Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(17):5187-5197. doi:10.7150/jca.41193 This issue Cite
Research Paper
Prognostic values of m6A RNA methylation regulators in differentiated Thyroid Carcinoma
Nizhen Xu*, Jian Chen, Gaofei He, Li Gao, Deguang Zhang ![]()
Department of Head and Neck surgery, Institute of Micro-Invasive Surgery of Zhejiang University, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, 3 East Qingchun Road, Hangzhou 310016, P.R. China.
*First author.
Received 2019-10-15; Accepted 2020-5-12; Published 2020-7-6
Abstract
N6-methyladenosine (m6A) is the most prevalent modification of RNA in mammals. m6A RNA methylation levels are dynamically regulated by m6A RNA methylation regulators. While increasing evidence has suggested that m6A RNA methylation is vital in the initiation and progression of human carcinoma, little is known about the expression and effect of m6A RNA methylation regulators in differentiated thyroid carcinoma (DTC). Herein, we demonstrate that most of the thirteen main m6A RNA methylation regulators are differentially expressed in DTC tissues and normal thyroid tissues. Based on consensus clustering of m6A RNA methylation regulators, DTC cases were divided into two subgroups (TC1 and TC2). Compared with the TC1 subgroup, the TC2 subgroup was associated with a poorer prognosis, older age, higher T grade, higher N grade and higher TNM stage. The results indicated that alteration of m6A RNA methylation regulators was closely related to DTC. We further established a risk signature of four m6A RNA methylation regulators that could evaluate prognosis and clinicopathological features in DTC. Finally, the results of the TCGA analysis were verified by other cohorts from Gene Expression Omnibus (GEO) database. In conclusion, m6A RNA methylation regulators play a crucial part in the progression of DTC and are potentially useful for evaluating the prognosis and providing potential novel insights into treatment strategies.
Keywords: thyroid carcinoma, m6A, RNA modification, prognosis, TCGA
Introduction
Thyroid carcinoma is the most common malignant endocrine tumor with an incidence that has remarkably increased in the past 10 years [1‑5]. Thyroid carcinoma ranks ninth in cancer incidence worldwide and accounts for 5.1% of the total estimated cancer burden in females in 2018 [6]. Differentiated thyroid carcinoma (DTC), which includes papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC), comprises more than 90% of all thyroid carcinomas [7, 8]. Although DTC generally presents indolent behavior and has a favorable prognosis, approximately 20%-30% of DTC patients develop recurrence or distant metastasis after primary treatment. Several cases have a poor response to conventional treatment, resulting in poor prognoses [9-11].
RNA modification have widely studied in the proliferation and metastasis of human carcinoma [12, 13]. The importance of messenger RNA (mRNA) plays in the post-transcriptional regulation of gene expression is well-established. In eukaryotes, N6-methyladenosine (m6A) is recognized as the most common internal chemical modification in mRNA, the process of which has been found to be dynamic and reversible [14-16]. During the processes of stem cell differentiation, embryo development, neural development or stress responses, m6A can regulate the key biological process of cells through modulating mRNA stability, splicing, intracellular distribution and translation [17-20]. m6A modification is dynamically regulated by methyl-transferases ('writers'), binding proteins ('readers'), and demethylases ('erasers'). The abundance, prevalence, and distribution of m6A are regulated by writers and erasers, while readers transform the m6A methylation information into a functional signal [21, 22]. Writers include WT1-associated protein (WTAP), methyltransferase like 3 (METTL3), methyltransferase like 14 (METTL14), RNA binding motif protein 15 (RBM15), zinc finger CCCH-type containing 13 (ZC3H13) and KIAA1429, and readers include YTH domain-containing 1 (YTHDC1), YTH domain-containing 1 (YTHDC2), YTH N6-methyl-adenosine RNA binding protein 1 (YTHDF1), YTH N6-methyladenosine RNA binding protein 2 (YTHDF2) and heterogeneous nuclear ribonucleoprotein C (HNRNPC). Erasers include fat mass- and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) [23-25]. Increasing evidence indicates that alterations of m6A regulatory genes are closely associated with obesity, infertility, immunological disease and neurological diseases. Recent studies have revealed that genetic changes and dysregulated expressions of m6A RNA methylation regulators play a crucial role in the progression of a variety of human cancers [26-28]. Although previous studies have found that m6A RNA methylation regulators were related to tumor progression in different types of cancers, the expression of m6A RNA methylation regulators as well as the prognosis value in DTC has not been completely explored.
The present study analyzed the expression of 13 m6A RNA regulators in DTC with RNA sequencing data from The Cancer Genome Atlas (TCGA) and evaluated the association between m6A RNA methylation regulators and clinicopathological characteristics in DTC patients. We further established a signature with four m6A RNA methylation regulators to predict the prognosis of DTC.
Materials and Methods
Data source
The RNA-seq transcriptome data and clinical information of DTC patients were obtained from the Genomic Data Commons Data Portal within TCGA (https://portal.gdc.cancer.gov/) in June 2019. First, the RNA‑seq data files were merged into a matrix file using the merge script of the Perl language (http://www.perl.org/). Gene names were converted from the Ensembl ID to the matrix of the gene symbol through the Ensembl database version 84 (http://asia.ensembl.org/index.html). The downloaded data included 509 thyroid carcinoma samples and 58 normal thyroid samples. The data of 13 m6A RNA modification regulators were extracted and analyzed. The R (version 3.6.0) package [29] edgeR version 3.27.6 (r-project.org/) was used to identify genes that were differentially expressed between tumor and normal thyroid samples, with a false discovery rate <0.05 and |log2 fold change| >2 set as the threshold. The gene expression level based on microarray data was calculated using R package limma (version 3.40.2; bioconductor.org/packages/release/bioc/html/limma.html) with robust multiarray average (RMA) correction.
Information of 13 m6A RNA methylation regulators and clinicopathological features
We excluded patients with incomplete clinicopathological parameters or those with missing prognostic follow-up data. A total of 425 DTC patients were enrolled in our study (Table 1). Information of 13 m6A RNA methylation regulators in DTC and normal thyroid were obtained from downloaded data, including 425 tumors and 57 normal samples. We systematically evaluated the association between clinicopathological features and the expression of the 13 m6A RNA methylation regulators in DTC.
Clinical features of 425 DTC patients
| Variables | DTC patients (N=425) | |
|---|---|---|
| n | % | |
| Gender | ||
| Male | 118 | 27.76 |
| Female | 307 | 72.24 |
| Age (year) | 0.689 | |
| <55 | 281 | 66.12 |
| ≥55 | 144 | 33.88 |
| TNM stage | ||
| Stage I | 236 | 55.53 |
| Stage II | 43 | 10.12 |
| Stage III | 99 | 23.29 |
| Stage IV | 47 | 11.06 |
| Focus type | ||
| Unifocal | 223 | 52.47 |
| Multifocal | 202 | 47.53 |
| Vital Status | ||
| Free | 360 | 84.71 |
| Recurrence | 65 | 15.29 |
DTC: Differentiated Thyroid Carcinoma.
Prognostic model
We clustered the DTC patients into different groups using R package ConsensusClusterPlus (version 1.49.0, resample rate of 80%, 50 iterations and Pearson correlation, bioconductor.org/packages/devel/bioc/html/ConsensusClusterPlus.html). To discover potential m6A RNA methylation regulators that affect the prognosis of DTC patients, least absolute shrinkage and selection operator (LASSO) Cox regression algorithm [30, 31] was applied using the R package survival (version 2.44; https://CRAN.R-project.org/view=Survival) and R package glmnet (version 2.0-18; https://CRAN.R-project.org/view=Glmnet) to identify optimal prognostic m6A RNA methylation regulators that impact progression-free survival (PFS) of DTC patients. An individual's risk score signature was established as follows:
Risk Score = ∑coefficient (GENEi) × expression (GENEi)
Here, GENEi is the identifier of the ith selected gene. The risk score signature was a measure of prognostic risk for each DTC patient.
Risk stratification and ROC curve
The risk score was calculated according to the predictive GENE signature model. Using the median risk score as the cutoff, DTC patients were classified into the high-risk group and low-risk group. Kaplan‑Meier analysis was used to generate PFS curves, and log-rank tests were performed to assess PFS differences between high-risk and low-risk groups. The prediction efficiency of the risk signature model was evaluated by calculating the area under the curve (AUC) of the receiver operating characteristic (ROC) curve using R package survival ROC (version 1; https://CRAN.R-project.org/view=Survival ROC). Univariate and multivariate analyses with Cox proportional hazards regression for PFS were performed to determine the prognostic value of the risk score and various clinical characteristics. Hazard ratios (HRs) and 95% confidence intervals (CIs) were estimated.
GEO database verification
The datasets of DTC patients were obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Three mRNA datasets of DTC (GSE33630, GSE35570, GSE60542) were selected in the present study and the expression profiles were normalized by log2‐conversion.
Statistical analysis
Statistical analyses were performed using SPSS v21.0 software (IBM Corp.). The edger function was used to analyze 13 m6A RNA modification regulators in DTC and normal thyroid tissues. Patients were clustered into different groups by consensus expression of m6A RNA methylation regulators. Chi-square tests were used to evaluate the distribution of clinicopathological characteristics between the two risk groups. In addition, the association between risk groups and the prognosis of patients with DTC was evaluated with the Kaplan‑Meier method and log‑rank test. Prognostic performance was estimated by ROC analysis. Univariate and multivariate Cox regression analyses were used to identify factors that were independently related to the prognosis of DTC patients. The Student's t-test were performed to calculate the results of GEO datasets. P<0.05 was considered statistically significant.
Results
Differentially expressed m6A RNA methylation regulators in DTC and normal thyroid
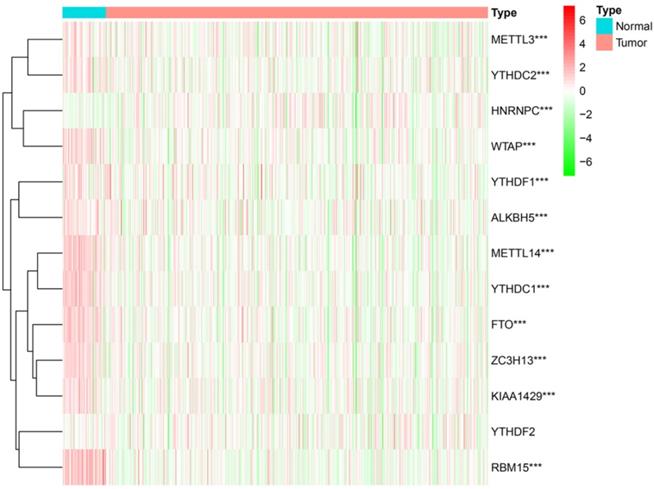
While m6A RNA methylation regulators play a crucial role in the evolution and progression of malignant tumors, little information is available on their expression in DTC. We thus systematically investigated the expressions of 13 m6A RNA methylation regulators in DTC tissue and normal thyroid tissue using gene expression information from TCGA database profiles. The results showed that the expressions of 12 m6A RNA methylation regulators were significantly different between normal thyroid and DTC tissue (P<0.001) (Figure 1).
The expression of 13 m6A RNA methylation regulators in DTC and normal thyroid. *P<0.05, **P<0.01, ***P<0.001.
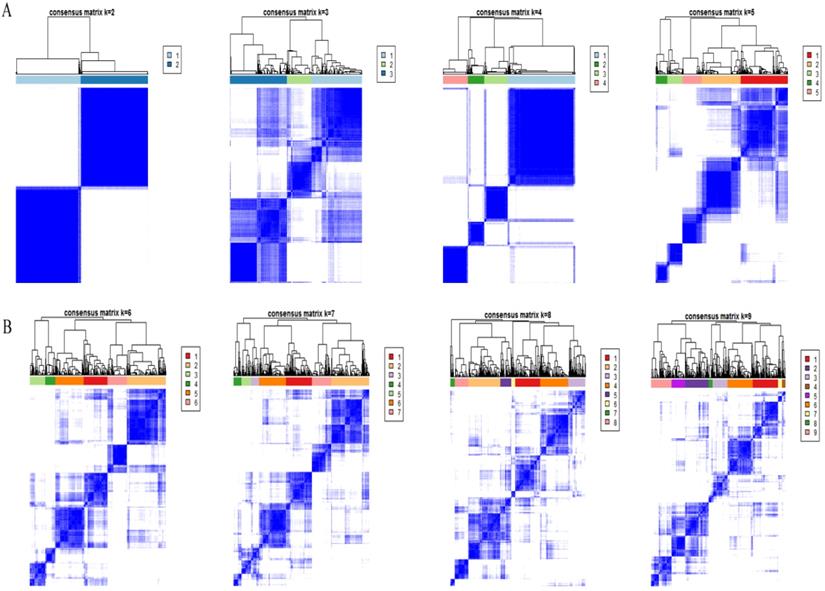
Consensus clustering of m6A RNA methylation regulators
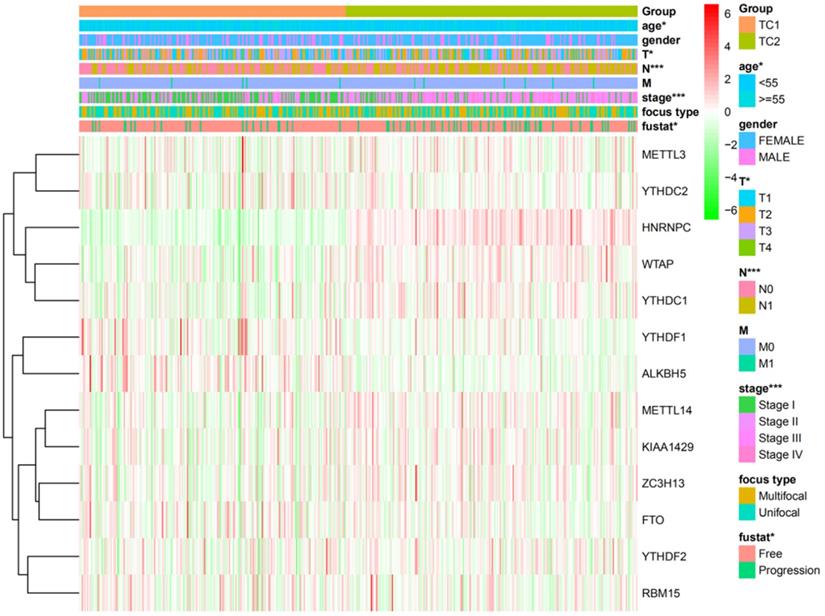
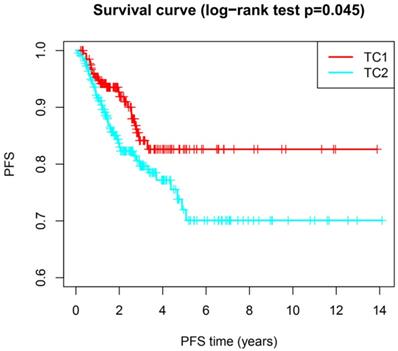
Based on the expression similarity of m6A RNA methylation regulators, high intra-group correlation and low inter-group correlation, k=2 was the most appropriate selection with clustering stability increasing from k=2 to 9 in our study (Figure 2A and 2B). Therefore, we divided DTC patients into TC1 or TC2 groups by applying consensus cluster k=2 and analyzed the clinicopathological characteristics between these two subgroups. The TC2 subgroup was significantly associated with an older age at diagnosis (P<0.05), higher T stage (P<0.05), lymph node metastasis (P<0.001), higher TNM stage (P<0.001) and disease-progression state (P<0.05). There were no correlations with other characteristics including gender, focus type, or metastasis stage (P>0.05) (Figure 3). We also investigated the prognosis of DTC patients in TC1 and TC2 subgroups using Kaplan‑Meier and log‑rank test and found that the TC2 subgroup had a shorter PFS compared with the TC1 subgroup (Figure 4; P=0.045).
Consensus clustering cumulative distribution feature. (A) Consensus clustering cumulative distribution feature for k = 2 to 5. (B) Consensus clustering cumulative distribution feature for k = 6 to 9.
Heatmap and clinicopathologic features of the two clusters (TC1/2).
The progression-free survival of DTC between TC1 and TC2. Log-rank test, P<0.05.
Risk model with four selected m6A RNA methylation regulators
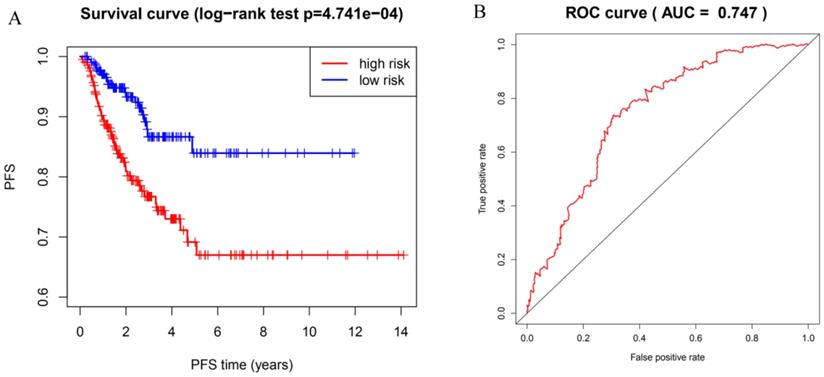
To identify potential prognostic m6A RNA methylation regulators, we examined the expression data of the twelve difference-expressed genes using the LASSO Cox regression algorithm. According to the minimum criteria, we selected four genes to build the risk signature. The signature was developed as a linear combination of the expression levels of the four genes weighted by their relative regression coefficients in the LASSO algorithm as follows: RS = (0.0341 × expression value of HNRNPC) + (-0.0184 × expression value of ZC3H13) + (-0.0128 × expression value of ALKBH5) + (-0.0877 × expression value of WTAP). We calculated the risk score for each DTC patient based on the risk score model and separated them into high-risk (n = 212) and low-risk groups (n = 213) using the median risk score as the cutoff point. The PFS of the two groups was significantly different; patients in the high-risk group had a shorter PFS compared with patients in low-risk group (Figure 5A; P=4.741e-04). The ROC curve was used to measure the predictive performance of the four-gene prognostic risk model. The AUC of the ROC for the four-gene prognostic model was 0.747 at 5 years of PFS (Figure 5B). These results indicate that the four-gene risk model can accurately predict the outcome of DTC patients.
Risk model with four selected m6A RNA methylation regulators. (A) Kaplan-Meier curve analysis of progression-free survival in high-risk and low-risk DTC patients. (B) Time-dependent ROC curve analysis of the four-gene prognostic risk model.
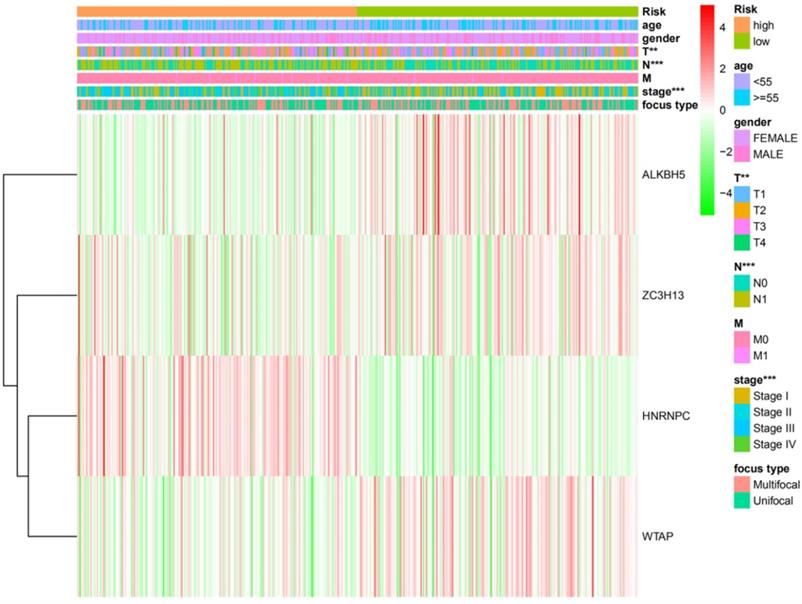
Association between prognostic risk scores and clinicopathological features of DTC
Next, χ2 and Fisher exact tests were performed to evaluate whether the prognostic risk scores were connected with clinicopathological features in DTC patients. We found significant differences between the high- and low-risk groups in relation to N grade (P<0.001), T grade (P<0.01), and TNM stage (P<0.001). However, there was no association with other clinicopathological features including gender, age, focus type or M grade (P>0.05). Furthermore, the expression of the four selected m6A RNA methylation regulators in high- and low-risk groups was shown in our study (Figure 6).
The clinicopathological features and the expression levels of the four m6A RNA methylation regulators in low- and high-risk groups. *P<0.05, **P<0.01, ***P<0.001.
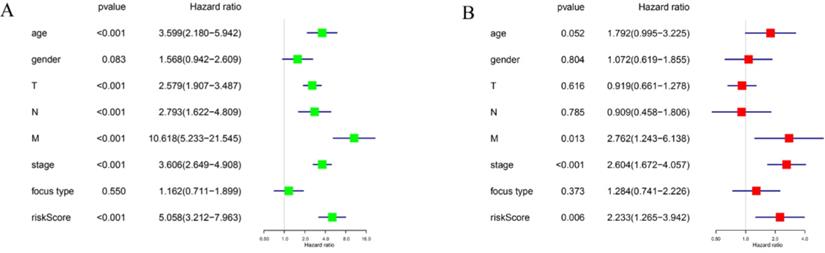
The four-gene risk signature is an independent prognostic indicator
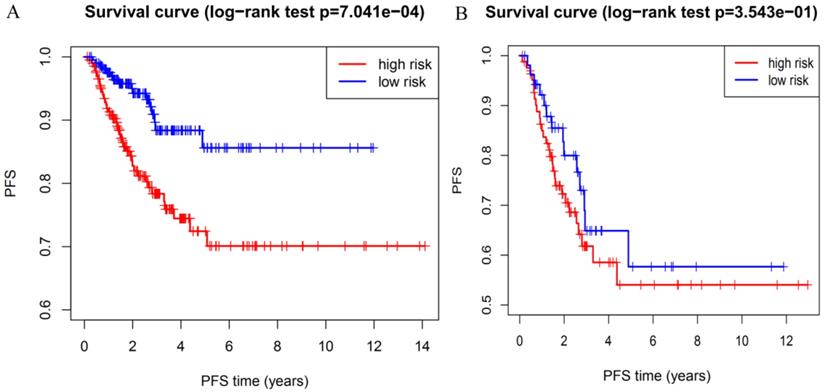
To determine whether the risk signature can independently predict the outcomes of DTC patients, univariate and multivariate Cox analyses were performed. Univariate analysis showed that age, T grade, N grade, M grade, TNM stage and the risk score were all related to PFS (Figure 7A). Including these factors into the multivariate analysis showed that the M grade, TNM stage and the risk score were significantly associated with PFS (Figure 7B). Moreover, DTC patients with high-risk scores had a shorter PFS than those with low risk scores in M grade 0 group (Figure 8A) and TNM stage III groups (Figure 8B). These results confirmed that the four-gene risk signature can predict prognosis of DTC patients independently.
The association between clinicopathological factors and PFS of DTC patients. (A) Univariate Cox regression analyses of the association between clinicopathological factors and PFS of DTC patients. (B) Multivariate Cox regression analyses of the association between clinicopathological factors and PFS of DTC patients.
Prognostic value of the risk signature in patients with M grade 0 and TNM grade III. (A) Kaplan-Meier PFS curves for patients with M grade 0. (B) Kaplan-Meier PFS curves for patients with TNM grade III.
GEO verification
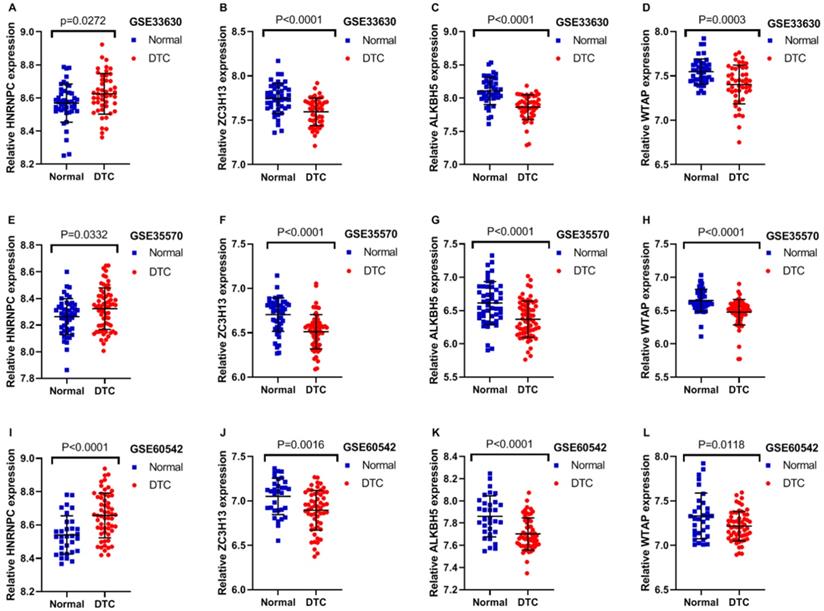
In order to further confirm the previous findings from TCGA analysis,we selected three datasets from GEO to verify the accuracy of the above results. The expression profile of these three mRNAs is showed in Table 2. The expression of HNRNPC was significantly higher in DTC than that in normal thyroid tissues as shown in GSE33630, GSE35570 and GSE60542. Furthermore, the results from these three GEO datasets also indicated that the expression of ZC3H13, ALKBH5 and WTAP were all evidently lower in DTC, compared with that in normal thyroid tissues (Figure 9A-L; P<0.05). The outcomes of GEO datasets were consistent with results of the abovementioned TCGA profiles. Unfortunately, no survival information of HNRNPC, ZC3H13, ALKBH5 and WTAP in DTC could be obtained from GEO datasets.
Expression data of three mRNA in GEO datasets
| GEO datasets | Year | Country | Platform | Sample | N | Relative expression of mRNA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HNRNPC | ZC3H13 | ALKBH5 | WTAP | ||||||||||
| X±S | P | X±S | P | X±S | P | X±S | P | ||||||
| GSE33630 | 2012 | Belgium | GPL570 | Normal | 44 | 8.568±0.1152 | 0.0272 | 7.746±0.1695 | <0.0001 | 8.108±0.2046 | <0.0001 | 7.549±0.1431 | 0.0003 |
| DTC | 47 | 8.625±0.1229 | 7.596±0.1549 | 7.863±0.1880 | 7.403±0.2190 | ||||||||
| GSE35570 | 2015 | Poland | GPL570 | Normal | 48 | 8.263±0.1357 | 0.0332 | 6.705±0.1896 | <0.0001 | 6.615±0.3214 | <0.0001 | 6.653±0.1673 | <0.0001 |
| DTC | 65 | 8.324±0.1558 | 6.513±0.1935 | 6.370±0.2733 | 6.477±0.1909 | ||||||||
| GSE60542 | 2015 | Belgium | GPL570 | Normal | 32 | 8.540±0.1142 | <0.0001 | 7.051±0.2058 | 0.0016 | 7.861±0.1845 | <0.0001 | 7.331±0.2567 | 0.0118 |
| DTC | 57 | 8.657±0.1343 | 6.894±0.2241 | 7.702±0.1441 | 7.216±0.1650 | ||||||||
GEO: Gene Expression Omnibus; DTC: Differentiated Thyroid Carcinoma.
Expression of m6A RNA regulators in different GEO datasets. (A-D) The expression levels of four m6A RNA regulators in normal and DTC tissues from the GSE33630. (E-H) The expression levels of four m6A RNA regulators in normal and DTC tissues from the GSE35570. (I-L) The expression levels of four m6A RNA regulators in normal and DTC tissues from the GSE60542.
Discussion
Thyroid carcinoma is the most common endocrine malignancy, with an increasing incidence around the world [32, 33]. Even though most thyroid carcinomas are DTCs with favorable prognosis, DTCs display heterogeneity in patients [34, 35]. Therefore, novel molecular indicators that could effectively predict and monitor the response to treatment and disease progression of DTCs should be identified to help improve patient care. Recent studies have revealed that m6A modification plays an essential role in various biological processes, including the initiation and progression of cancers [36-38]. However, little information has been available regarding the role of m6A in DTCs.
In this study, we found that most known m6A RNA methylation regulators were dysregulated in DTCs. Using consensus cluster, we classified DTC patients into two subgroups (TC1 and TC2). The prognosis and clinicopathological features were significantly different between TC1 and TC2 subgroups. We further developed a risk signature with four m6A RNA methylation regulators that categorized DTC patients into high- and low-risk groups with significantly different PFS.
In eukaryotes, m6A is the most abundant and evolutionarily conserved modification in mRNA and plays a crucial role in several aspects of RNA metabolism [39, 40]. Increasing numbers of studies have shown that m6A modifications are controlled by dynamic changes of m6A RNA methylation regulators. Furthermore, dysregulated expression of m6A RNA methylation regulators has been associated with several types of cancers [41-43]. For instance, the m6A writer protein METTL3 is a reported oncogene for liver cancer [44], bladder cancer [45] and acute myeloid leukemia [46], but a tumor suppressor for breast cancer [38]. Downregulation of the m6A eraser protein ALKBH5 was correlated with poor prognosis in pancreatic cancer [47]. However, Zhang et al [48] reported that ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells. These findings suggested that m6A RNA methylation regulators may likely have different pathological implications in different diseases.
To explore the role of m6A RNA methylation regulators in DTC patients, we comprehensively analyzed the expression of thirteen regulators in our study. All writers (WTAP, METTL3, METTL14, RBM15, ZC3H13, KIAA1429) were downregulated in DTC compared with normal thyroid tissues. The m6A methylation readers YTHDC1, YTHDC2, and YTHDF1 were expressed significantly lower in DTCs, but the expression of HNRNPC was increased. Both FTO and ALKBH5 m6A methylation erasers were decreased in DTCs. Furthermore, we identified four m6A RNA methylation regulators (WTAP, ZC3H13, HNRNPC and ALKBH5) that were related to the prognosis of DTCs and used these to develop a prognostic signature. In addition, we found that the high-risk group was associated with high N grade, high T grade and high TNM grade, which indicated poor outcomes of DTCs. Univariate and multivariate Cox regression analyses suggested that the four-gene risk signature was an independent prognostic factor for PFS in DTCs. We also found that DTCs with M grade 0 were categorized into high-risk group with shorter PFS or low-risk groups with longer PFS. A similar situation was found in DTCs with TNM stage III, and PFS was significantly different between high- and low-risk groups.
Zinc finger proteins are involved in the regulation of transcription or translation by specific binding of the target molecules. Different combined with DNA, RNA, DNA-RNA, protein, or zinc finger motifs, which result in zinc finger proteins present multifunctional in biological processes [49, 50]. ZC3H13 is a classical CCCH zinc finger protein and the encoding gene is located in human chromosome 13q14.13 [51]. Increasing evidence has shown that ZC3H13 plays an important role in inhibiting tumor progression. In previous research, it has been reported that somatic frame-shift mutation in ZC3H13 gene is detected in colon carcinoma, which suggests that ZC3H13 may be a tumor suppressor [52]. Zhu et al [53] found that ZC3H13 acts as a tumor suppressor by regulating activation of the Ras-ERK signaling pathway. ALKBH5 is a demethylase that is associated with the regulation of mRNA translation and mRNA metabolism. ALKBH5 is a demethylase that is associated with the regulation of mRNA translation and mRNA metabolism. Increasing evidence indicate that ALKBH5 is implicated in the development of multiple cancers. Overexpression of ALKBH5 has been reported in glioblastoma stem cells and it by stabilizing FOXM1 to promote the proliferation of glioblastoma [48, 54]. Zhang C et al [55] found that ALKBH5 enhances m6A stability, which increases the levev of m6A in breast carcinoma. Moreover, the number of breast cancer ctem cells can be reduced by ALKBH5 knockdown in breast cancer. He et al [47] revealed that ALKBH5 inhibits the motility of pancreatic cancer by downregulating long non-coding RNA KCNK15-AS1 methylation. In our study, we revealed that the expression of ALKBH5 was lower in DTC compared with that in normal thyroid tissues, suggesting that further research is needed to understand the role of ALKBH5 in DTC. WTAP is a nuclear protein widely expressed in cells and tissues, plays an essential role in cellular function and cancer progression, which was first been found for its specific interaction with Wilms' tumor 1 [56, 57]. WTAP is a conserved nuclear protein that shows decreased expression in breast cancer [38]. The reduced expression of WTAP correlated with the accelerated proliferation and invasion of breast cancer. However, a number of recent studies have also shown that WTAP was act as an oncogene and was associated with malignant tumors closely. WTAP negatively regulate WT1.9 to promote tumorigenesis in colorectal cancer [58]. HNRNPC is an RNA-binding protein located in the nucleus and the cytoplasm. It is closely related to mRNA metabolism [59], including mRNA splicing, mRNA stabilization and translation.
HNRNPC is aberrantly up-regulated in melanoma [60], glioblastoma [61] and breast cancer [62]. In the present study, we demonstrated that the expression of HNRNPC was increased and that of m6A methylases (ZC3H13, ALKBH5 and WTAP) were significantly decreased in DTC tissues compared with normal thyroid tissue. Additionally, we developed a risk signature that divided DTC patients into high-risk and low-risk groups with significantly different PFS times. These results may allow clinicians to determine individualized treatment for DTC patients with different clinical features.
In conclusion, here we revealed the expression and prognostic value of m6A RNA methylation regulators in DTC. This study also provides crucial evidence supporting further research of the role of RNA m6A methylation in DTC.
Acknowledgements
Funding
Natural Science Foundation of Zhejiang Province, Grant/Award Number: LGF18H160001; Department of Education of Zhejiang Province, Grant/Award Number: Y201737945.
Availability of data and materials
All the data collected and analyzed in the study are available from the corresponding author on reasonable request.
Authors' Contributions
Deguang Zhang and Nizhen Xu designed the study. Nizhen Xu, Jian Chen, Gaofei He and Li Gao performed the data collection and analysis. All authors participated in the writing of the manuscript. All the authors have read and approved the final version of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang L, Lian R, Zhao J. et al. IGFBP7 inhibits cell proliferation by suppressing AKT activity and cell cycle progression in thyroid carcinoma. Cell Biosci. 2019;9:44
2. Gambardella C, Patrone R, Di Capua F. et al. The role of prophylactic central compartment lymph node dissection in elderly patients with differentiated thyroid cancer: a multicentric study. BMC Surg. 2019;18(Suppl 1):110
3. Lee J, Park HL, Jeong CW. et al. CYFRA 21-1 in Lymph Node Fine Needle Aspiration Washout Improves Diagnostic Accuracy for Metastatic Lymph Nodes of Differentiated Thyroid Cancer. Cancers (Basel). 2019;11:4
4. Zhang TT, Li CF, Wen SS. et al. Effects of tumor size on prognosis in differentiated thyroid carcinoma smaller than 2 cm. Oncol Lett. 2019;17:4229-36
5. Yu ST, Ge JN, Luo JY. et al. Treatment-related adverse effects with TKIs in patients with advanced or radioiodine refractory differentiated thyroid carcinoma: a systematic review and meta-analysis. Cancer Manag Res. 2019;11:1525-32
6. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
7. Yamazaki H, Iwasaki H, Takasaki H. et al. Efficacy and tolerability of initial low-dose lenvatinib to treat differentiated thyroid cancer. Medicine (Baltimore). 2019;98:e14774
8. Carling T, Udelsman R. Thyroid cancer. Annu Rev Med. 2014;65:125-37
9. Grant CS. Recurrence of papillary thyroid cancer after optimized surgery. Gland Surg. 2015;4:52-62
10. Lin HC, Liou MJ, Hsu HL. et al. Combined analysis of circulating epithelial cells and serum thyroglobulin for distinguishing disease status of the patients with papillary thyroid carcinoma. Oncotarget. 2016;7:17242-53
11. Haugen BR, Alexander EK, Bible KC. et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133
12. Leighton LJ, Bredy TW. Functional Interplay between Small Non-Coding RNAs and RNA Modification in the Brain. Noncoding RNA. 2018;4:2
13. Liu F, Clark W, Luo G. et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell. 2016;167:1897
14. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31-42
15. Du K, Zhang L, Lee T. et al. m6A RNA Methylation Controls Neural Development and Is Involved in Human Diseases. Mol Neurobiol. 2019;56:1596-606
16. Wang S, Sun C, Li J. et al. Roles of RNA methylation by means of N6-methyladenosine (m6A) in human cancers. Cancer Lett. 2017;408:112-20
17. Ke S, Pandya-Jones A, Saito Y. et al. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31:990-1006
18. Maity A, Das B. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. 2016;283:1607-30
19. Yoon KJ, Ringeling FR, Vissers C. et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell. 2017;171:877-89.e17
20. Engel M, Eggert C, Kaplick PM. et al. The Role of m6A/m-RNA Methylation in Stress Response Regulation. Neuron. 2018;99:389-403.e9
21. Zhou J, Wang J, Hong B. et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging (Albany NY). 2019;11:1633-47
22. Zhou Y, Yin Z, Hou B. et al. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res. 2019;11:3921-31
23. Yang Y, Hsu PJ, Chen YS. et al. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616-24
24. Li A, Chen YS, Ping XL. et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444-7
25. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343-55
26. Zhang C, Zhi WI, Lu H. et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527-42
27. Pan Y, Ma P, Liu Y. et al. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol. 2018;11:48
28. Dai D, Wang H, Zhu L. et al. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124
29. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2019
30. Sauerbrei W, Royston P, Binder H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat Med. 2007;26:5512-28
31. Bøvelstad HM, Nygård S, Størvold HL. et al. Predicting survival from microarray data-a comparative study. Bioinformatics. 2007;23:2080-7
32. Manzella L, Massimino M, Stella S. et al. Activation of the IGF Axis in Thyroid Cancer: Implications for Tumorigenesis and Treatment. Int J Mol Sci. 2019;20:13
33. Vigneri R, Malandrino P, Vigneri P. The changing epidemiology of thyroid cancer: why is incidence increasing. Curr Opin Oncol. 2015;27:1-7
34. George N, Agarwal A, Kumari N. et al. Molecular Profiling of Follicular Variant of Papillary Thyroid Cancer Reveals Low-Risk Noninvasive Follicular Thyroid Neoplasm with Papillary-Like Nuclear Features: A Paradigm Shift to Reduce Aggressive Treatment of Indolent Tumors. Indian J Endocrinol Metab. 2018;22:339-46
35. Ho AS, Davies L, Nixon IJ. et al. Increasing diagnosis of subclinical thyroid cancers leads to spurious improvements in survival rates. Cancer. 2015;121:1793-9
36. Liu ZX, Li LM, Sun HL. et al. Link Between m6A Modification and Cancers. Front Bioeng Biotechnol. 2018;6:89
37. Cui Q, Shi H, Ye P. et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622-34
38. Wu L, Wu D, Ning J. et al. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326
39. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science. 2016;352:1408-12
40. Roignant JY, Soller M. m6A in mRNA: An Ancient Mechanism for Fine-Tuning Gene Expression. Trends Genet. 2017;33:380-90
41. Visvanathan A, Patil V, Arora A. et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522-33
42. Liu J, Eckert MA, Harada BT. et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074-83
43. Lin S, Choe J, Du P. et al. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335-45
44. Chen M, Wei L, Law CT. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254-70
45. Cheng M, Sheng L, Gao Q. et al. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667-80
46. Barbieri I, Tzelepis K, Pandolfini L. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552:126-31
47. He Y, Hu H, Wang Y. et al. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell Physiol Biochem. 2018;48:838-46
48. Zhang S, Zhao BS, Zhou A. et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591-606.e6
49. Addepalli B, Hunt AG. Ribonuclease activity is a common property of Arabidopsis CCCH-containing zinc-finger proteins. FEBS Lett. 2008;582:2577-82
50. Zhang P, Huang A, Morales-Ruiz M. et al. Engineered zinc-finger proteins can compensate genetic haploinsufficiency by transcriptional activation of the wild-type allele: application to Willams-Beuren syndrome and supravalvular aortic stenosis. Hum Gene Ther. 2012;23:1186-99
51. Ouna BA, Stewart M, Helbig C. et al. The Trypanosoma brucei CCCH zinc finger proteins ZC3H12 and ZC3H13. Mol Biochem Parasitol. 2012;183:184-8
52. Kim YR, Chung NG, Kang MR. et al. Novel somatic frameshift mutations of genes related to cell cycle and DNA damage response in gastric and colorectal cancers with microsatellite instability. Tumori. 2010;96:1004-9
53. Zhu D, Zhou J, Zhao J. et al. ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. J Cell Physiol. 2019;234:8899-907
54. Chai RC, Wu F, Wang QX. et al. m6A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging (Albany NY). 2019;11:1204-25
55. Zhang C, Samanta D, Lu H. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6 A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047-56
56. Yu HL, Ma XD, Tong JF. et al. WTAP is a prognostic marker of high-grade serous ovarian cancer and regulates the progression of ovarian cancer cells. Onco Targets Ther. 2019;12:6191-201
57. Little NA, Hastie ND, Davies RC. Identification of WTAP, a novel Wilms' tumour 1-associating protein. Hum Mol Genet. 2000;9:2231-9
58. Zhang J, Tsoi H, Li X. et al. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut. 2016;65:1482-93
59. Lee EK, Kim HH, Kuwano Y. et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol. 2010;17:732-9
60. Mulnix RE, Pitman RT, Retzer A. et al. hnRNP C1/C2 and Pur-beta proteins mediate induction of senescence by oligonucleotides homologous to the telomere overhang. Onco Targets Ther. 2013;7:23-32
61. Park YM, Hwang SJ, Masuda K. et al. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol Cell Biol. 2012;32:4237-44
62. Wu Y, Zhao W, Liu Y. et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 2018;37:23
Author contact
![]() Corresponding author: Deguang Zhang, Department of Head and Neck surgery, Institute of Micro-Invasive Surgery of Zhejiang University, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, 3 East Qingchun Road, Hangzhou 310016, P.R. China. E-mail: 3409086edu.cn.
Corresponding author: Deguang Zhang, Department of Head and Neck surgery, Institute of Micro-Invasive Surgery of Zhejiang University, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, 3 East Qingchun Road, Hangzhou 310016, P.R. China. E-mail: 3409086edu.cn.