Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):1013-1022. doi:10.7150/jca.29527 This issue Cite
Research Paper
LncRNA SNHG16 Functions as an Oncogene by Sponging MiR-135a and Promotes JAK2/STAT3 Signal Pathway in Gastric Cancer
Xinguo Wang1*, Jie Kan1*, Jun Han1, Wenyan Zhang1, Liyan Bai1, Hao Wu1,2 ![]()
1. Department of Oncology, Qinghai Provincial People's Hospital, 2 Gonghe Road, Xining 810007, Qinghai Province, PR China
2. Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, Jiangsu Province, PR China.
*Equal contributors.
Received 2018-8-27; Accepted 2018-12-18; Published 2019-1-29
Abstract
lncRNA can serve as a miRNA sponge and block the function of miRNA. High expression of lncRNA SNHG16 (small nucleolar RNAhostgene16) was discovered in gastric cancer (GC) and many other tumors. However, the mechanism of SNHG16 in GC is still unclear. In this research, we detected the expression level of SNHG16 in GC tissues and cell lines by qRT-PCR and FISH assay. RIP and Dual Luciferase Reporter Assay revealed that miR-135a is a target of SNHG16. SNHG16 gene knockdown experiment indicated that the expression level of SNHG16 can influence GC cells proliferation, colony formation, invasion ability and apoptosis in a miR-135a dependent manner. Western Blot assay showed that knockdown of SNHG16 decreased the expression of JAK2 and p-STAT3 in GC cells while miR-135a can offset the facilitated impact. Then the expression level of SNHG16 and miR-135a in the si-STAT3 GC cells was detected by qRT-PCR and the results showed that SNHG16 may be a target gene of p-STAT3. Collectively, it was suggested that SNHG16 can serve as a miR-135a sponge and block the function of miR-135a in JAK2/STAT3 pathway.
Keywords: SNHG16, miR-135a, JAK2/STAT3, gastric cancer, ceRNA
Introduction
Gastric cancer is one of the leading causes of cancer deaths worldwide. The latest statistics show that the incidence of gastric cancer is the fourth in the malignant tumor and the case fatality rate has become the third (1-2). The mortality of gastric cancer is closely linked with tumor lymphatic metastasis, bloodstream metastasis and peritoneal infiltration (3). Therefore, exploring the mechanisms involved in gastric cancer development to discovery a way to inhibit the proliferation and metastasis of gastric cancer will become the key to decrease cancer mortality.
Non-coding RNA is a type of single-stranded RNA that does not encode proteins. According to its length degrees, non-coding RNA can be divided into two groups. One is more than 50 bases in length, including long-chain non-coding RNA (lncRNA), circRNA, tRNA and so on (4). The other is less than 50 bases in length, including small RNA (miRNA) and small interference (siRNA) (5). Most researchers focused on two types of molecules, miRNA and lncRNA. A large number of studies have shown that non-coding RNAs can participate in the regulation of the occurrence and development of gastric cancer (6-8).
According to the literature, many miRNAs have been found to participate in proliferation, migration, invasion, and apoptosis of GC cells (9-12). Many studies have also shown that lncRNA can competitively bind to target mRNA against miRNA and affect the occurrence and development of tumor. For example, the HOX transcription antisense RNA (HOTAIR) and miR-331-3p competitively bind human epithelial growth factor receptor 2(HER2) to alter HER2 transcription, therefore accelerate the proliferation, migration and invasion of gastric cancer cells (13). Long non-coding RNA GAPLINC can be used as miR-211-3p molecular bait and promoted gastric cancer cell proliferation and invasion (14). In addition, MEG3 works as a competing endogenous RNA to promote gastric cancer progression (15). In conclusion, lncRNA can serve as a “miRNA sponge”, which can restore the expression level of target genes by blocking the function of miRNA.
LncRNA SNHG16, which is known as small nucleolar RNA host gene 16, was rarely studied. It was discovered in neuroblastoma and any other tumors. Recent studies have found that SNHG16 contributed to breast cancer, glioma, cervical cancer, and ovarian cancer (16-19). Wei's group demonstrated that SNHG16 contributes to gastric cancer progression and metastasis (20). However, the precise mechanism of SNHG16 in GC is still unclear.
In this investigation, we want to explore the biological function and mechanism of SNHG16 in GC cells. All the results indicated that SNHG16 functioned as an oncogene by sponging miR-135a-5p and up-regulated JAK2 expression in gastric cancer. This discovery demonstrated the mechanism of SNHG16 in GC which has not been reported yet.
1. Materials and Methods
1.1 Clinical Tissues Collection
32 pairs of surgically resected gastric cancer patients with tumor and corresponding non-tumor normal tissues samples were collected from the First Affiliated Hospital of Nanjing Medical University between Jan 2014 and Dec 2014. All patients were confirmed as gastric cancer by histopathological analysis and followed up until Dec 2017 without preoperative radiotherapy or chemotherapy. All fresh specimens were stored in liquid nitrogen immediately at -80 °C until use. The detailed criteria for the selection of the patient enrolled into the study was that the pathological type is adenocarcinoma, age and gender not to limit. The clinical parameters of the enrolled patients such as gender, age, TNM stage, tumor size and location were recorded and analyzed to correlate the in vitro findings with the clinical presentations (Table 1). Patient and their families have been fully informed that their specimens would be used for scientific research and signed the relevant informed consent.
clinicopathological characteristics of gastric cancer patients (n=32)
| Characteristics | All patients enrolled |
|---|---|
| Age | |
| <65 | 14 |
| ≥65 | 18 |
| Gender | |
| Male | 24 |
| Fmale | 8 |
| Tumor size | |
| <5cm | 16 |
| ≥5cm | 16 |
| TNM stage | |
| I-II | 10 |
| Ⅲ-Ⅳ | 22 |
| tumor location | |
| Antrum | 28 |
| body | 4 |
1.2 Cell cultures and plasmids design
Four gastric cancer cell lines (BGC823, MGC803, MKN45, SGC7901) and normal GC cell line GES-1 were purchased from Chinese Academy of Sciences (Shanghai Institute of Biochemistry and Cell Biology, Shanghai, China). All cultured cells were maintained routinely in RPMI-1640 media (Gibco, 11875-093) supplemented with 10% fetal bovine serum (Gibco, 10049-141) in incubator containing 5% CO2 at 37℃.The siRNA SNHG16 plasmids were designed to knock down the SNHG16 gene expression. PmirGLO-SNHG16-WT and PmirGLO-SNHG16-MUT were designed for dual luciferase reporter assay. Has-miR-135a-5p inhibitors were purchased from Invitrogen.
1.3 Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from GC tissues specimens and GC cell lines with Trizol reagent (Invitrogen, 15596026) according to the manufacturer's instruction. Reverse transcription of lncRNA and mRNA were performed using Prime Script RT Reagent Kit (Takara, RR047). MiRNA was reverse transcribed using One-step miRNA reverse transcription kit (Haigene, china). qRT-PCR was carried out with SYBR® Premix Ex Taq (Takara, RR42LR) on VII™7 System according to the manufacturer's protocol (Applied Biosystems, USA). The levels of lncRNA, miRNA and mRNA were separately calculated with the 2-ΔΔCt method, which were normalized to U6 and GAPDH mRNA. The primers for qRT-PCR were as follows:
SNHG16 (F:GTGCCTCAGGAAGTCTCTTGCC; R:ATCCAAACAAGTTATCACACAGCAC); miR135a (F:TATGGCTTTTTATTCCTATGTGA;R:GCTGTCAACGATACGCTACCTA); GAPDH (F:ATGGGGAAGGTGAAGGTCG;R:TTACTCCTTGGAGGCCATGTG); U6 (F:CTCGCTTCGGCAGCACA;R:AACGCTTCACGAATTTGCGT).
1.4 Cell Viability Assay
Cell viability was detected using the cell counting Kit-8(CCK8, beyotime, china) according to the manufacturer's guide. After 24h of transfection, cells were collected and plated into a 96-well plate (2,000 cells/well). The CCK8 regent (10 µL) was added to each well at 0, 24, 48,72h after cells were seeded. Following incubation for 2 h, the cell viability was calculated by measuring the optical density of cell lysates at a wavelength of 450 nm. All assays were repeated in triplicate and three replicate wells were performed in each group.
1.5 Colony Formation Assay
Transfected GC cells were plated in 6-well plates and incubated for 14 days (500 cells/well). Then the cells in the well were fixed and stained with 0.1% of crystal violet. The colony formation ability of cells was calculated by counting the number of visible colonies.
1.6 Cell apoptosis analysis
Cells that were transfected with indicated plasmid or negative control were reaped after 48 h and washed twice with PBS. Then the cells were incubated in binding buffer containing Annexin V-FITC and PI according to the manufacturer's protocol (BD Biosciences, USA). Stained cells were analyzed by flow cytometry using the CELLQUEST program (Becton Dickinson, USA). Three independent experiments were performed.
1.7 Cell invasion
Transwell chamber (8 μm pore size, Corning) which is coated with Matrigel (BD Biosciences, USA) was used to measure cell invasion. 48 h after transfection, cells in serum-free media were put into the upper chamber. Then, media containing 10% of bovine calf serum was added into the lower chamber. 48 h after incubation, cells remaining in the upper membrane were wiped off, while migrated cells were fixed in methanol, stained with 0.1% of crystal violet and counted under a microscope. Three independent experiments were implemented.
1.8 Western Blot Analysis
Transfected GC cells were collected and lysed with RIPA lysis buffer (epizyme, china, PC101) containing protease inhibitors. The protein concentration was measured by bicinchoninic acid (BCA) protein assay Kit (epizyme, china, ZJ101). Equal amounts of protein samples were separated on sodium dodecyl sulphate-polyacrylamide gel (SDS-PAGE) electrophoresis (epizyme, china, PG110) and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, USA). The membranes were blocked with 1× blocking buffer (epizyme, china, PS108) for 30 mins, followed by the incubation with primary antibodies at 4℃ overnight. The following primary antibody were used: JAK2 (Cell Signaling, USA, 3230), P-STAT3 (Cell Signaling, USA, 9145), GAPDH (ProteinTech, USA, 10494). After incubating with secondary-HRP-antibodies (ProteinTech, USA, SA00001) for 2h at room temperature, signal on the membranes were visualized by using an enhanced chemiluminescence kit (epizyme, china, SQ201). The pictures of protein bands were taken by Tanon 5200 Biotanon and the results were analyzed with image J software.
1.9 Dual Luciferase Reporter Assay
SNHG16 fragment containing putative miR-135a binding site was cloned into pmirGLO vector (Promega, USA) and named PmirGLO-SNHG16-WT. Then we constructed a PmirGLO-SNHG16-MUT which the putative miR-135a binding site was mutated. The two reporter plasmids and empty vector plasmid were co-transfected with miR-135a mimics or miR-NC into HEK293T cells respectively. The relative luciferase activities were measured by a dual-luciferase reporter assay system (promega, USA, E1910) at 48h after transfection. Firefly luciferase activity was normalized against Renilla luciferase activity.
1.10 RNA Immunoprecipitation
RNA immunoprecipitation was performed by using RIP kit (Millipore, USA, 17-700) according to the manufacturer's instructions. Ago2 antibodies and IgG negative control were both purchased from Abcam. qRT-PCR was used to detect lncRNA SNHG16 and miR-135a in the RISC complex.
1.11 In situ hybridization of lncRNA SNHG16
Gastric cancer tissue sections were hybridized with the lncRNA SNHG16 probes (Exiqon, JAPAN). Probe mix was denatured at 85℃ and hybridization was carried out at 65℃ overnight. Sections were washed using reducing concentrations of SSC. Then slides were treated with 5% blocking solution for 30min at room temperature. Each section was incubated with 100μlHRP-labelled anti-DIG antibody (Perkin-Elmer, USA, NEF832001EA) at 1:500 in blocking buffer overnight at 4℃. Wash 3×10 min with TBS and make up TSA staining solution by using Perkin-Elmer TSA Plus kit (Perkin-Elmer, USA, NEL753001KT) according to the manufacturer's instructions. Incubate in DAPI-containing TBS, rinse in water, air dry, and mount in aqueous fluorescence mounting media. Images were captured using Leica SP8 laser scanning confocal microscope.
1.12 Statistical Analysis
All data are presented as mean ±standard deviation (SD) from three independent experiments. The software GraphPad Prism 6 was used for statistical analysis. Significance of differences between two groups or among multiple groups were determined by using student's t-test or one-way ANOVA analysis. The level of significance was set at p < 0.05, p < 0.01 and p < 0.001 in which p < 0.05 were considered statistically significant.
2. Results
2.1 SNHG16 was up-regulated in GC cell lines and GC tissues
The expression level of SNHG16 in GC cell lines (BGC823, MGC803, MKN45, SGC7901) were all up regulated in different degrees. The expression level in MGC-803 is almost three times as high as GES-1 (Fig. 1A). However, the expression level of miR-135a in GC cell showed opposite trend of change which proved that there may be interaction between SNHG16 and miR-135a (Fig. 1B). The GC cell line MGC-803 was used for the following experiments.
Expression level of lncRNA SNHG16 in GC cell lines and tissues was detected by qRT-PCR. A, Expression level of lncRNA SNHG16 in GC cell lines; B, Expression level of lncRNA miR-135a in GC cell lines; C, Expression level of lncRNA SNHG16 in GC tissues; D, Kaplan-Meier survival curves of patients with GC based on SNHG16 expression. Error bars represented the mean ± SD of more than two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001vs. control group. Data were presented as 2-∆∆Ct.
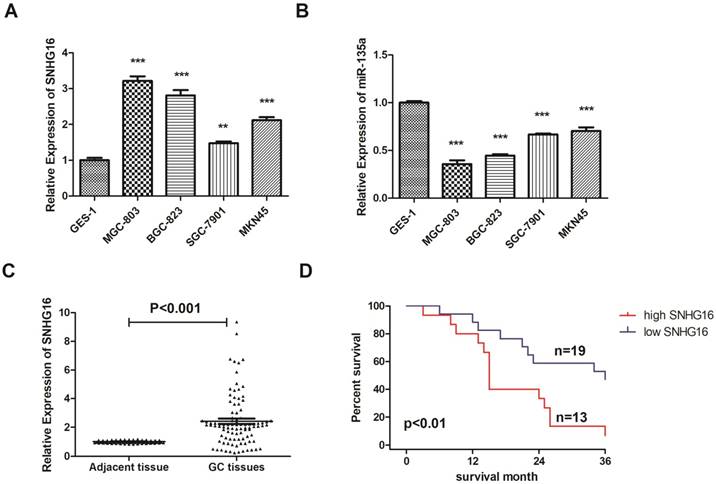
In addition, the expression level of SNHG16 in GC tissues was measured by qRT-PCR (Fig. 1C) and results showed that SNHG16 was up-regulated in GC tissues compared with normal tissues. Three pairs of GC tissues and adjacent tissues were used for FISH assay and the results indicated that SNHG16 can be detected in GC tissues while in adjacent tissues was barely detectable (Fig. 2B).
A. Relationship between lncRNA SNHG16 expression level and GC clinicopathological features. Data were presented as 2-∆∆Ct. p < 0.05 were considered statistically significant. B. Expression level of lncRNA SNHG16 in GC tissues and Adjacent GC tissues was detected by FISH. Tissues from three patients were used for detection. Green fluorescence represents the expression pattern of SNHG16 and blue fluorescence is DAPI.
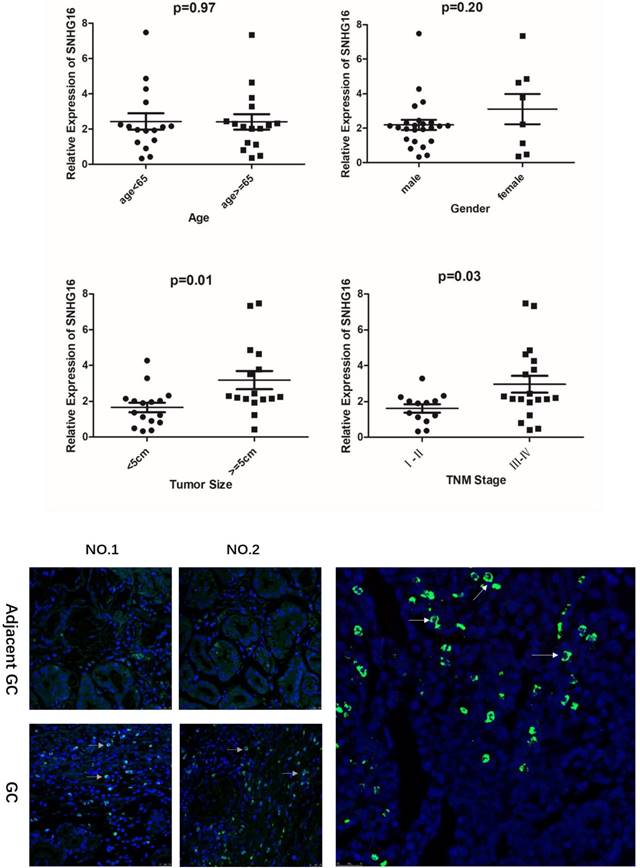
2.2 Relationship between lncRNA SNHG16 expression level and clinicopathological features of Gastric Cancer
As shown in the results that the lncRNA SNHG16 expression have no clearly relevance to gender or age (P >0.05) but was closely connected to the GC tumor size and TNM staging (P<0.05), indicating that severer invasion and more advanced stage may lead to higher SNHG16 expression level in GC tissues (Fig. 2A). Kaplan-Meier analysis and log-rank test were used to evaluate the effect of SNHG16 expression on survival to explore the potential relationship between SNHG16 and the patients' prognosis (Fig. 1D). The results indicated that patients with higher SNHG16 expression had a significantly poorer survival.
2.3 Mir-135a is a target of SNHG16
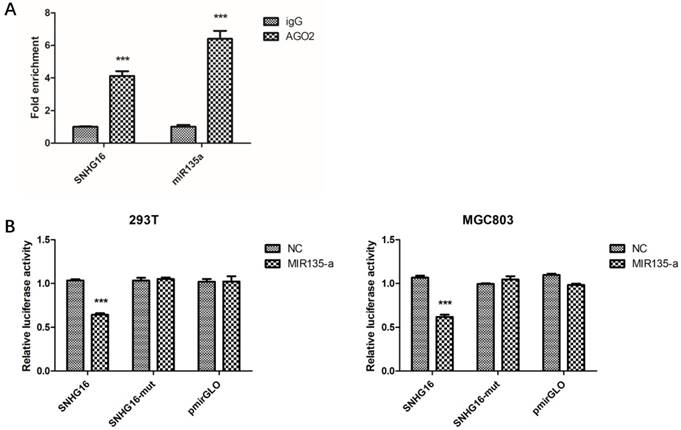
Preliminary study had proven lncRNA can work as ceRNA and sponge miRNAs (21-22). Bioinformatics analysis showed that SNHG16 may interact with miR-135a. In order to verify this, RIP assay was performed to confirm the interaction. The result of RIP revealed that SNHG16 RNA can be found in Ago2 immunoprecipitates which may participate in RISC complex (Fig. 3A). The most direct evidence of interaction was from dual-luciferase assay. The dual-luciferase assay indicated that the relative luciferase activity in cells transfected with pmirGLO-SNHG16-WT and miR-135a mimic recombinant vector was significantly decreased compared with that in cells transfected with pmirGLO-SNHG16-MUT vector. The FISH results showed that the SNHG16 was mainly expressed in the cytoplasm of GC cells where mature miRNA located in (Fig.2B). All these data demonstrated the fact that miR-135a is a target of SNHG16 (Fig. 3B).
The interaction between SNHG16 and miR-135a. A, The enrichment of SNHG16 bound to Ago2 or IgG was measured by RT-qPCR after RIP, in which IgG was used as a negative control; B, Luciferase assays of the cells transfected with pmirGLO-SNHG16-WT or pmirGLO-SNHG16-MUT reporter and NC or miR-miR135a-5p mimic. Error bars represented the mean ± SD of more than two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001vs. control group.
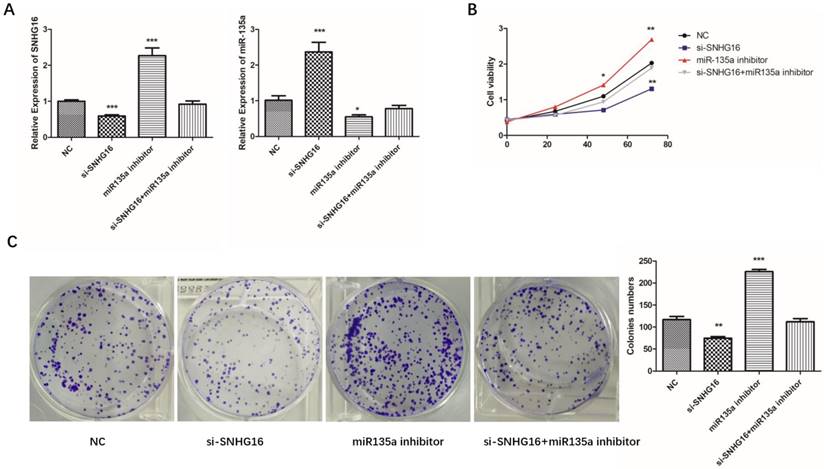
In order to explore the relationship of SNHG16 and miR-135a in GC cells, SNHG16 gene knockdown experiment was performed. As shown in Fig. 4A, there is a negative relationship between SNHG16 and miR-135a expression level in GC cells.
Effects of si-SNHG16 and miR135a inhibitor on cell proliferation and colony formation. A, Expression level of SNHG16 and miR-135a in each group were detected by qRT-PCR; B, Ability of proliferation of each group were measured by CCK-8 assay; C, colony formation assay. Error bars represented the mean ± SD of more than two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001vs. control group.
2.4 Expression level of SNHG16 can influence GC cells physiology
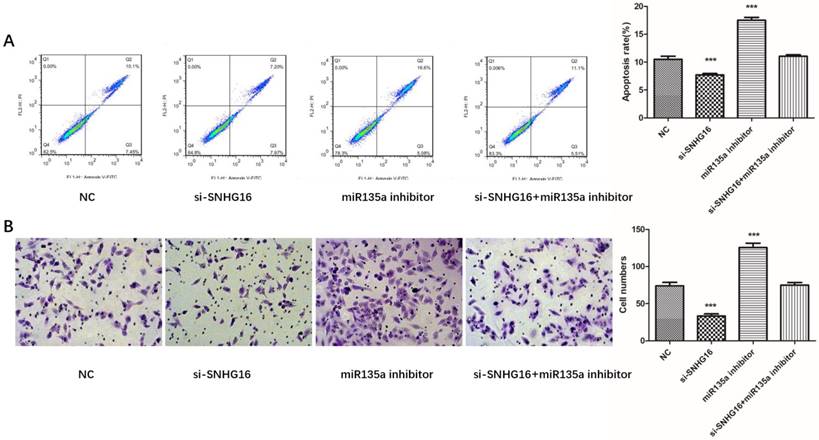
To further investigated the biological function of SNHG16 on GC cells. MGC803 cells were transfected with si-SNHG16 in combination with miR-135a inhibitor. As shown in the Fig. 4B and Fig. 4C, knock down of SNHG16 can decrease GC cells proliferation rate and colony formation. Meanwhile, the cell invasion ability and apoptosis level were also decreased by SNHG16 knockdown (Fig. 5A and Fig. 5B). However, these results can be reversed by additional transfection of miR-135a inhibitor. All the results indicated that the expression level of SNHG16 can influence GC cells physiology in a miR-135a-dependent manner.
Effects of si-SNHG16 and miR135a inhibitor on cell invasion and apoptosis of GC cells. A, Apoptosis of each group analyzed by flow cytometry; B, Comparisons of cell invasion in each group. Error bars represented the mean ± SD of more than two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001vs. control group.
2.5 SNHG16 mediates up-regulation of JAK2 and STAT3 by sponging miR-135a in GC cells
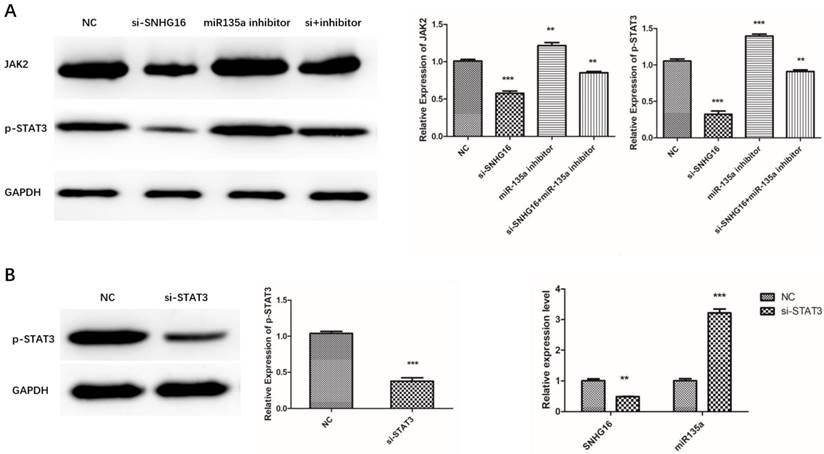
Our previous study had proved that miR-135a targets JAK2/STAT3 pathway and inhibits GC cell proliferation (23). This study indicated SNHG16 can sponge miR135a and influence GC cell physiology. Therefore, we assumed that SNHG16 may mediate the regulation of JAK2 and p-STAT3 by sponging miR135a in GC cells. To verify this hypothesis, the expression level of JAK2 and p-STAT3 in MGC803 cells transfected with siRNA SNHG16 in combination with miR-135a inhibitor was measured by Western Blot (Fig. 6A). The results showed that knockdown of SNHG16 decreased the expression of JAK2 and p-STAT3 in GC cells while miR-135a can offset the facilitated impact.
Effects of si-SNHG16 and si-STAT3 on expression level of related gene in GC cells. A, Effects of si-SNHG16 and miR-135a inhibitor on expression level of JAK2 and p-STAT3; B, Effects of si-STAT3 on expression level of SNHG16 and miR-135a; Error bars represented the mean ± SD of more than two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001vs. control group.
2.6 STAT3 may affect the expression of SNHG16
As is known to all, STAT3 is a transcription factor which can be activated by JAK2 mediated phosphorylation in cancer cells. In order to test whether p-STAT3 can regulate the transcription of SNHG16, STAT3 was knocked down in GC cells. Then the expression level of SNHG16 and miR-135a in the si-STAT3 GC cells was detected by qRT-PCR (Fig. 6B). The results indicated that when the STAT3 is degraded, the SNHG16 is decreased and expression level of miR-135a is increased. All the findings suggested that p-STAT3 may affect the expression of SNHG16 in a direct or indirect manner. More evidence will be discovered from CHIP and any other experiments.
3. Discussion
Further exploration about the molecular mechanism of the initiation and progression of Gastric Cancer is helpful to find out novel therapeutic targets of GC. LncRNA has been known as important regulators in biological processes by modulating gene expression at transcriptional, post-transcriptional and epigenetic level. In recent years, more and more lncRNAs were identified as being involved in development of cancers. SNHG16 which was known as a new identified lncRNA was found to have close relationships with many cancers. However, the relationship between SNHG16 and GC was rarely reported. In this research, we carried out an in-depth study on SNHG16. The results of qRT-PCR have showed that the expression level of SNHG16 in GC tissues and cell lines is much higher than normal control. The analysis of clinicopathological features and SNHG16 expression level of patients' samples indicated that SNHG16 expression level have a positive correlation with tumor size and stage in GC tissues and negative correlation with survival of GC patients. Cellular experiments uncovered that SNHG16 promotes the proliferation, colony formation and cell invasion ability. All these results indicated that SNHG16 and Gastric Cancer are closely related. However, the molecular mechanisms of these phenomena are still unclear.
The JAK2/STAT3 signaling pathway plays an important role in cell proliferation, differentiation and apoptosis (24). Once JAK2 protein was activated by cytokine, the STAT3 protein can be phosphorylated. The p-STAT3 protein has a DNA binding domain and works as a transcription factor to regulate downstream genes. The JAK2/STAT3 pathway has been intensely investigated in gastric and other cancers (25-26). Previously studies have demonstrated that miR-135a worked as a tumor suppressor by targeting JAK2 to repress p-STAT3 activation and inhibit gastric cancer cell proliferation (27). In this research, a series of experiments have proved that lncRNA SNHG16 can be targeted by miR-135a. We verify that SNHG16 could serve as a ceRNA by competitively sponging miR-135a with JAK2. The inhibition effect of miR-135a on JAK2 was suppressed and JAK2/STAT3 pathway was promoted. Therefore, JAK2/STAT3 signal pathway can be elevated by SNHG16 and then the development of tumor is promoted. This interaction between SNHG16 and miR135a enhanced our comprehension on the role of RNA-RNA interaction and lncRNA-based mechanisms of GC tumorigenesis.
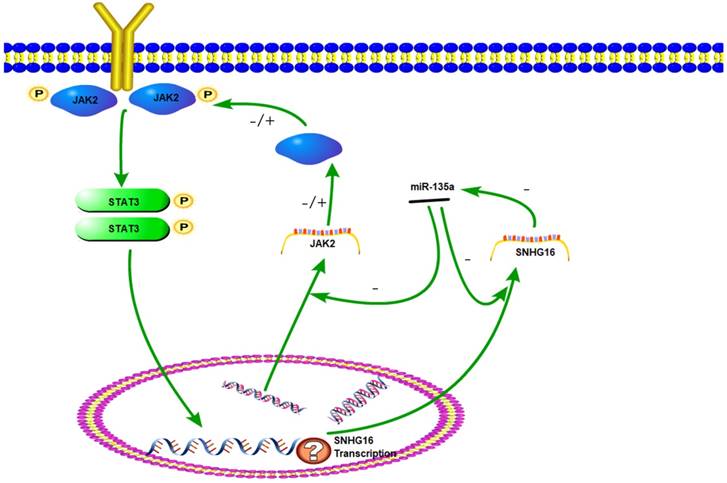
As a key component of JAK2/STAT3 signaling pathway, STAT3 could be phosphorylated and activated. Then p-STAT3 was dimerizated and worked as a transcription factor. Studies have found that the downstream target genes regulated by STAT3, such as bcl-2, survivin and cyclinD1, are closely related to tumor cell cycle, proliferation, differentiation, and apoptosis (28-29). In this study, STAT3 knock down experiment indicated that expression level of SNHG16 is positively correlated with p-STAT3. Based on these results, we hypothesized that SNHG16 is a target gene of p-STAT3. Therefore, JAK2, miR-135a, SNHG16 and STAT3 could form a signal circuit (Fig 7). If these assumptions are true, we will have a deeper understanding of the function of SNHG16 and the JAK2/STAT3 signaling pathway. However, more work needs to be done to verify the hypothesis.
The sketch of the mechanism of lncRNA SNHG16 on JAK2/STAT3 signaling pathway.
Acknowledgements
This work was supported by a grant from Project of science and Technology Department of Qinghai Province (No. 2015-ZJ-751), and the National Natural Science Foundation of China (No. 81301898).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liang D, Liang S, Jin J. et al. Gastric cancer burden of last 40 years in North China (Hebei Province): A population-based study. Medicine (Baltimore). 2017;96:e5887
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
3. Hamashima C, Shabana M, Okada K. et al. Mortality reduction from gastric cancer by endoscopic and radiographic screening. Cancer Sci. 2015;106:1744-9
4. Volders PJ, Helsens K, Wang X. et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246-51
5. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-31. Review. Erratum in: Nat Rev Genet. 2004;5:631
6. Li D, Chen Y, Mei H. et al. Ets-1 promoter-associated noncoding RNA regulates the NONO/ERG/Ets-1 axis to drive gastric cancer progression. Oncogene. 2018;37:4871-4886
7. Zhang M, Du X. Noncoding RNAs in gastric cancer: Research progress and prospects. World J Gastroenterol. 2016;22:6610-8
8. Li T, Mo X, Fu L. et al. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget. 2016;7:8601-12
9. Duan J, Zhang H, Qu Y. et al. Onco-miR-130 promotes cell proliferation and migration by targeting TGFβR2 in gastric cancer. Oncotarget. 2016;7:44522-44533
10. Zhang H, Duan J, Qu Y. et al. Onco-miR-24 regulates cell growth and apoptosis by targeting BCL2L11 in gastric cancer. Protein Cell. 2016;7:141-51
11. Wang F, Liu J, Zou Y. et al. MicroRNA-143-3p, up-regulated in H. pylori-positive gastric cancer, suppresses tumor growth, migration and invasion by directly targeting AKT2. Oncotarget. 2017;8:28711-28724
12. Mi Y, Zhang D, Jiang W. et al. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer Lett. 2017;389:11-22
13. Liu XH, Sun M, Nie FQ. et al. LncRNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92
14. Hu Y, Wang J, Qian J. et al. Long noncoding RNA GAPLINC regulates CD44-dependent cell invasiveness and associates with poor prognosis of gastric cancer. Cancer Res. 2014;74:6890-902. Erratum in: Cancer Res. 2015;75:3683
15. Yan H, Rao J, Yuan J. et al. Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate ischemic neuronal death by targeting miR-21/PDCD4 signaling pathway. Cell Death Dis. 2017;8:3211
16. Yan L, Jiao D, Hu H. et al. Identification of lymph node metastasis-related microRNAs in lung adenocarcinoma and analysis of the underlying mechanisms using a bioinformatics approach. Exp Biol Med (Maywood). 2017;242:709-717
17. Cai C, Huo Q, Wang X. et al. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485:272-278
18. Duan W, Du L, Jiang X. et al. Identification of a serum circulating lncRNA panel for the diagnosis and recurrence prediction of bladder cancer. Oncotarget. 2016;7:78850-78858
19. Christensen LL, True K, Hamilton MP. et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10:1266-82
20. Lian D, Amin B, Du D. et al. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21:151-160
21. Ding J, Yeh CR, Sun Y. et al. Estrogen receptor β promotes renal cell carcinoma progression via regulating LncRNA HOTAIR-miR-138/200c/204/217 associated CeRNA network. Oncogene. 2018;37:5037-5053
22. Xu J, Feng L, Han Z. et al. Extensive ceRNA-ceRNA interaction networks mediated by miRNAs regulate development in multiple rhesus tissues. Nucleic Acids Res. 2016;44:9438-9451
23. Wu H, Huang M, Cao P. et al. MiR-135a targets JAK2 and inhibits gastric cancer cell proliferation. Cancer Biol Ther. 2012;13:281-8
24. Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18:545-51
25. Hajimoradi M, Mohammad Hassan Z, Ebrahimi M. et al. STAT3 is Overactivated in Gastric Cancer Stem-Like Cells. Cell J. 2016;17:617-28
26. Vera J, Rateitschak K, Lange F. et al. Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol. 2011;106:426-34
27. Xiong H, Du W, Wang JL. et al. Constitutive activation of STAT3 is predictive of poor prognosis in human gastric cancer. J Mol Med (Berl). 2012;90:1037-46
28. Kanda N, Seno H, Konda Y. et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921-9
29. Deng JY, Sun D, Liu XY. et al. STAT-3 correlates with lymph node metastasis and cell survival in gastric cancer. World J Gastroenterol. 2010;16:5380-7
Author contact
![]() Corresponding author: Hao Wu, Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, Jiangsu Province, PR China. Tel: +86-25-68306393; E-mail: whdactorcom
Corresponding author: Hao Wu, Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, Jiangsu Province, PR China. Tel: +86-25-68306393; E-mail: whdactorcom