Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(8):1870-1878. doi:10.7150/jca.28299 This issue Cite
Research Paper
AMPK Inhibition Suppresses the Malignant Phenotype of Pancreatic Cancer Cells in Part by Attenuating Aerobic Glycolysis
Mingyue Hu1, Xiangxu Chen1, Li Ma1, Yu Ma1, Yuan Li1, Huihui Song1, Jiajia Xu1, Lingna Zhou1, Xiaoxue Li1, Yuhui Jiang1, Bo Kong2,3, Peilin Huang1 ![]()
1. Medical School of Southeast University, Nanjing 210009, China
2. Department of Surgery, Klinikum rechts der Isar, School of Medicine, Technical University of Munich (TUM), Munich 81675, Germany
3. Department of Gastroenterology, the Affiliated Drum Tower Hospital of Nanjing University, Medical School, Nanjing, China.
Received 2018-7-7; Accepted 2019-2-21; Published 2019-4-21
Abstract
Pancreatic cancer is a highly aggressive tumor characterized by enhanced aerobic glycolysis. AMP-activated protein kinase (AMPK), which is identified as a well-known regulator of glycolysis, plays an essential role in tumorigenesis. In the present study, we aim to explore the function of AMPK in pancreatic cancer cells and attempt to clarify the possible underlying mechanism. The Cancer Genome Atlas (TCGA) data showed that elevated AMPK expression highly correlated with lower median survival time. In an in vitro study, inhibition of AMPK blocked the proliferation, migration, and invasion ability of four cell lines under normoxia and hypoxia. Additionally, AMPK suppression led to cell cycle arrest and remarkably induced apoptosis. Furthermore, the lactic acid content, ATP content, and the glucose consumption rate were significantly reduced in all four cell lines under different conditions, accompanied by down-regulation of glycolytic biomarkers including phosphorylated mammalian target of rapamycin (p-mTOR)/total mTOR (t-mTOR), Pyruvate kinase M2 (Pkm2), and Hexokinase 2 (Hk2). Collectively, our data showed that AMPK activation is highly involved in pancreatic cancer progression and exerts its pro-tumorigenic functions partly by sustaining glycolytic activity. Hence, AMPK is expected to be a potential therapeutic target for pancreatic cancer.
Keywords: pancreatic cancer, aerobic glycolysis, AMPK, mTOR, hypoxia
Introduction
Pancreatic cancer is one of the most lethal cancers worldwide with an overall 5-year survival rate as low as 8% [1]. Surgery is regarded as the only potential cure for pancreatic cancer; however, this promising therapy is limited by complicated tumor anatomic location, late diagnosis, early metastasis, and various complications [2]. For unresectable cases, palliative chemotherapies are applied (e.g. FOLFIRINOX). However, the effectiveness of these therapies is still limited [3]. Hence, investigation of novel therapies for effective treatment is urgently needed, and a sound understanding of the molecular mechanism underlying pancreatic cancer development will help to provide some crucial answers.
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of most malignancies, and in this effect, tumor cells utilize glucose to rapidly generate lactate and ATP via glycolysis even in the presence of oxygen [4]. It is worth noting that enhanced aerobic glycolysis facilitates tumor development and progression, regarding not only aggressive growth but also malignant transformation, and predicts poor prognosis [5-8]. Moreover, both in vivo and in vitro studies have demonstrated that inhibition of glycolysis not only impaired tumorigenesis and metastasis but also promoted drug sensitivity in different tumors [9-12]. Therefore, aerobic glycolysis has the potential to be an efficient therapeutic target in tumor treatment.
AMP-activated protein kinase (AMPK), a serine/threonine kinase, is an important cellular energy sensor induced by the AMP/ATP ratio [13]. To cope with stress such as hypoxia and nutrient deprivation in normal cells, activated AMPK decreases the ATP-consuming anabolic processes (such as protein and lipid synthesis) and increases the ATP-producing catabolic processes (such as glycolysis and oxidation) to sustain energy homeostasis [14]. More importantly, emerging research has reported that AMPK also plays a regulatory role in aerobic glycolysis in various tumors [15-20]. Acting as a tumor suppressor, AMPK blocked tumorigenesis of liver cancer and lymphoma [21, 22]. Meanwhile, AMPK also showed contextual pro-tumor effects in breast cancer and lung cancer [23, 24]. Moreover, in our previous study, we found that pancreatic cancer cells that highly express AMPK exhibit strong glycolytic phenotypes and have more aggressive behaviors [25]. However, the mechanism underlying this effect still needs further exploration.
Based on these previous findings, we employed an in vitro model to explore the function of AMPK in pancreatic carcinoma, discussed the correlation between AMPK and aerobic glycolysis, and attempted to elucidate the underlying molecular mechanism.
Materials and Methods
Cell culture and treatment
The two pancreatic cancer cell lines, KrasG12D (hereafter denoted as 399 and 403) and KrasG12D-LOH (exhibiting a loss of heterozygosity of KrasG12D, hereafter denoted as 907 and 897), were generous gifts from the Technical University of Munich. All four cell lines were isolated from transgenic p48Cre/+; LSL-KrasG12D/+; Tsc1fl/+ mice as previously described [26]. All cells were incubated in Dulbecco's Modified Eagle's Medium (DMEM, HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA) and 100 U/ml penicillin and streptomycin (Beyotime Biotechnology Corporation, Shanghai, China) at 37ºC under normoxia (95% air, 5% CO2) or hypoxia (1% O2, 94% N2 and 5% CO2). For Compound C stimulation, the cells were incubated with Dorsomorphin 2HCl (10 μM, Selleck, Houston, TX, USA) for 24 hours before further measurement, and cells treated with the same volume of the vehicle were used as the control group.
Cell vitality assay
After Compound C treatment, the cells were seeded in 96-well plates at 2×103 cells/well (100 μL/well) and were incubated for 1, 2, 3, 4, and 5 days. Then ten μL of Cell Counting Kit-8 solution (Dojindo, Tokyo, Japan) was added to each well. All plates were incubated for two hours at 37ºC in 5% CO2. The OD value was measured at 450 nm using a spectrophotometric plate reader.
Colony formation assay
Cells pretreated with Compound C were seeded in 6-well plates at a density of 200 cells/well. The cells were incubated in 4 mL of medium containing 10% FBS at 37ºC for ten days. The visible colonies were fixed with 75% paraformaldehyde for 30 min and were stained with 0.5% crystal violet for 30 min. After washing twice with phosphate buffer solution (PBS), fresh colonies containing more than 50 cells were counted. Colony formation efficiency (%) = (number of colony/number of seeded cell) × 100%.
Cell invasion and migration assay
For cell invasion and migration assay, the transwell chambers (8 μm pores, Corning, NY, USA) were precoated with or without 50 μL of matrigel (1:3 mixed with FBS-free medium, BD Bioscience, Bedford, NY, USA) and were dried at 37ºC for six hours. The cells were suspended in FBS-free medium at a density of 50×104 cells/mL. Next, 200 μL of the cell suspension was seeded into the upper chamber, and 600 μL of medium containing 10% FBS was added into the bottom chamber. After incubating for 24 hours under normoxia or hypoxia, the Transwell chambers were removed and fixed with paraformaldehyde for 30 min and were stained with 0.5% crystal violet for 30 min. For counting the cell numbers, five fields of the membrane were randomly selected, and the cell numbers were averaged. The assays were repeated three times independently.
Glucose uptake, intracellular ATP and lactic acid content assay
The cells were seeded in 6-well plates and were treated with Compound C for 24 hours in advance. The glucose uptake rate and lactic acid production in the culture supernatant were measured via a glucose assay kit and lactic acid assay kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. The intracellular ATP content was detected via absorption at 630 nm using the ATP assay kit (Solarbio Science and Technology, Beijing, China) according to the manufacturer's instructions.
Cell apoptosis and cell cycle analysis
Cell cycle and cell apoptosis were detected using a cell cycle detection kit and an Annexin V-FITC apoptosis detection kit (KeyGEN BioTECH, Nanjing, China) via flow cytometry (BD Biosciences, Philadelphia, PA, USA). For cell cycle detection, 100×104 cells were harvested and washed twice with PBS before being fixed using 70% cold ethanol at 4ºC overnight. After the ethanol was washed away using PBS, 100 μL of RNase A was added to the cells for a 30-min incubation at 37ºC. Next, the cells were treated with 400 μL of propidium iodide (PI), incubated for 30 min at 4ºC and analyzed via flow cytometry. For cell apoptosis analysis, the harvested cells were suspended in 500 μL of binding buffer. Next, the suspension was mixed with five μL of Annexin V-FITC and five μL of PI in turn. After incubating in the dark for 15 min, the cells were analyzed via flow cytometry.
Western blotting
The cells were lysed on ice using RIPA buffer with PMSF and a protease inhibitor (KeyGEN BioTECH, Nanjing, China). Protein concentrations were detected via a BCA protein assay kit (Beyotime Biotechnology Corporation, Shanghai, China). Proteins were electrophoresed via 8-10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA). Next, the membranes were blocked in 5% nonfat milk dissolved in TBST (a mixture of Tris-buffered saline and Tween 20) at room temperature for one hour. Subsequently, the membranes were incubated with primary antibodies against Hif1α (1:1000, Cat: NB100-105, Novus Biologicals, Cambridge, UK), phosphorylated Ampk (p-Ampk, 1:1000, Cat: 2535, Cell Signaling Technology, Beverly, MA, USA), total Ampk (t-Ampk, 1:1000, Cat: 2603, CST), phosphorylated mammalian target of rapamycin (p-mTOR, 1:1000, Cat: 5536, CST), total mTOR (t-mTOR, 1:1000, Cat: 2983, CST), Pyruvate kinase M2 (Pkm2, 1:1000, Cat: 3198, CST), Hexokinase II (Hk2, 1:2000, Cat:2867, CST) and β-actin (1:5000, Cat: AP0060, Bioworld Technology, Louis Park, MN, USA) at 4ºC overnight. After washing three times with TBST, the membranes were incubated with the HRP-conjugated goat anti-rabbit IgG secondary antibody (1:5000, Cat: SA00001-2, Proteintech, Chicago, IL, USA) for one hour at 25ºC. After washing three times with TBST again, the membranes were visualized using enhanced chemiluminescence (Millipore, Billerica, MA, USA). The band intensities were analyzed using Image-Pro Plus (Media Cybernetics, Rockville, MD, USA).
Quantitative Real-Time PCR
Total RNA was isolated using RNAiso Plus reagent (Takara Biotechnology, Dalian, China) and reverse transcribed to cDNA using PrimerScriptTM RT Master kit (Takara) according to the product instructions. Real-Time PCR was performed on the Step One Plus Real-Time PCR System using SYBR Premix Ex TaqTM kit (Takara) according to its instructions. The primer sequences were described as follows: Pkm2, 5'-GAGGCTGCCATCTACCACTT-3' (forward) and 5'-CACTCCTGCCAGACTTGGTG-3' (reverse); Hk2, 5'-AGCTGCCGACTAGCCTTTTT-3' (forward) and 5'-AGCGAACCGTGTTGGTAGAG-3' (reverse); B2m, 5'-CGGCCTGTATGCTATCCAGAAAACC-3' (forward) and 5'-TGTGAGGCGGGTGGAACTGTG-3' (reverse). The relative expression level of each gene was calculated using the 2-ΔΔCt method.
TCGA Database analysis
The address of TCGA (The Cancer Genome Atlas) is https://cancergenome.nih.gov/. The mRNA expressions data and the clinical patient information were downloaded from the TCGA Data Portal.
Statistical analysis
All assays were repeated three times independently. Statistical analyses were performed using one-way ANOVA and Student's t-tests using SPSS 21.0 software (International Business Machines Corp, NY, USA). The Kaplan-Meier survival curve analysis and linear correlation analysis with data from TCGA were performed using GraphPad Prism7 (GraphPad Software Inc., San Diego, CA, USA). The results were expressed as the mean ± SD, and P < 0.05 was considered statistically significant.
Results
Elevated AMPK expression in pancreatic cancer predicted poor prognosis
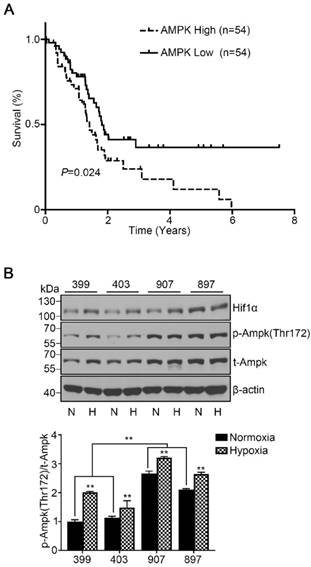
To assess the role of AMPK in pancreatic cancer development, we used data from TCGA and examined the overall survival based on AMPK (PRKAA1) mRNA expression of 176 patients. All patients were classified into two cohorts, the AMPK high-expression cohort (the highest 30% of AMPK expression, n = 54) and the AMPK low-expression cohort (the lowest 30% of AMPK expression, n = 54). The survival curve showed that patients with high expression of AMPK exhibited a lower median survival time of 1.4 years than patients with low expression of AMPK (P < 0.05, Fig. 1A). To understand the basic AMPK expression level of our experimental cell lines, we detected the phosphorylated AMPK expression level of the four cell lines under normoxia and hypoxia separately. Under normoxia, the phosphorylated AMPK levels in the 907 and 897 cells were significantly higher than that in the 399 and 403 cells (P < 0.01). Notably, the phosphorylated AMPK expressions of all four cell lines were strongly increased when they were incubated under hypoxic conditions (P < 0.01, Fig. 1B).
High AMPK expression in pancreatic cancer patients correlated with reduced median survival time and basic AMPK expression in experimental cell lines. (a) Kaplan-Meier survival curve showing the correlation of the mRNA expression level of AMPK with overall survival in 176 patients. The median survival time of patients in the AMPK high-expression cohort (n = 54, median survival time = 1.4 years) was significantly lower than that in patients in the AMPK low-expression cohort (n = 54, median survival time = 1.8 years). (b) Relative protein expression of AMPK in four experimental pancreatic cancer cell lines (399, 403, 907, and 897 cells) under normoxia and hypoxia. N: Normoxia, H: Hypoxia. The bar graphs show the mean ± SD. *, P < 0.05, and **, P < 0.01.
Inhibition of AMPK attenuated malignant phenotypes of pancreatic cancer cells
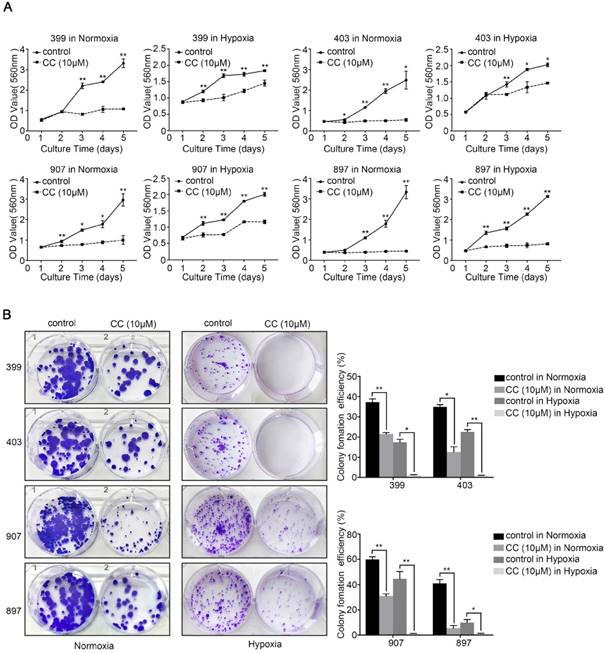
To explore the role of AMPK in the malignant behavior of pancreatic cancer cells, all four cell lines were treated with the selective AMPK inhibitor Compound C. After inhibitor dose screening, the dose of 10 μM was selected for the further study (Supplementary figure 1). Then, the CCK-8, colony-formation, migration, and invasion assays were performed. As shown in Fig. 2A, under normoxia and hypoxia, the AMPK-inhibited groups of 399, 403, 907, and 897 cells exhibited lower proliferative ability than the control groups (P < 0.05). A similar trend was observed in the colony-formation assays. The colony-formation ability of all four cell lines that were subjected to AMPK suppression was markedly reduced compared with the control groups (P < 0.05, Fig. 2B).
AMPK inhibition suppressed the proliferative ability of pancreatic cancer cells. Selective AMPK inhibitor Compound C (CC, 10 μM) was applied to inhibit the AMPK activation in the 399, 403, 907, and 897 cell lines before performing CCK-8 assays and colony-formation assays. (a) CCK-8 assays showing reduced cell proliferation rate in the four cell lines after AMPK inhibition. (b) Colony-formation assays showing decreased colony-formation ability of the four cell lines after AMPK inactivation under normoxia and hypoxia. Each experiment was independently repeated at least three times. The bar graphs show the mean ± SD. *, P < 0.05, and **, P < 0.01.
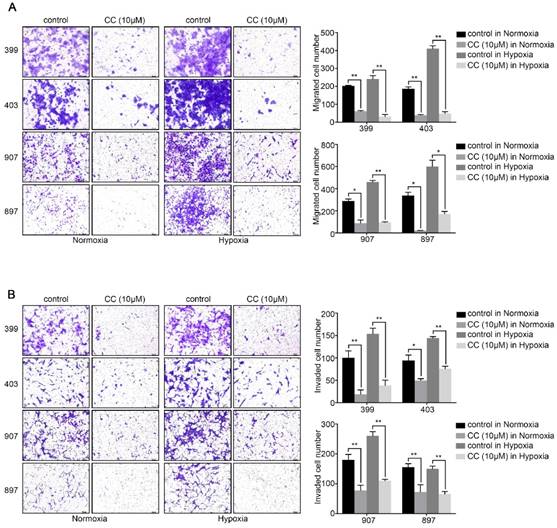
Also, we attempted to investigate whether AMPK inhibition affected the invasion and migration ability of pancreatic cancer cells. The migration assays illustrated that AMPK inhibition remarkably reduced the migrated cells number of 399, 403, 907, and 897 cells under normoxic and hypoxic conditions (P < 0.05, Fig. 3A). Similarly, as displayed in Fig, 3b, the numbers of invaded cells in the AMPK-inhibited groups of the four cell lines were lower than that in the control groups both under normoxia and hypoxia (P < 0.05, Fig. 3B). These results indicated that inhibition of AMPK by Compound C significantly blocks the proliferation and metastasis of pancreatic cancer cells.
AMPK inhibition decreased the migration and invasion ability of pancreatic cancer cells. (a) Migration assays showing a remarkable decrease of migrated cells number of 399, 403, 907, and 897 cells under normoxia and hypoxia. (b) Invasion assays exhibiting similar results of invaded cells reduction in response to AMPK suppression in four cell lines. Each experiment was independently repeated at least three times. The bar graphs show the mean ± SD. *, P < 0.05, and **, P < 0.01.
AMPK suppression induced cell cycle arrest and increased apoptosis
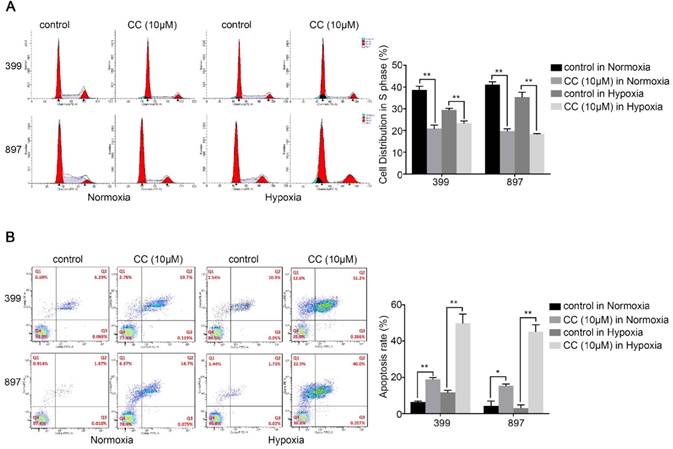
We further selected 399 and 897 cells to examine the effect of AMPK suppression on cell cycle and apoptosis in pancreatic cancer cells. Cell cycle analysis showed that fewer cells were distribution in S-phase in the AMPK-inhibited 399 and 897 cells than in the control groups (P < 0.01, Fig. 4A). As presented in Fig, 4b, suppression of AMPK led to an obvious increase of apoptosis rate in both types of pancreatic cancer cells compared with that in the control groups under normoxia and hypoxia (P < 0.05, Fig. 4B). In summary, these results were consistent with the impaired proliferative ability in pancreatic cancer cells.
Decreased AMPK activity led to cell cycle arrest and facilitated apoptosis. 399 and 897 cells were treated with Compound C (CC, 10 μM) before flow cytometry analysis. (a) Cell cycle analysis using flow cytometry showing that AMPK inhibition in the 399 and 897 cells drastically reduced the S-phase distribution under normoxic and hypoxic conditions. (b) Alteration in the apoptosis rate in response to AMPK suppression. Decreased apoptosis rates were detected in AMPK-inhibited cell lines under normoxia and hypoxia. Each experiment was independently repeated at least three times. *, P < 0.05, and **, P < 0.01.
Reduced AMPK expression impaired aerobic glycolysis in vitro
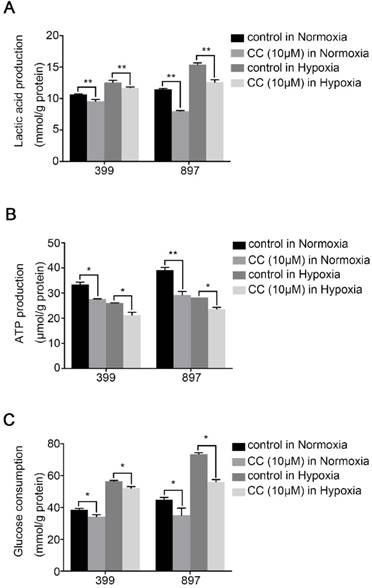
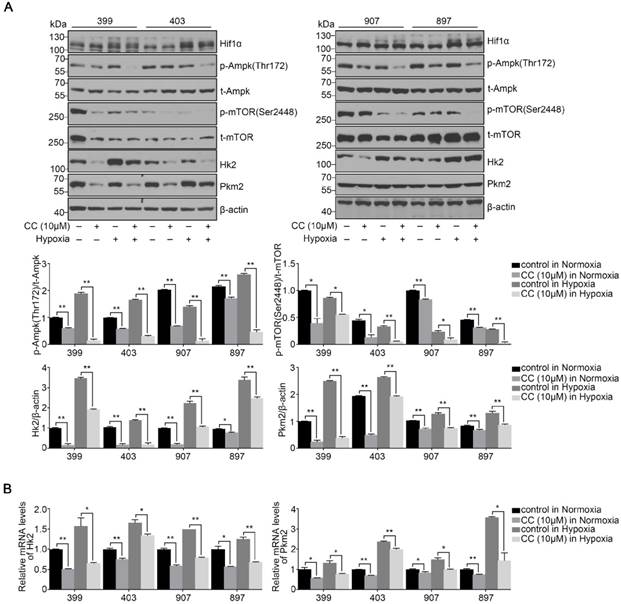
To determine the influence of AMPK inhibition on aerobic glycolysis in pancreatic cancer cells, we tested the levels of lactic acid, ATP production and glucose consumption in 399 and 897 cells. Compared with the control groups, the levels of lactic acid, ATP production and glucose consumption dramatically decreased in the experimental groups of 399 and 897 cells both under normoxia and hypoxia (P < 0.01, P < 0.05, and P < 0.05, Fig. 5A-5C). To further explore the possible mechanism underlying glycolysis limitation, we analyzed the correlation between different subunits of AMPK and 49 glycolytic biomarkers based on TCGA database. The correlation analysis showed that all six AMPK subunits (α1, α2, β1, β2, γ1 and γ2) are closely related to different glycolytic biomarkers. (Supplementary figure 2). Then we analyzed protein and mRNA expression levels of glycolytic biomarkers in 399, 403, 907, and 897 cells, including Hif1α, p-Ampk/t-Ampk, p-Mtor/t-Mtor, Pkm2, and Hk2, via Western Blot analysis and qPCR. Western Blot analysis showed that following AMPK inhibition in 399, 403, 907, and 897 cells under normoxia and hypoxia, a remarkable down-regulation was observed in p-Mtor/t-Mtor, Pkm2, and Hk2 protein expression (P < 0.05, Fig. 6A). Similarly, there was a drastic decrease in the mRNA expression of glycolytic enzymes, including Pkm2 and Hk2 in all four cell lines (P < 0.05, Fig. 6B). Moreover, correlation analysis of TCGA data revealed that except AMPKβ1, all AMPK subunits had a strong positive correlation with Rictor, which is one vital complex of mTOR (Supplementary figure 3). Collectively, these data revealed that AMPK suppression to attenuate the glycolytic phenotypes and decrease the expression of glycolytic biomarkers.
AMPK suppression attenuated the glycolytic phenotypes. The glycolytic phenotypes of 399 and 897 cells were measured for lactic acid and ATP production and glucose consumption. (a) Alteration of cellular lactic acid content with AMPK inhibition. (b) Changes in cellular ATP content in response to AMPK inactivation. (c) Variation of glucose consumption after inhibiting AMPK. Limited lactic acid and ATP production and glucose consumption were detected in two cell lines with AMPK suppression in normoxia and hypoxia. Each experiment was independently repeated at least three times. The bar graphs show the mean ± SD. *, P < 0.05, and **, P < 0.01.
Reduced AMPK expression altered the activities of glycolytic biomarkers. After treating with the AMPK inhibitor Compound C (CC, 10 μM), several glycolytic biomarkers were detected in 399, 403, 907, and 897 cells under normoxia and hypoxia. (a) Relative protein expression of p-Ampk/t-Ampk, p-mTOR/t-mTOR, Hk2/β-actin, and Pkm2/β-actin in four cell lines in response to AMPK reduction. Decreased expression levels of p-Ampk/t-Ampk, p-mTOR/t-mTOR, Hk2/β-actin, and Pkm2/β-actin were detected in AMPK-inhibited groups. (b) Relative mRNA expression of Pkm2 and Hk2 after inactivating AMPK. AMPK suppression led to the decline of Pkm2 and Hk2 mRNA expression in all four cell lines. Each experiment was independently repeated at least three times. The bar graphs show the mean ± SD. *, P < 0.05, and **, P < 0.01.
Discussion
In the present study, we explored the role of AMPK in pancreatic cancer cells under normoxia and hypoxia and observed that pharmacological inhibition of AMPK drastically impaired the proliferative and invasive capacity of pancreatic tumor cells. Meanwhile, AMPK suppression also blocked aerobic glycolysis including ATP production, lactic acid production and the glucose uptake rate. Collectively, these results indicated that AMPK activation plays an important role in protecting tumor cells partly by sustaining aerobic glycolysis and helped to understand the novel functions of AMPK in pancreatic cancer progression.
Previously, AMPK was regarded as a tumor suppressor because it promotes the activation of tumor suppressor p53 and inhibits angiogenesis via down-regulating the mTOR pathway, as well as negatively regulates the Warburg effect in vivo [21, 22, 27, 28]. However, with an ever-growing understanding of AMPK, its function becomes increasingly controversial. One study previously reported that AMPK facilitates tumor cell growth via sustaining NADPH homeostasis during energy stress [24]. Another study found that the absence of AMPK drastically attenuates tumorigenicity in vivo [23]. Similarly, Daurio et al. declared that the loss of the AMPKα2 isoform leads to an increase in the death of breast cancer cells [29]. In agreement with these studies, our data from TCGA suggested that pancreatic cancer patients with low AMPK activation exhibited a better prognosis. After pharmacological inhibition of AMPK, malignant phenotypes including proliferation and invasion were severely blocked, accompanied by cell-cycle arrest and an increase in apoptosis. In summary, AMPK is involved in the progression of cancer and exerts pro-tumor effects.
To investigate the mechanism underlying the protective function of AMPK, we continued to explore the correlation between AMPK and aerobic glycolysis. Enhanced aerobic glycolysis not only provides ATP rapidly but also creates a suitable tumor environment, and it is closely associated with tumorigenesis[4]. As mentioned before, AMPK has been found to play critical roles in negatively modulating glycolysis [22]. However, we observed an opposing role of AMPK in pancreatic cancer cells. Pharmacological loss of AMPK led to a decrease in glycolytic flux and a reduction in glycolytic biomarker expression. Moreover, correlation analysis also showed that all AMPK subunits are closely related to aerobic glycolysis. These results emphasize that AMPK plays a protective role in maintaining aerobic glycolysis in pancreatic cancer cells. Moreover, two previous studies revealed that the absence of AMPK reduces glycolysis, which fully agrees with our findings [30, 31]. However, the mechanism by which AMPK facilitates aerobic glycolysis remains unclear. We inferred that AMPK suppression might be able to directly reduce the activity of glycolytic enzymes or their regulators, such as PKM2, which subsequently impairs aerobic glycolysis.
mTOR has been defined as the core of signaling networks implicated in aerobic glycolysis through increasing the expression of glycolytic enzymes and promoting the glycolytic shift [32-34]. Also, it is generally recognized that AMPK negatively regulates mTOR activation directly by inactivating mTOR complex 1 (mTORC1) and indirectly by phosphorylating the mTOR inhibitor Tuberous sclerosis complex 1/Tuberous sclerosis complex 2 (TSC1/TSC2) complex [35, 36]. Intriguingly, our study showed that mTOR expression exhibited a remarkable decrease in response to AMPK inhibition. Moreover, TCGA data even pointed out Rictor had a strong positive correlation with most subunits of AMPK. The explanation for these paradoxical results lies mainly in the regulatory mechanism indirectly caused by AMPK. AMPK inhibition in tumor cells caused significant glycolysis reduction, which led to growth limitation. We hypothesized that in order to maintain energy support, the anabolic pathways mediated by mTOR might be inhibited [37]. Although mTOR inhibition synergetic with AMPK suppression can reduce glycolytic flux in pancreatic cancer cells, further studies focusing on the mTOR regulation mechanism are urgently needed.
Regarding our study, AMPK inhibition under hypoxia also remarkably undermines the malignant phenotypes and glycolysis in cancer cells. Hypoxia is ubiquitously detected in solid tumors, and it predicts poor prognosis due to its effect of promoting angiogenesis, facilitating drug resistance, and boosting metabolism reprogramming [38, 39]. Generally, AMPK is highly activated in response to low-oxygen stress [40]. However, its role in hypoxic conditions is still under investigation. From a mechanistic perspective, activated AMPK induced by hypoxia can directly promote the expression of glycolytic enzymes and glucose transporters [41]. Another possible explanation is that AMPK can sustain the stabilization of Hypoxia-inducible factor 1-α (Hif1α), which is the major target of hypoxia and can facilitate glycolysis [42]. Collectively, it is clear that AMPK activation is indispensable for protecting cancer cells under hypoxia in part by maintaining glycolysis.
Conclusion
In conclusion, our study underscored the critical role of AMPK in the progression of pancreatic cancer and indicated that the pro-tumorigenic function of AMPK might partly rely on promoting glycolysis both in normoxia and hypoxia.
Abbreviations
AMPK, AMP-activated protein kinase; TCGA, The Cancer Genome Atlas; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; TSC1/TSC2, Tuberous sclerosis complex 1/Tuberous sclerosis complex 2; Pkm2, Pyruvate kinase M2; Hk2, Hexokinase 2; Glut4, Glucose transporter type 4; DMEM, Dulbecco's Modified Eagle's Medium; FBS, fetal bovine serum; PBS, phosphate buffer solution; TBST, a mixture of Tris-buffered saline and Tween 20; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; PI, propidium iodide; CC, Compound C; mRNA, messenger RNA.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by research grants from the National Natural Science Foundation of China (Grant No.81372152).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7-30
2. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK. et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318-348
3. Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15(6):333-348
4. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211-218
5. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-1033
6. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-464
7. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926
8. Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q. et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med. 2017;21(9):2055-2067
9. Xu W, Zhang Z, Zou K, Cheng Y, Yang M, Chen H. et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017;8(5):e2761
10. Chen Z, Zuo X, Zhang Y, Han G, Zhang L, Wu J. et al. MiR-3662 suppresses hepatocellular carcinoma growth through inhibition of HIF-1alpha-mediated Warburg effect. Cell Death Dis. 2018;9(5):549
11. Torres MP, Rachagani S, Purohit V, Pandey P, Joshi S, Moore ED. et al. Graviola: a novel promising natural-derived drug that inhibits tumorigenicity and metastasis of pancreatic cancer cells in vitro and in vivo through altering cell metabolism. Cancer Lett. 2012;323(1):29-40
12. Kim SM, Yun MR, Hong YK, Solca F, Kim JH, Kim HJ. et al. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol Cancer Ther. 2013;12(10):2145-2156
13. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356(2):165-170
14. Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf). 2009;196(1):55-63
15. Chuang HC, Chou CC, Kulp SK, Chen CS. AMPK as a potential anticancer target - friend or foe? Curr Pharm Des. 2014;20(15):2607-2618
16. Gu DN, Jiang MJ, Mei Z, Dai JJ, Dai CY, Fang C. et al. microRNA-7 impairs autophagy-derived pools of glucose to suppress pancreatic cancer progression. Cancer Lett. 2017;400:69-78
17. Li Q, Qin Y, Wei P, Lian P, Li Y, Xu Y. et al. Gas1 Inhibits Metastatic and Metabolic Phenotypes in Colorectal Carcinoma. Mol Cancer Res. 2016;14(9):830-840
18. Fumarola C, Caffarra C, La Monica S, Galetti M, Alfieri RR, Cavazzoni A. et al. Effects of sorafenib on energy metabolism in breast cancer cells: role of AMPK-mTORC1 signaling. Breast Cancer Res Treat. 2013;141(1):67-78
19. Zhang L, Yang H, Zhang W, Liang Z, Huang Q, Xu G. et al. Clk1-regulated aerobic glycolysis is involved in glioma chemoresistance. J Neurochem. 2017;142(4):574-588
20. Liu Y, Tong L, Luo Y, Li X, Chen G, Wang Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J Cell Biochem. 2018;119(7):6162-6172
21. Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM. et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72(17):4394-4404
22. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17(1):113-124
23. Laderoute KR, Calaoagan JM, Chao WR, Dinh D, Denko N, Duellman S. et al. 5'-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J Biol Chem. 2014;289(33):22850-22864
24. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661-665
25. Shen X, Chang LG, Hu MY, Yan D, Zhou LN, Ma Y. et al. KrasG12D-LOH promotes malignant biological behavior and energy metabolism of pancreatic ductal adenocarcinoma cells through the mTOR signaling pathway. Neoplasma. 2018;65(1):81-88
26. Kong B, Wu W, Cheng T, Schlitter AM, Qian C, Bruns P. et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2016;65(4):647-657
27. Pisanti S, Picardi P, Ciaglia E, Margarucci L, Ronca R, Giacomini A. et al. Antiangiogenic effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, mediated by AMPK activation. FASEB J. 2014;28(3):1132-1144
28. Head SA, Shi WQ, Yang EJ, Nacev BA, Hong SY, Pasunooti KK. et al. Simultaneous Targeting of NPC1 and VDAC1 by Itraconazole Leads to Synergistic Inhibition of mTOR Signaling and Angiogenesis. ACS Chem Biol. 2017;12(1):174-182
29. Daurio NA, Tuttle SW, Worth AJ, Song EY, Davis JM, Snyder NW. et al. AMPK Activation and Metabolic Reprogramming by Tamoxifen through Estrogen Receptor-Independent Mechanisms Suggests New Uses for This Therapeutic Modality in Cancer Treatment. Cancer Res. 2016;76(11):3295-3306
30. Domenech E, Maestre C, Esteban-Martinez L, Partida D, Pascual R, Fernandez-Miranda G. et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol. 2015;17(10):1304-1316
31. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L. et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30(13):2719-2733
32. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G. et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J Biol Chem. 2015;290(28):17495-17504
33. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V. et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684
34. Toschi A, Lee E, Thompson S, Gadir N, Yellen P, Drain CM. et al. Phospholipase D-mTOR requirement for the Warburg effect in human cancer cells. Cancer Lett. 2010;299(1):72-79
35. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829-1834
36. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214-226
37. Pelletier J, Roux D, Viollet B, Mazure NM, Pouyssegur J. AMP-activated protein kinase is dispensable for maintaining ATP levels and for survival following inhibition of glycolysis, but promotes tumour engraftment of Ras-transformed fibroblasts. Oncotarget. 2015;6(14):11833-11847
38. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851-864
39. Jawhari S, Ratinaud MH, Verdier M. Glioblastoma, hypoxia and autophagy: a survival-prone 'menage-a-trois'. Cell Death Dis. 2016;7(10):e2434
40. Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J. et al. 5'-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26(14):5336-5347
41. Zhdanov AV, Dmitriev RI, Golubeva AV, Gavrilova SA, Papkovsky DB. Chronic hypoxia leads to a glycolytic phenotype and suppressed HIF-2 signaling in PC12 cells. Biochim Biophys Acta. 2013;1830(6):3553-3569
42. Smith TA, Zanda M, Fleming IN. Hypoxia stimulates 18F-fluorodeoxyglucose uptake in breast cancer cells via hypoxia inducible factor-1 and AMP-activated protein kinase. Nucl Med Biol. 2013;40(6):858-864
Author contact
![]() Corresponding author: Peilin Huang, hpledu.cn, Medical School of Southeast University, Dingjiaqiao 87, Gulou District, Nanjing 210009, China
Corresponding author: Peilin Huang, hpledu.cn, Medical School of Southeast University, Dingjiaqiao 87, Gulou District, Nanjing 210009, China