed by Spearman correlation to identify the potential subtypes of infiltrating immune cells. Immune gene markers were selected from the website of R&D Systems.
Immune factors correlation analysis
Different immune factors lists were obtained from the Tumor Immune System Interactions Database [20], which includes immunoinhibitory and immunostimulatory factors. The Spearman method was used to determine the correlation coefficients.
GO functional analysis and KEGG pathway enrichment analysis of DEGs
GO functional analysis is a useful method for annotating genes and identifying characteristic biological attributes from high-throughput genome or transcriptome data [21]. KEGG incorporates a wide range of databases, including those on genomes, biological pathways, diseases, drugs and chemical substances [22]. The Database for Annotation, Visualization and Integrated Discovery, an online bioinformatics database [23], was used for the GO functional analysis and KEGG pathway enrichment analysis, with FDR<0.05 as the cut-off criterion.
PPI network and TF regulatory network
A PPI network was developed using the online database STRING (http://string-db.org) [24]. Cytoscape software was used to construct a PPI network and analyze the interactions between APBB1IP and associated proteins [25]. The iRegulon Cytoscape plugin was used to predict the TF regulatory network.
Transcription analysis
We downloaded miRNA data and the location of potential binding sites on the 3′UTR of APBB1IP from miRWalk [26]. The Spearman correlation between the expression of these miRNAs and APBB1IP was investigated using STARBASE v3.0 [27].
Genetic and epigenetic analysis
GSCALite consists of analytical modules for multi-omics data from TCGA 11 160 samples across 33 cancer types (TCGA Cancer), 746 drug data from Genomics of Drug Sensitivity in Cancer (GDSC) and the Cancer Therapeutics Response Portal (CTRP), as well as normal tissue expression data of 11 688 samples from GTEx (GTEx Normal Tissue). We used GSCALite to analyzed the single nucleotide variation, copy number variation, methylation and pathway activity [28].
Construction of a Prognostic Model
APBB1IP and associated genes were evaluated by step-wise multivariate Cox regression analysis. Risk scores were calculated based on gene expression multiplied by a linear combination of a regression coefficient obtained from the multivariate Cox analysis. Patients were assigned to high- and low-risk groups according to the median risk score. The survival analysis of patients in the high- and low-risk groups was conducted using the “survival” R package. The receiver operating characteristic (ROC) curve was implemented by the R software package “survival ROC”.
Results
Pan-cancer Analysis of APBB1IP mRNA Expression Levels
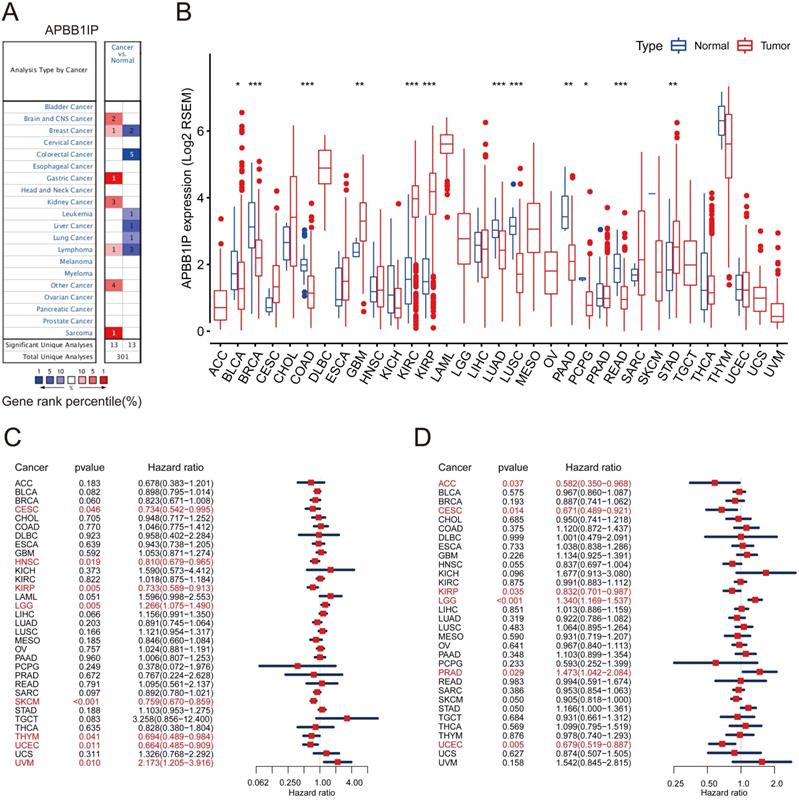
To explore its role in cancer, the APBB1IP mRNA expression levels were analyzed over a cancer-wide range in Oncomine. The results revealed that the expression of APBB1IP was inconsistently up- or down-regulation in different cancer types (Figure 1A). The details of APBB1IP expression in multiple cancers are summarized in Table S1. To further evaluate APBB1IP expression in different cancers, we examined the expression levels of APBB1IP in all 33 cancer types available in TCGA the pan-cancer database (Summery of TCGA data are in Table S2). The differential expression patterns of APBB1IP in tumors and adjacent normal tissues are shown in Figure 1B, APBB1IP expression was lower in some cancers, including BLCA, BRCA, COAD, LUAD, LUSC, PAAD and READ, while others were characterized by high APBB1IP expression (GBM, KIRC, KIRP and STAD). These findings demonstrate the intrinsic differences in the expression of APBB1IP between different tumor types, and detailed analyses of APBB1IP expression were considered for further analysis.
mRNA Expression Levels Prognostic Significance of APBB1IP in different Cancers. (A) High or low expression of APBB1IP in different human cancer tissues compared with normal tissues using the Oncomine database. The number in each cell is the amount of datasets. (B) The level of APBB1IP expression in different tumor types from the TCGA database. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Correlation of APBB1IP mRNA expression with OS for different cancer types in TCGA. (D) Correlation of APBB1IP mRNA expression with RFS for different cancer types in TCGA. Red squares represent the hazard ratios. Short bars appear due to limited sample size due to which the parameters and hazard ratio could not be calculated. OS: overall survival; PFS: progression-free survival. Red Font represented P < 0.05.
Prognostic Significance of APBB1IP Expression in Human Cancers
We next investigated the prognostic value of APBB1IP via pan-cancer analysis in different databases. First of all, we used univariate Cox proportional hazard regression models to analyze the association between APBB1IP expression with the overall survival and progression-free survival in various cancers in the TCGA. The criterion for significant association was a P-value of less than 0.05. As shown in the Figure 1C, APBB1IP was associated with poor prognosis in LGG (OS: HR = 1.266, 95% CI from 1.075 to 1.490, p= 0.005) and UVM (OS: HR = 2.173, 95% CI from 1.205 to 3.916, p= 0.010), while increased expression of APBB1IP was primarily associated with a survival advantage in patients with CESC, HNSC, KIRP, SKCM, THYM, and UCEC. Figure 1D indicated that high expression of APBB1IP predicted shorter RFS in patients with LGG and PRAD. In patients with ACC, CESC, KIRP and UCEC, high APBB1IP expression predicted better disease‐free survival. Overall- and progression-free survival curves stratified by high- and low-expression of APBB1IP in different types of cancer are shown in the Figure S1A and S1B, respectively.
To further examine the prognostic potential of APBB1IP in different cancers, the Kaplan-Meier plotter database was used to evaluate the prognostic value of APBB1IP based on gene chip and RNA-seq data from the GEO and EGA databases. Low APBB1IP expression levels were associated with poorer OS in BRCA, CESC, HNSC, KIRP, READ, SARC, THYM and UCEC. Conversely, low expression of APBB1IP was correlated with better OS in ESCA, LIHC, STAD and TGCT (Figure S2A-F). These results demonstrate the prognostic significance of APBB1IP expression in several human cancers, although their correlation may vary depending on the cancer type.
Correlation of APBB1IP Expression with Immune Infiltration and Various Subsets of Immune Cells
It has long been recognized that lymphocytes are intimately associated with tumor cells. For instance, it was reported that the presence of TILs is associated with a more favorable prognosis in patients with breast cancer [29]. Multiple correlation-based studies have implicated APBB1IP in signaling events critical for integrin-mediated control of immune function [4, 6], and these findings support a prognostic role of APBB1IP in cancer. It is likely that APBB1IP might influence the progression of cancer by influencing the regulation of immune infiltration. To determine the role of APBB1IP expression in TIL abundance, TIMER was used to obtain the Spearman correlation values for APBB1IP expression and the infiltration levels of various immune cells. These correlations were presented as heat maps in Figure 2A. The results revealed that APBB1IP expression was significantly negatively correlated with tumor purity in most cancer types, except CHOL, DLBC, KIRC, KIRP, MESO, THCA, THYM, UCS and UVM, indicating that APBB1IP expression in tumor tissues might be induced by the infiltrating immune cells. Higher APBB1IP expression in most cancers markedly increased the infiltration of immune cells, especially in BRCA, CESC, HNSC, PRAD, SKCM, TGCT and UCEC. However, In CHOL, DLBC, MESO, and UVM, the expression of APBB1IP showed no significant correlation with the infiltration of various immune cells.
Correlation of APBB1IP Expression with Immune Infiltration and Various Subsets of Immune Cells. (A) The correlation between APBB1IP expression and abundance of infiltrating immune cells across cancer types. (B) Correlation of APBB1IP expression with markers of CD8+ T cells in SKCM and LGG. (C) Correlation of APBB1IP expression with markers of NK cells in SKCM and LGG.
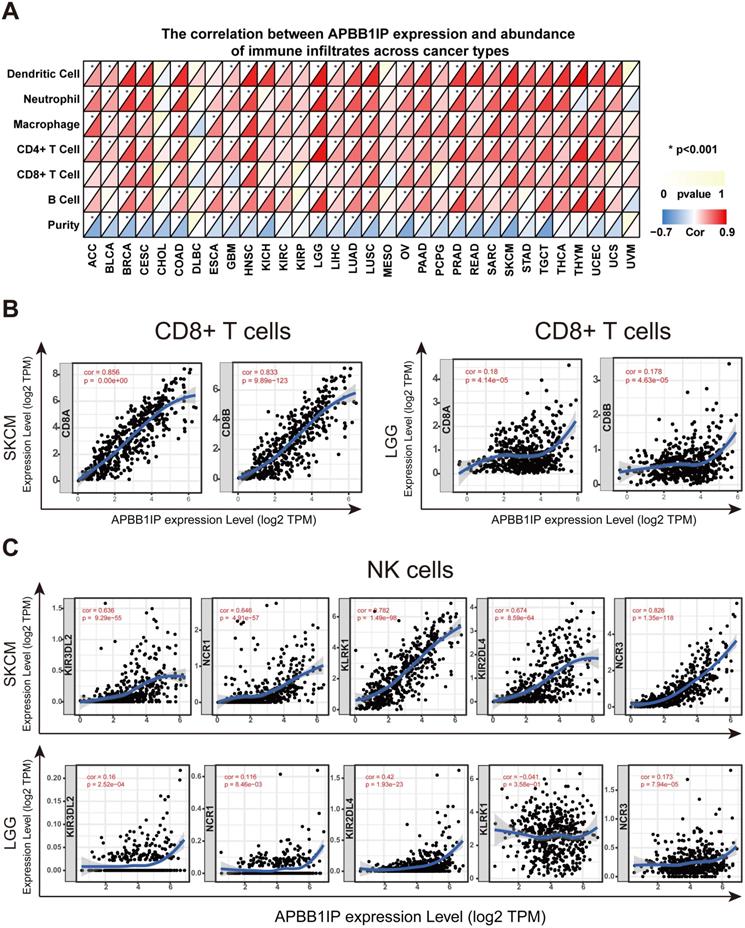
These results demonstrate that there is a positive correlation between APBB1IP expression and immune cell infiltration levels in different cancer types, such as BRCA, CESC, HNSC, SKCM and UCEC, in which APBB1IP expression levels are correlated with good prognosis. Since APBB1IP expression is related to poor prognosis in LGG, STAD, and TGCT, we explore the clinical relevance of immune subsets using the “Survival” module in the TIMER database. Kaplan-Meier curves showed that patients with higher abundance of tumor-infiltrating B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophils and dendritic cells had shorter overall survival in LGG (Figure S2M). Similarly, in patients with STAD, high abundance of macrophages was associated with shorter survival time (Figure S2M), and the infiltration levels of macrophages and neutrophils in TGCT were correlated with shorter survival time (Figure S2M). These results indicated that APBB1IP might affect patient survival by interacting with tumor-infiltrating immune cells, but the anti-cancer and pro-cancer effects depend on the host environment and cancer types.
Most immune system components are implicated in the initiation and progression of melanoma [30, 31]. To further determine the correlation between APBB1IP expression levels and various subsets of infiltrating immune cells in SKCM, we analyzed the correlation between APBB1IP and immune cell markers comprising subsets of T cells, B cells, monocytes, M1 and M2 macrophages, neutrophils, NK cells, and dendritic cells (DCs) in SKCM and LGG, in which APBB1IP expression was correlated with a poor prognosis. As shown in Table 1 and Figure 2B, after adjustments for tumor purity, APBB1IP expression was significantly correlated with most immune cell markers in SKCM, in addition to several markers of M1 macrophages and neutrophils. By contrast, APBB1IP was not significantly correlated with gene markers of CD8+ T cells and NK cells in LGG (Figure 2B, 2C). CD8+ T cells constitute the majority of TILs and directly induce cell death in tumors. Thus, a large number of CD8+ T cells in the tumor environment are considered to be associated with a favorable prognosis [10, 32, 33]. It is also well known that NK cells have spontaneous killing activity against tumor cells [34]. These findings could partly explain why APBB1IP is related to a bad prognosis in LGG.
Correlation between APBB1IP and markers of various immune cells in the Tumor Immune Estimation Resource (TIMER)
| Description | Gene Markers | SKCM | LGG | ||||||
|---|---|---|---|---|---|---|---|---|---|
| None | Purity | None | Purity | ||||||
| Cor | P | Cor | P | Cor | P | Cor | P | ||
| CD8+ T cell | CD8A | 0.8560732 | *** | 0.7694524 | *** | 0.1797299 | *** | 0.0526211 | 0.2508652 |
| CD8B | 0.8331593 | *** | 0.7260344 | *** | 0.1783245 | *** | 0.0817846 | 0.0740368 | |
| T cell (general) | CD3D | 0.8765923 | *** | 0.7843417 | *** | 0.3926254 | *** | 0.341427 | *** |
| CD3E | 0.8909953 | *** | 0.81022 | *** | 0.417972 | *** | 0.3857384 | *** | |
| CD2 | 0.8975331 | *** | 0.8229912 | *** | 0.4173085 | *** | 0.3923904 | *** | |
| B cell | CD19 | 0.6846681 | *** | 0.5602065 | *** | 0.3834718 | *** | 0.3352988 | *** |
| CD79A | 0.724277 | *** | 0.5894023 | *** | 0.324637 | *** | 0.3542463 | *** | |
| Monocyte | CD86 | 0.9296171 | *** | 0.8931612 | *** | 0.9074751 | *** | 0.8943717 | *** |
| CD115 (CSF1R) | 0.905323 | *** | 0.867535 | *** | 0.9069494 | *** | 0.8913472 | *** | |
| TAM | CCL2 | 0.6663165 | *** | 0.5284483 | *** | 0.4837392 | *** | 0.4375916 | *** |
| CD68 | 0.5852682 | *** | 0.450489 | *** | 0.829085 | *** | 0.8148534 | *** | |
| IL10 | 0.716315 | *** | 0.614492 | *** | 0.6101679 | *** | 0.5730547 | *** | |
| M1 Macrophage | INOS (NOS2) | 0.0510847 | 0.268534 | 0.0559468 | 0.232609 | -0.1511358 | *** | -0.1781116 | *** |
| IRF5 | 0.7086537 | *** | 0.5635679 | *** | 0.8953138 | *** | 0.8761291 | *** | |
| COX2 (PTGS2) | 0.0642153 | 0.164114 | -0.0220597 | 0.638106 | 0.1411687 | ** | 0.070951 | 0.1213558 | |
| M2 Macrophage | CD163 | 0.7727474 | *** | 0.7078413 | *** | 0.3657231 | *** | 0.3589378 | *** |
| VSIG4 | 0.7660752 | *** | 0.7124946 | *** | 0.7985103 | *** | 0.7744972 | *** | |
| MS4A4A | 0.8297304 | *** | 0.7714602 | *** | 0.5797796 | *** | 0.5803033 | *** | |
| Neutrophils | CD66b (CEACAM8) | -0.0298919 | 0.5175338 | 0.0014881 | 0.9746913 | 0.0356518 | 0.4190064 | 0.0322264 | 0.4821109 |
| CD11b (ITGAM) | 0.7985079 | *** | 0.7504803 | *** | 0.9015692 | *** | 0.8829104 | *** | |
| CCR7 | 0.7806993 | *** | 0.6267265 | *** | 0.2095446 | *** | 0.1908896 | *** | |
| Natural killer cell | KIR2DL1 | 0.3978069 | *** | 0.2549098 | *** | 0.018379 | 0.6770348 | 0.0617757 | 0.1775405 |
| KIR2DL3 | 0.5712978 | *** | 0.4038145 | *** | 0.1169197 | ** | 0.1191511 | ** | |
| KIR2DL4 | 0.6744853 | *** | 0.5261334 | *** | 0.4197599 | *** | 0.4394195 | *** | |
| KIR3DL1 | 0.53491 | *** | 0.3765632 | *** | -0.0278331 | 0.5281487 | -0.0256187 | 0.5763447 | |
| KIR3DL2 | 0.6360165 | *** | 0.4703593 | *** | 0.160454 | *** | 0.1668523 | *** | |
| KIR3DL3 | 0.1615421 | *** | 0.0930881 | *** | 0.0763603 | 0.0831139 | 0.0825424 | 0.071389 | |
| KIR2DS4 | 0.4411108 | *** | 0.3118604 | *** | 0.1341571 | ** | 0.1188675 | ** | |
| KLRK1 (NKG2D) | 0.7824953 | *** | 0.6765392 | *** | -0.0405603 | 0.357716 | 0.0365504 | 0.4252862 | |
| NCR1 (NKp46) | 0.6462555 | *** | 0.5564235 | *** | 0.1158136 | ** | 0.1606459 | *** | |
| NCR2 (NKp44) | 0.3058505 | *** | 0.2514162 | *** | 0.0304199 | 0.490514 | 0.0647637 | 0.1574441 | |
| NCR3 (NKp30) | 0.8255278 | *** | 0.7169654 | *** | 0.172838 | *** | 0.1768626 | *** | |
| Dendritic cell | HLA-DPB1 | 0.868107 | *** | 0.7793636 | *** | 0.6505592 | *** | 0.6235519 | *** |
| HLA-DQB1 | 0.8030186 | *** | 0.6833435 | *** | 0.4962863 | *** | 0.4617414 | *** | |
| HLA-DRA | 0.8805606 | *** | 0.8017141 | *** | 0.7050379 | *** | 0.6814707 | *** | |
| HLA-DPA1 | 0.8493377 | *** | 0.7661858 | *** | 0.6513003 | *** | 0.6270917 | *** | |
| BDCA1 (CD1C) | 0.6545897 | *** | 0.4916537 | *** | 0.3206443 | *** | 0.3254187 | *** | |
| BDCA4 (NRP1) | 0.4935402 | *** | 0.4514923 | *** | 0.1058552 | * | 0.1603316 | *** | |
| CD11c (ITGAX) | 0.6902283 | *** | 0.5453348 | *** | 0.7899023 | *** | 0.7551692 | *** | |
| Th1 | T-bet (TBX21) | 0.8701187 | *** | 0.7846811 | *** | 0.2129897 | *** | 0.2334805 | *** |
| STAT4 | 0.7799773 | *** | 0.671695 | *** | -0.1770701 | *** | -0.2653241 | *** | |
| STAT1 | 0.643486 | *** | 0.5645606 | *** | 0.3297585 | *** | 0.3193543 | *** | |
| IFNγ (IFNG) | 0.7535284 | *** | 0.6265012 | *** | 0.2479673 | *** | 0.2208263 | *** | |
| TNFα (TNF) | 0.6784416 | *** | 0.5088296 | *** | 0.3810268 | *** | 0.3364843 | *** | |
| Th2 | GATA3 | 0.7460884 | *** | 0.5580142 | *** | 0.3701424 | *** | 0.3215647 | *** |
| STAT6 | 0.064225 | 0.1640502 | 0.120339 | * | 0.5565766 | *** | 0.4636034 | *** | |
| STAT5A | 0.2495179 | *** | 0.3128001 | *** | 0.7509945 | *** | 0.7019067 | *** | |
| IL13 | 0.2016526 | *** | 0.1185491 | * | -0.0110191 | 0.8028152 | -0.0236849 | 0.6054741 | |
| Tfh | BCL6 | 0.3600216 | *** | 0.3065975 | *** | 0.203324 | *** | 0.2535651 | *** |
| IL21 | 0.5664735 | *** | 0.4559844 | *** | 0.1068566 | * | 0.098485 | * | |
| Th17 | STAT3 | 0.3361749 | *** | 0.3549276 | *** | 0.4724055 | *** | 0.5021622 | *** |
| IL17A | -0.0944109 | * | -0.191403 | *** | -0.0189334 | 0.6678661 | -0.0356259 | 0.4370972 | |
| Treg | FOXP3 | 0.7678603 | *** | 0.6220553 | *** | -0.1794219 | *** | -0.1521192 | *** |
| CCR8 | 0.7853676 | *** | 0.6941604 | *** | 0.1045328 | * | 0.0986246 | * | |
| STAT5B | 0.3224104 | *** | 0.4384412 | *** | -0.0256525 | 0.5609735 | 0.0785814 | 0.0861259 | |
| TGF β (TGFB1) | 0.5557131 | *** | 0.447552 | *** | 0.8019722 | *** | 0.7831079 | *** | |
| T cell exhaustion | PD1 (PDCD1) | 0.8308004 | *** | 0.7229568 | *** | 0.3971191 | *** | 0.3693027 | *** |
| CTLA4 | 0.557273 | *** | 0.390242 | *** | 0.3747752 | *** | 0.3282445 | *** | |
| LAG3 | 0.8049163 | *** | 0.6924997 | *** | 0.1894906 | *** | 0.2319562 | *** | |
| TIM3 (HAVCR2) | 0.9274436 | *** | 0.8854098 | *** | 0.9365782 | *** | 0.9274279 | *** | |
Immune Factors and Functional Analysis
To clarify the mechanisms underlying the involvement of APBB1IP in the enhancement of immune-cell infiltration, we calculated the Spearman correlations of APBB1IP expression with immune factors in the TISIDB database, including immunoinhibitory and immunostimulatory factors. These results are presented as heat maps in Figure 3A, and 3B. The results indicated that APBB1IP expression is positively correlated with both immunoinhibitory and immunostimulatory factors in the majority of cancers. We chose a correlation coefficient > 0.5 and FDR> 0.001 as cut-off criteria. According to this analysis, APBB1IP was not significantly correlated with majority immune factors in CHOL, DLBC, KIRP, LAML, MESO, PCPG and THYM. Especially in the latter, most immune factors showed a negative correlation with APBB1IP, which was opposite to other cancer types, suggesting that the immune microenvironment of THYM is different from other cancers. Accordingly the mechanism by which APBB1IP affects the prognosis of THYM might be different.
Immune Factors and Functional Analysis. (A) The correlation between the expression of APBB1IP and immunostimulatory factors. (B) The correlation between the expression of APBB1IP and immunoinhibitory factors. (C) GO analysis of significantly correlated immune factors. (D) KEGG pathway analysis of correlated immune factors.
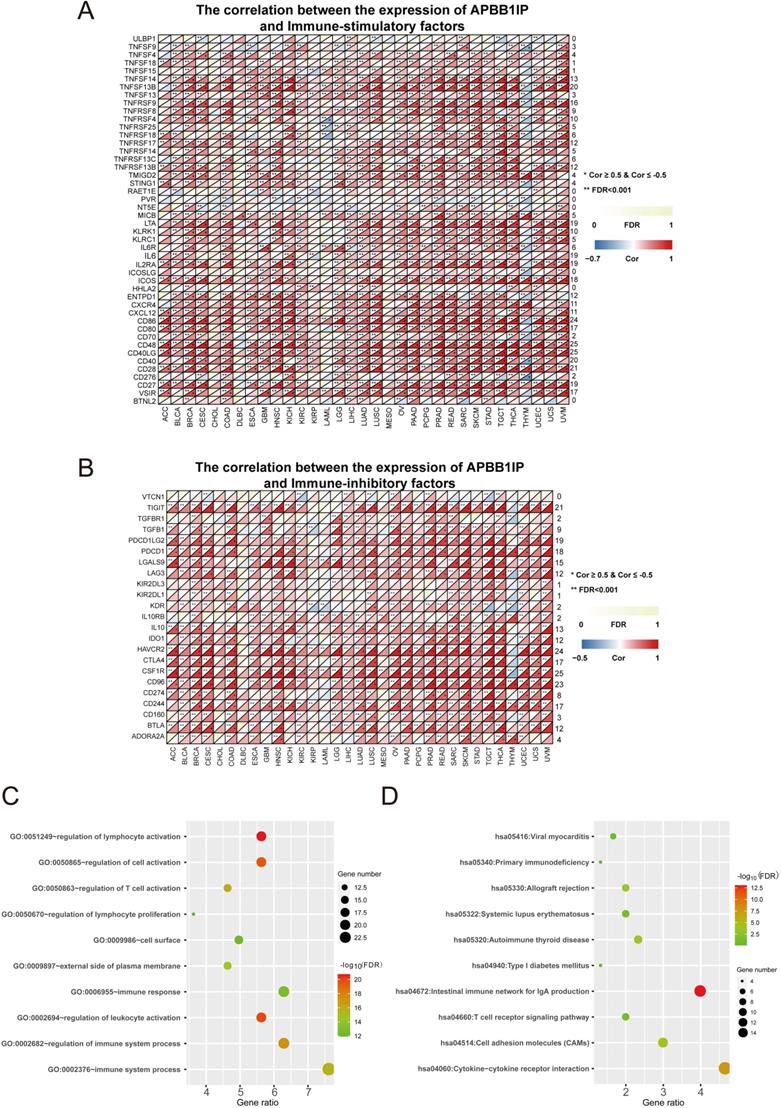
Next, 35 genes (Table S3) that were significantly associated with APBB1IP expression according to the cut-off standard in greater than or equal to 15 cancers, were selected as candidate genes for GO and KEGG pathway analysis in DAVID (https://david.ncifcrf.gov/summary.jsp). Figure 3C shows the top 10 most highly enriched GO items. Specifically, the immune factors were mainly enriched in biological processes (BPs) related to the regulation of lymphocyte activation, regulation of cell activation, regulation of T cell activation, regulation of lymphocyte proliferation, immune response, regulation of leukocyte activation, regulation of immune system processes, and immune system processes. In terms of function, the immune factors were predicted to be associated with the cell surface and the external side of plasma membrane. More specific information is listed in Table S4. Additionally, the most significantly enriched KEGG pathways are displayed in Figure 3D. The immune factors were enriched in the categories of viral myocarditis, primary immunodeficiency, allograft rejection, systemic lupus erythematosus, autoimmune thyroid disease, type I diabetes mellitus, intestinal immune network for IgA production, T cell receptor signaling pathway, cell adhesion molecules (CAMs) and cytokine-cytokine receptor interaction. The detailed information is shown in Table S5.
PPI Network Establishment and Transcription and Epigenetics Analysis of APBB1IP in Pan-cancer analysis
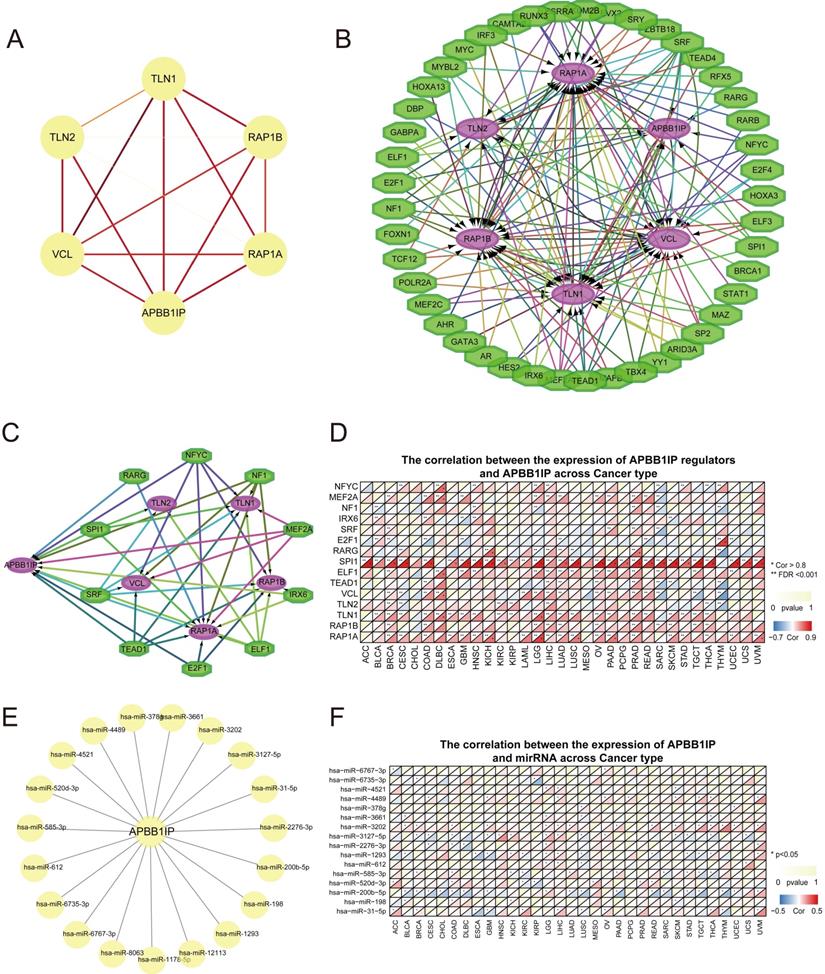
Protein-protein interaction (PPI) analysis can reflect the molecular mechanisms of physiological and pathological changes that drive cancer progression. The PPI network of APBB1IP and its protein partners was constructed using the STRING database (https://string-db.org/) and Cytoscape software (Figure 4A). The network contained 6 nodes and 15 edges, with the 5 predicted proteins interacting with APBB1IP including members of the RAS oncogene family (RAP1A and RAP1B), talin 1 (TLN1), talin 2 (TLN2), and vinculin (VCL). These genes were further considered for downstream analysis of the APBB1IP network. Subsequently, a TF regulatory network with 45 nodes was predicted using the iRegulon Cytoscape plugin (Figure 4B). Among these TFs, NFYC, MEF2A, NF1, IRX6, SRF, E2F1, RARG, SPI1, ELF1 and TEAD1 were identified as targeting APBB1IP. Additionally, they were also predicted to regulate the interacting proteins (Figure 4C). Subsequently, we explored the Spearman correlations of APBB1IP and these transcriptional regulators together with the protein partners of APBB1IP, as shown in Figure 4D. Among the transcriptional regulators, the expression of SPI1 was significantly positively associated with APBB1IP in the majority of cancers. Furthermore, it was found to target TLN1, TLN2 and RAP1A as well. SPI1/PU.1 is a member of the ETS family that is critical for specifying cell fate and proper hematopoietic differentiation [35]. SPI1 plays a crucial part in the self-renewal of hematopoietic stem cells (HSCs) as well as in myeloid and B lymphoid differentiation [36, 37]. Based on these findings, SPI1 might function by regulating the transcription of APBB1IP and other associated proteins. Subsequently, using miRWalk (including miRBase, TargetScan, miRDB, and miRTarBase), we predicted 19 miRNAs that could potentially target the 3′UTR of APBB1IP (Figure 4E). Using STARBASE v3.0 with p < 0.05 as screening criteria, we investigated the Spearman correlation between miRNAs and the expression of APBB1IP in 32 kinds of cancers from TCGA (Figure 4F). The hsa-miR-200b-5p was predicted to have a significant inhibitory effect on APBB1IP in BLCA, BRCA, CESC, CHOL, COAD, DLBC, GBM, LUAD, LUSC, PAAD, READ, SARC, STAD, TGCT and THYM.
Establishment of a PPI Network and Pan-cancer Analysis of Transcription and Epigenetic Factors of APBB1IP. (A) PPI network of APBB1IP and its interacting proteins. The PPI consists of 6 nodes and 15 edges, with an average node degree of 5 and average local clustering coefficient of 1 at a PPI enrichment p-value of 3.69 × 10-4. The color and thickness of the solid line represents the strength of the relationship. (B) TF-target regulatory network of APBB1IP and its interacting proteins. Green polygons represent TFs, the purple ellipses represent the target proteins. (C) The TFs that target APBB1IP. Green polygons represents TFs, the purple ellipses represent the target proteins. (D) Correlation between the expression of APBB1IP regulators and APBB1IP across cancer types. (E) MiRNAs that target APBB1IP. (F) The correlation between the expression of APBB1IP and miRNAs across cancer types.
Analysis of Genetic Mutations and Methylation of APBB1IP
To further understand how APBB1IP expression is altered across different cancers, we explored the Single Nucleotide Variation (SNV) profile of APBB1IP and its protein partners using GSCALite [28] (Figure 5A). All of the 677 analyzed tumors showed at least 1 mutation. TLN1 had the highest SNV frequency (43%) among the analyzed tumors, followed by TLN2 (35%). For APBB1IP, the SNV frequency was 20%, at the cancer level. Additionally APBB1IP-associated SKCM exhibited the highest number of mutations (32), followed by LUSC (22) and UCEC (21). The most frequent DNA alterations of these 6 genes in the pan-cancer analysis were missense mutations. CNVs were reported as frequent pathogenic events in cancers, which could contribute to increased DNA instability and occurrence of genomic imbalance [38]. Figure 5B shows the heterozygous/homozygous CNV status of each gene in each cancer. Next, the Pearson correlation was between gene expression and CNV was analyzed in different cancers to identify the genes significantly affected by CNV. As shown in Figure 5C, the expression of TLN1 and TLN2 was positively associated with CNV in most cancers, except for APBB1IP, RAP1B, and RAP1A.
Genetic Mutation and Methylation Analysis of APBB1IP. (A) SNV frequency of APBB1IP and its protein partners. Each gray vertical bar represents a patient. The side and top column diagrams show the numbers of variants in each sample or each gene. (B) Pan-cancer analysis of heterozygous/homozygous CNV of APBB1IP and its protein partners. Hete Amp: heterozygous amplification; Hete Del: heterozygous deletion; Homo Amp: homozygous amplification; Homo Del: homozygous deletion; None: no CNV. (C) Pan-cancer analysis of the CNV correlation with mRNA of APBB1IP and its protein partners. Genes whose mRNA expression significantly correlates with CNV percentage (FDR<=0.05) are shown in the figure. Blue bubbles represent a negative correlation, and red bubbles represent positive correlation (genes having a high frequency of CNV are shown in a deeper color, indicating a higher correlation. The size of each bubble represents statistical significance. (D) Bubble map of the differential methylation of APBB1IP and its protein partner s between normal and cancer samples in TCGA. The significance of differences was analyzed using Student's t-test. Blue dots represent down-regulation of methylation in tumors, and red dots represent up-regulation of methylation in tumors; the darker the color, the greater the difference. The size of the bubble represents statistical significance. (E) Correlation between methylation, APBB1IP and its protein partners in cancer samples from TCGA. The data were subjected to Person correlation analysis. Blue bubbles indicate that the gene methylation level is up-regulated and gene expression is down-regulated. Red bubbles indicate that the gene methylation level and gene expression are up-regulated. The darker the color, the higher the correlation. The size of the bubble represents the statistical significance.
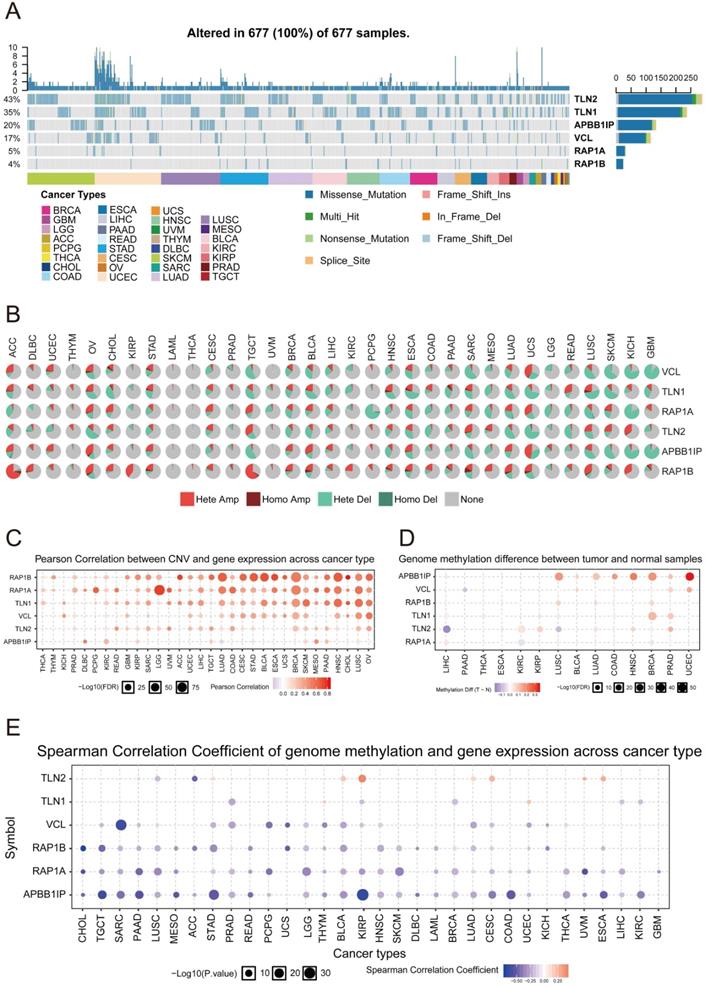
Since DNA methylation is involved in gene regulation and cell differentiation [39], we next explored whether methylation is involved in the regulation of APBB1IP. We found that the methylation of APBB1IP was significantly up-regulated in LUSC, BLCA, COAD, HNSC, BRCA, PAAD, and UCEC, while co-methylation patterns of APBB1IP and the protein partners of APBB1IP were not observed (Figure 5D). In addition, the expression of APBB1IP and protein partners was mainly negatively correlated with methylation, with only a few positive correlations (Figure 5E), providing clues for the deregulation of APBB1IP.
Cox Progression Analysis and Identification of a Prognostic Signature in Lung Cancer
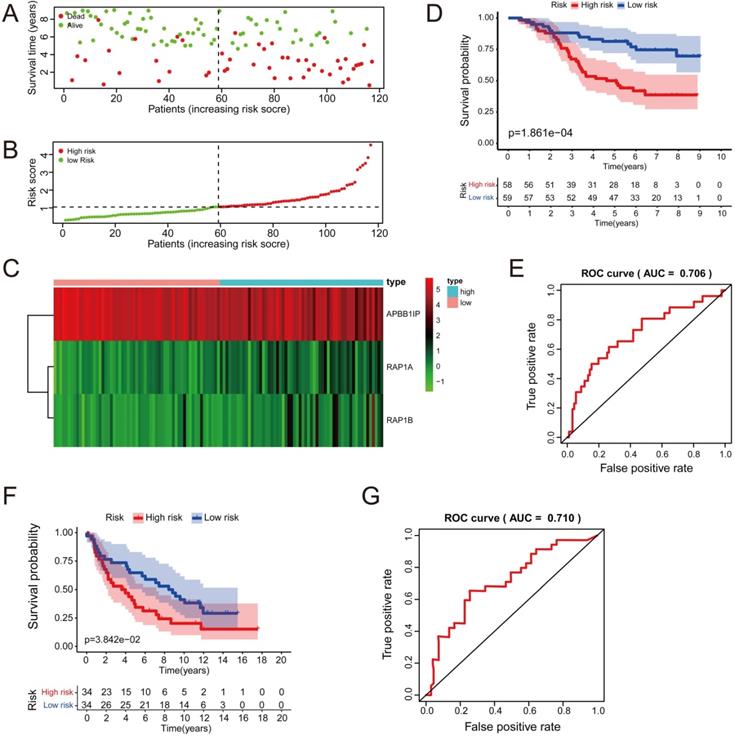
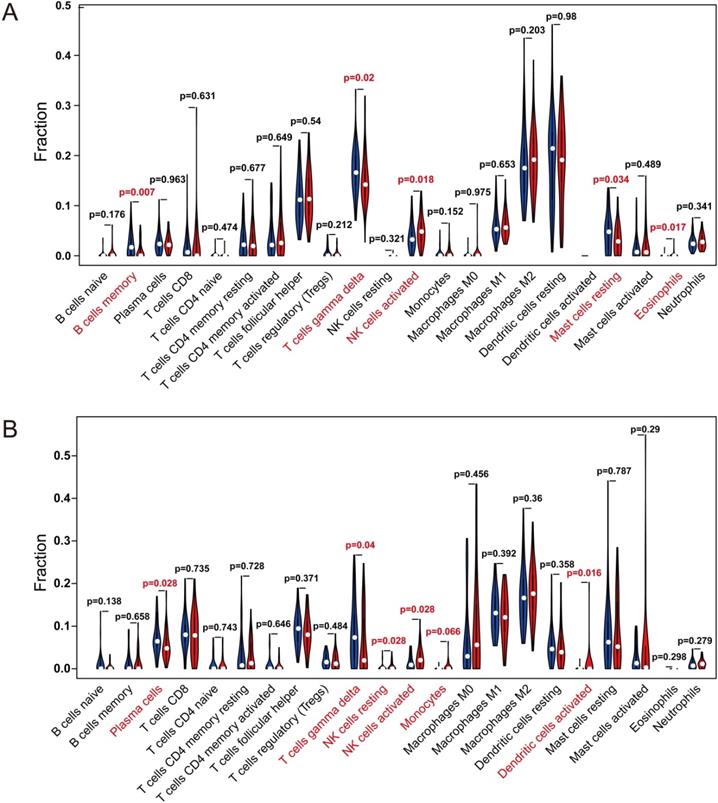
Multivariate Cox analysis was performed for APBB1IP, RAP1A, RAP1B, TLN1, TLN2 and VCL in GSE13213. Then, APBB1IP, RAP1B and RAP1B were finally selected to establish a prognostic model. The model was described using the formula risk score = (-0.85392 × expression level of APBB1IP)+(0.422303 × expression level of RAP1A) +(0.377711 × expression level of RAP1B). All three genes were prognostic for increased risk, including APBB1IP (HR = 0.43, 95% CI = 0.25 to 0.73, p value= 0.002), RAP1A (HR = 1.53, 95% CI = 1.00 to 2.33, p value= 0.049), and RAP1B (HR = 1.46, 95% CI = 1.08 to 1.96, p value= 0.013). Multivariate Cox regression was used to calculate regression coefficients. Risk scores were based on gene expression levels multiplied by the corresponding regression coefficients. Subsequently, 117 lung cancer samples were divided into a high-risk group (n = 59) and a low-risk group (n = 58) based on the median risk score (Figure 6B). The survival status and survival time in the model group are shown in Figure 6A, while Figure 6C shows a gene expression heatmap of the low- and high-risk groups. Survival analysis indicated that patients in the high-risk group showed markedly poorer overall survival than those in the low-risk group (p=1.861e-04; Figure 6D). According to the ROC curve of 5-year OS (Figure 6E), an area under curve (AUC) value of 0.706 (>0.7), indicating that this prognostic model exhibited good sensitivity and specificity. To identify the relationship between risk score model and clinicopathological characteristics in lung cancer patients, we further analyzed the risk score level in lung cancer patients at different clinical stages. As revealed in Table 2, risk score based on this prognostic model was significantly associated with T stage, N stage, TNM stage and relapse (both P < 0.05). Subsequently, the GSE29016 dataset was used as the testing cohort. The Kaplan-Meier analysis shown in Figure 6F indicated that the high-risk group had poorer overall survival (p=3.842e-02). Time-dependent ROC curves showed that the model had good accuracy with a value of 0.710 in 5 years (Figure 6G). Moreover, analysis of the correlation of risk stratification with clinicopathological data for patients with lung cancer also showed that TNM stage was significant associated with risk stratification in the testing cohort (Table S6). Next, the relationships between the risk score model and immune cell infiltration was investigated. As shown in Figure 7A, NK cells activation and eosinophil infiltration were positively correlated with the risk score. However, negative correlations were observed between the risk score and memory B cells, gamma delta T cells and resting mast cells. Similarly, the high- and low-risk groups also showed differential immune cell abundance in GSE29106 (Figure 7B).
Association between the clinicopathologic parameters and the risk score levels in GSE13213
| Clinical variables | Total | Risk score levels | p value | |
|---|---|---|---|---|
| Low | High | |||
| Gender | 0.228 | |||
| Male | 60 | 27 | 33 | |
| Female | 57 | 32 | 25 | |
| Age (years) | 0.779 | |||
| ≤60 | ||||
| >60 | ||||
| Smoking | 0.164 | |||
| NO | 56 | 32 | 24 | |
| YES | 61 | 27 | 34 | |
| T Stage | ||||
| T1+T2 | 104 | 56 | 48 | 0.036 |
| T3+T4 | 13 | 3 | 10 | |
| N Stage | 0.039 | |||
| N0 | 87 | 48 | 39 | |
| >N0 | 30 | 10 | 20 | |
| TNM Stage | 0.048 | |||
| I+II | 92 | 50 | 42 | |
| III+IV | 25 | 8 | 17 | |
| Relapse | 0.002 | |||
| NO | 59 | 38 | 21 | |
| YES | 58 | 21 | 37 | |
| EGFR status | 0.619 | |||
| WT | 72 | 35 | 37 | |
| MUT | 45 | 24 | 21 | |
| KRAS status | 0.755 | |||
| WT | 102 | 52 | 50 | |
| MUT | 15 | 7 | 8 | |
Cox Regression Analysis and Identification of a Prognostic Signature in Lung Cancer. (A) The risk score distribution of HCC patients in the GSE13213 dataset. (B) Patients' survival status distribution. (C) A heatmap of APBB1IP and RAP1B in low- and high-risk groups. (D) The survival curves of GSE13213 patients in low- and high-risk groups. (E) Receiver operating characteristic curve (ROC) analysis predicting overall survival using the risk score in GSE13213. (F) The survival curves of GSE29016 patients in low- and high-risk groups. (G) Receiver operating characteristic curve (ROC) analysis predicting overall survival using the risk score in GSE29016.
The Relationships Between the Risk Score Model and Immune Cell Infiltration. (A, B) Violin plot showing the relationship between the risk score with the abundances of different types of infiltrating immune cells. Red color represents the high-risk group while blue color represents the low-risk group. Differential abundance of immune cell types was observed between the high and low-risk groups. (A) GSE13213. (B) GSE29016.
Discussion
APBB1IP contains Ras association (RA) and pleckstrin homology (PH) domains and proline-rich regions, which are defining features of the Mig-10/RIAM/Lamellipodin (MRL) family of adapter proteins. It was identified as a Rap1-binding protein important for integrin-mediated migration and activation of leukocytes [2]. In spite of the important roles of APBB1IP in the immune system, APBB1IP has not been well-studied in immuno-oncology. Here, we conducted a pan-cancer analysis of the expression profile and prognostic significance of APBB1IP and revealed its potential role in tumor immunology via bioinformatics analysis.
In this study, the expression levels of APBB1IP were examined and the prognostic landscape in pan-cancer analysis were visualized using independent datasets in Oncomine and Kaplan-Meier plotter, as well as TCGA data for 33 types of cancer. According to the combined results from the Oncomine and TCGA data, APBB1IP was highly expressed in KIRC, KIRP, STAD, sarcoma, as well as brain and CNS cancers compared to corresponding normal tissues, while it had lower expression levels in BLCA, BRCA, COAD, LUAD, LUSC, PAAD, PCCG, READ, liver cancer and leukemia. From the comprehensive TCGA and Kaplan-Meier plotter data, we found consistent prognostic correlations of APBB1IP. Specifically, decreased APBB1IP expression was correlated with poor prognosis in most tumor types (CESC, HNSC, KIRP, THYM, UCEC). However, STAD was an exception where high levels of APBB1IP expression indicated a poorer prognosis. The discrepancies in APBB1IP levels and prognosis in different cancer types in different databases might be a reflection of different data collection approaches and underlying mechanisms pertinent to different biological properties. Notably, APBB1IP was up‐regulated in KIRP, but high APBB1IP expression indicated a better prognosis in this cancer. This counterintuitive finding can be explained by the small sample size of the normal group (n = 32), which might have led to unreliable data on the expression of APBB1IP in KIRP. Hence, in future studies, researchers need to collect more samples to verify the expression of APBB1IP in KIRP. In addition, APBB1IP might play distinct roles in the initiation and progression of KIRP. To confirm this, further studies are needed to explore the expression of APBB1IP in tumors with different stages or grades and analyze the relationship between APBB1IP expression and survival of KIRP patients with different tumor stages or grades.
Another important finding of this study is that APBB1IP expression is correlated with diverse immune-cell infiltration levels in most cancer types (Figure 2A). Moreover, the correlation between APBB1IP and the expression of immune regulators indicate a role of APBB1IP in regulating tumor immunology in different cancers (Figure 3A). Interestingly, although the prognostic implications of APBB1IP were not the same in different cancer types, APBB1IP expression was consistently positively correlated with the immune-cell infiltration levels in these cancers. Accordingly, tumor infiltration by the same immune cells may have a different effect on the prognosis in different cancer types. For example, high infiltration of B cells, CD8+ T cells, neutrophils and dendritic cells is associated with poor prognosis in LGG, but also with better prognosis in SKCM (Figure S2M). Furthermore, multiple studies have reported differences in the correlation between intratumoral immune-cell activity and survival across different cancer types [40-44]. Notably, the strongest positive correlation was observed between the expression of specific markers for CD8+ T cells and NK cells and APBB1IP expression in SKCM; while the correlation in LGG was lower (Figure 2B-C). CD8+ T cells and NK cells are well known as effector cells in the tumor microenvironment via their cytolytic activity [45-48]. This might be another reason for the different prognostic implications of APBB1IP in SKCM and LGG.
RAP1A, RAP1B, TLN1, TLN2, and VCL were predicted to interact with APBB1IP. Talin1 and talin2 are large (270 kDa) cytoplasmic adapter proteins (Figure 4A) [49, 50]. In resting cells, a large portion of talin proteins resides in the cytoplasm in a closed, auto-inhibited conformation [51, 52]. Following stimulation, talin is efficiently recruited to the plasma membrane and transformed from its auto-inhibitory conformation to trigger integrin activation. Talin-mediated integrin activation is highly dependent on the membrane-anchored small GTPase Rap1 protein family [53, 54]. It was suggested that talin membrane recruitment is triggered through APBB1IP, which binds the talin rod domain and thereby links it to the plasma membrane [55, 56]. This Rap1-RIAM-talin pathway is crucial for leukocyte β2 integrin activation [6, 7]. Vinculin (VCL) is a key adaptor molecule that links adhesion complexes to actin filaments in integrin-based cell extracellular matrix (ECM) adhesions or cell-cell junctions [57]. APBB1IP was reported to promote the migration and invasion of melanoma cells [7], and we speculated that APBB1IP might interact with VCL to achieve this function. According to our results, high APBB1IP expression indicated poor prognosis in SKCM, and we deduced that the promoting effect of APBB1IP on the migration ability of SKCM might not be associated with its prognosis, while the influence on immune infiltration might play a leading role.
In this study, we found that RAP1A/B and TLN1 expression is positively associated with APBB1IP (Figure 4D). Thus, we displayed the TFs which might target APBB1IP and its interacting partners (Figure 4B). We observed that TFs targeting APBB1IP could target its interacting partners at the same time (Figure 4C). In addition to for TFs, methylation and miRNAs can also regulate the protein expression levels. According to our results, the methylation levels of APBB1IP and RAP1A/B were negatively correlated with gene expression (Figure 5E). More specifically, APBB1IP methylation was higher in LUSC, BLCA, LUAD, COAD, BRCA, HNSC, PRAD, and UCEC tumor tissues than in normal control tissues (Figure 5D). In addition, miR-200b-5p was identified as possibly targeting APBB1IP and it was negatively associated with the expression of APBB1IP in most cancers.
Nevertheless, although we integrated information across multiple databases, this study still has some limitations. Since large-scale microarray and sequencing data were initially collected by analyzing tumor tissue information, the cell-level analysis of immune cell markers could have introduced systematic bias. To overcome this problem, future studies relying on methods with a higher resolution, such as single-cell RNA sequencing, should be performed [58, 59]. Next, we could not prove that APBB1IP affected patient survival through immune infiltration even though we found that APBB1IP expression was correlated with both immune cell infiltration and patient survival in cancers. Hence, future prospective studies are needed to explore the relationship between APBB1IP expression and immune infiltration in a cancer patient population. Finally, we only conducted a bioinformatic analysis of APBB1IP expression and patient survival across different databases, and further mechanistic studies on APBB1IP at the cellular and molecular levels could help clarify its functions in cancer initiation and progression.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the State Key Project on Infectious Diseases of China (2018ZX10723204-003), the National Nature Science Foundation of China (No's. 81874065, 81500565, 81874149, 81572427, and 81401997), the Hepato-Biliary-Pancreatic Malignant Tumor Investigation Fund of Chen Xiao-ping Foundation for the Development of Science and Technology of Hubei Province (CXPJJH11800001-2018356).
Authors' contribution
All authors searched the literature, designed the study, interpreted the findings and revised the manuscript. Qianyun Ge and Ganxun Li carried out data management and statistical analysis and drafted the manuscript. Jin Chen, Jia Song, Guanzhen Cai, Yi He, Xuewu Zhang and Huifang Liang helped with cohort identification and data management. Zeyang Ding and Bixiang Zhang performed project administration.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T. et al. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. The Journal of biological chemistry. 1997;272:32869-77
2. Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A. et al. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Developmental cell. 2004;7:585-95
3. Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA. et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Current biology: CB. 2006;16:1796-806
4. Medraño-Fernandez I, Reyes R, Olazabal I, Rodriguez E, Sanchez-Madrid F, Boussiotis VA. et al. RIAM (Rap1-interacting adaptor molecule) regulates complement-dependent phagocytosis. Cellular and molecular life sciences: CMLS. 2013;70:2395-410
5. Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends in cell biology. 2006;16:453-60
6. Klapproth S, Sperandio M, Pinheiro EM, Prünster M, Soehnlein O, Gertler FB. et al. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired β2 integrin function in mice. Blood. 2015;126:2704-12
7. Hernández-Varas P, Coló GP, Bartolomé RA, Paterson A, Medraño-Fernández I, Arellano-Sánchez N. et al. Rap1-GTP-interacting adaptor molecule (RIAM) protein controls invasion and growth of melanoma cells. The Journal of biological chemistry. 2011;286:18492-504
8. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782-95
9. Lee KH, Kim EY, Yun JS, Park YL, Do SI. The prognostic and predictive value of tumor-infiltrating lymphocytes and hematologic parameters in patients with breast cancer. BMC Cancer. 2018;18:938
10. Lee N, Zakka LR, Mihm MC Jr, Schatton T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology. 2016;48:177-87
11. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-9
12. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450-61
13. Goldman MJ, Craft B. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675-8
14. Tomida S, Takeuchi T, Shimada Y, Arima C, Matsuo K, Mitsudomi T. et al. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:2793-9
15. Staaf J, Jönsson G, Jönsson M, Karlsson A, Isaksson S, Salomonsson A. et al. Relation between smoking history and gene expression profiles in lung adenocarcinomas. BMC medical genomics. 2012;5:22
16. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB. et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia (New York, NY). 2007;9:166-80
17. Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L. et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast cancer research and treatment. 2016;160:439-46
18. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS. et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer research. 2017;77:e108-e10
19. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J. et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome biology. 2016;17:174
20. Ru B, Wong CN, Tong Y, Zhong JY, Zhong SSW, Wu WC. et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics (Oxford, England). 2019;35:4200-2
21. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25-9
22. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research. 1999;27:29-34
23. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
24. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447-52
25. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498-504
26. Dweep H, Sticht C, Pandey P, Gretz N. miRWalk-database: prediction of possible miRNA binding sites by "walking" the genes of three genomes. Journal of biomedical informatics. 2011;44:839-47
27. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids research. 2014;42:D92-7
28. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics (Oxford, England). 2018;34:3771-2
29. Moore OS Jr, Foote FW Jr. The relatively favorable prognosis of medullary carcinoma of the breast. Cancer. 1949;2:635-42
30. Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Current opinion in immunology. 2013;25:261-7
31. Buonaguro FM, Pauza CD, Tornesello ML. Cancer Diagnostic and Predictive Biomarkers. BioMed Research Int. 2016. 2017;2017:7362721
32. Reissfelder C, Stamova S, Gossmann C, Braun M, Bonertz A, Walliczek U. et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. The Journal of clinical investigation. 2015;125:739-51
33. Seo AN, Lee HJ, Kim EJ, Kim HJ, Jang MH, Lee HE. et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. British journal of cancer. 2013;109:2705-13
34. Konjević GM, Vuletić AM, Mirjačić Martinović KM, Larsen AK, Jurišić VB. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine. 2019;117:30-40
35. DeKoter RP, Kamath MB, Houston IB. Analysis of concentration-dependent functions of PU.1 in hematopoiesis using mouse models. Blood cells, molecules & diseases. 2007;39:316-20
36. Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S. et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590-600
37. Staber PB, Zhang P, Ye M, Welner RS, Nombela-Arrieta C, Bach C. et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Molecular cell. 2013;49:934-46
38. Shlien A, Tabori U, Marshall CR, Pienkowska M, Feuk L, Novokmet A. et al. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11264-9
39. Compere SJ, Palmiter RD. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell. 1981;25:233-40
40. Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D. et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell reports. 2017;18:248-62
41. Danaher P, Warren S. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer. 2018;6:63
42. Tamborero D, Rubio-Perez C, Muiños F, Sabarinathan R, Piulats JM, Muntasell A. et al. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:3717-28
43. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH. et al. The Immune Landscape of Cancer. Immunity. 2018;48:812-30.e14
44. Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer research. 2017;77:1271-82
45. Chiossone L, Dumas PY. Natural killer cells and other innate lymphoid cells in cancer. 2018; 18: 671-88.
46. Church SE, Galon J. Regulation of CTL Infiltration Within the Tumor Microenvironment. Advances in experimental medicine and biology. 2017;1036:33-49
47. Martínez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:5047-56
48. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8⁺ T cells. Nature reviews Immunology. 2011;11:645-57
49. Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nature reviews Molecular cell biology. 2013;14:503-17
50. Klapholz B, Brown NH. Talin - the master of integrin adhesions. J of Cell Science. 2017;130:2435-46
51. Banno A, Goult BT, Lee H, Bate N, Critchley DR, Ginsberg MH. Subcellular localization of talin is regulated by inter-domain interactions. The Journal of biological chemistry. 2012;287:13799-812
52. Goult BT, Xu XP, Gingras AR, Swift M, Patel B, Bate N. et al. Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: implications for talin activation. Journal of structural biology. 2013;184:21-32
53. Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Current biology: CB. 2000;10:974-8
54. Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nature immunology. 2002;3:251-8
55. Lagarrigue F, Kim C. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood. 2016;128:479-87
56. Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. The Journal of biological chemistry. 2009;284:5119-27
57. Atherton P, Stutchbury B, Jethwa D, Ballestrem C. Mechanosensitive components of integrin adhesions: Role of vinculin. Experimental cell research. 2016;343:21-7
58. Giladi A, Amit I. Single-Cell Genomics: A Stepping Stone for Future Immunology Discoveries. Cell. 2018;172:14-21
59. Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nature reviews Immunology. 2018;18:35-45
Author contact
![]() Corresponding authors: Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang avenue 1095, Wuhan, China. E-mail address: zydingtjmu.edu.cn (Z. Ding) and bixiangzhangedu.cn (B. Zhang).
Corresponding authors: Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang avenue 1095, Wuhan, China. E-mail address: zydingtjmu.edu.cn (Z. Ding) and bixiangzhangedu.cn (B. Zhang).