Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(14):4206-4218. doi:10.7150/jca.117290 This issue Cite
Research Paper
Extrachromosomal circular DNA of multiple myeloma
Fangfang Li1, Xinyi Long1, Sishi Tang1, Jing Liu1 ![]() , Yunfeng Fu2
, Yunfeng Fu2 ![]()
1. Department of Hematology, The Third Xiangya Hospital, Central South University, Changsha, 410013, China.
2. Department of Blood Transfusion, The Third Xiangya Hospital, Central South University, Changsha, 410013, China.
Received 2025-5-10; Accepted 2025-9-13; Published 2025-10-1
Abstract

Background: Extrachromosomal circular DNA (EccDNA) is widespread in various heterogeneous tumors and closely associated with tumor resistance and progression.
Methods: Circle-seq and mRNA-seq were done on samples from three multiple myeloma (MM) patients, one when they had a complete response and one when they relapsed.
Results: A large number of EccDNA molecules were detected with high heterogeneity among the six samples. Circle-seq combined with mRNA-seq analyses revealed that there is no linear relationship between mRNA expression and EccDNA quantity. Chromosome 19 presented the highest density of differentially expressed EccDNA genes, followed by chromosome 17. Only the T3 sample showed del(17p) by fluorescence in situ hybridization at the time of relapse. Approximately 92% of all upregulated EccDNA genes from chromosome 17 (137) were present in the T3 sample. By integrating Circle-seq and mRNA-seq data, we obtained several potentially functional candidate protein-coding genes and miRNAs from chromosome 17. We further assessed the prognostic value of the three protein-coding genes in the MMRF-COMPASS clinical trial and found that all three genes were poor prognostic indicators for MM. Furthermore, WB, CCK8 and Annexin V-FITC/PI assays revealed that the overexpression of 2',3'-cyclic nucleotide 3' phosphodiesterase (CNP) downregulated the apoptotic pathway and increased bortezomib tolerance in MM cells.
Conclusion: We performed a Circle-seq analysis of MM and investigated the heterogeneity of EccDNA. Analysis of data from clinical studies and basic experiments revealed that the gene carried on EccDNA most likely contributed to the increased tolerance of bortezomib in MM with del(17p).
Keywords: Multiple myeloma, Extrachromosomal circular DNA (EccDNA), Circle-seq, del (17p), Heterogeneity, CNP
Introduction
Multiple myeloma (MM), a type of clonal plasma cell tumor, is the second most prevalent hematological cancer [1,2]. The development of novel drugs such as proteasome inhibitors has prolonged the survival of patients with MM by up to 3-6 years. Although significant progress has been made in MM treatment strategies, the 10-year survival rate is only 17% [3]. The clonal evolution of MM is eventually induced under various therapeutic pressures, ultimately resulting in drug resistance, disease progression, and relapse [4]. Treatment is often challenging when the disease progresses to drug resistance. Hence, identification of the underlying mechanism of relapsed refractory MM is urgently needed to identify novel and effective treatment strategies.
Extrachromosomal circular DNA (EccDNA) is a group of circular DNA molecules that can be double- or single-stranded. These molecules vary in size and sequence, and they exist in the nucleus. They are derived from chromosomes, but they are independent of them [5,6]. EccDNA was first confirmed in 1965, and research increased with the development of Circle-seq in 2015 [7]. EccDNA is widely distributed in eukaryotic organisms [5], including plants [7-9], ciliates [10], yeasts [11], xenopus [12], drosophila [13], mammals [14], and pigeons [15]. Numerous physiological and pathological processes are impacted by EccDNA, as confirmed by several studies, including gene amplification, telomere length restoration, genome plasticity, and molecular sponges [5]. Almost all the genes carried on EccDNA can be matched to human autosomes, indicating that EccDNA originates from autosomes rather than another independent set of genes. Although the mechanism of EccDNA production is still unclear, four widely accepted hypothesis models exist: the breakage-fusion-bridge (BFB) cycle, chromothripsis, episome model, and translocation-deletion-amplification model [5,16]. In conclusion, EccDNA is the product of a highly unstable genome. EccDNAs can be categorized into five types according to their size and sequence: small polydispersed DNA (spcDNA), microDNA, t-circle/c-circle, ERC, and EcDNA. Tumor cells are the main source of microDNA and EcDNA. MicroDNA can transcribe noncoding RNAs and act as regulators in a series of biological processes, whereas EcDNA is large enough to carry complete genes, thus participating in oncogene amplification and genomic heterogeneity [5]. Yi et al. established the CRISPR-based ecTag method to tag EcDNA with fluorescent markers in living cells and reported an uneven distribution of EccDNA during mitosis [17]. EccDNA does not have centromeres; therefore, EccDNA is unevenly distributed in offspring cells during cell division, mediating intercellular heterogeneity. EccDNA has been studied in various highly heterogeneous tumors and is involved in tumor progression and drug resistance [18-23]. EccDNA carrying oncogenes is associated with gastric cancer carcinogenesis and participates in gastric cancer progression [20]. DNMT1circle10302690-10302961 is associated with metastasis and poor prognosis in advanced high-grade serous ovarian cancer [21]. Amplification of the RAB3B-encoding gene on EccDNA leads to high expression of RAB3B, which induces cisplatin resistance in hypopharyngeal squamous cell carcinoma through autophagy [19]. High expression of PLCG2 is caused by the amplification of the PLCG2-encoding gene on EccDNA, and metastasis of non-small cell lung cancer is mediated by this high expression through enhanced mitochondrial respiration [22]. Amplification of the KIF3C-encoding gene on EccDNA downregulates MUC20, which weakens copper-mediated cell death, thereby inducing proteasome inhibitor resistance in multiple myeloma [24]. Zou et al. synthesized DNA circles harboring the miR-17-92 cluster and verified that artificial DNA circles harboring the miR-17-92 cluster can significantly promote the proliferation and promotion of hepatocellular carcinoma [25]. Paulsen et al synthesized microDNA molecules mimicking known microDNA sequences and discovered that they could create short regulatory RNA molecules that can repress gene expression without relying on the usual promoters [26]. It is evident that the efficient transcription of coding genes carried on EccDNA is essential for EccDNA to function in tumors.
The differential expression of EccDNA in completely relieved and relapsed MM was investigated to identify the mechanisms and potential therapeutic targets of EccDNA in relapsed refractory MM. Therefore, we first explored the differential expression profiles of EccDNA between completely relieved and relapsed MM. We found that EccDNA heterogeneity was present not only between groups but also among the six samples. We also found that chromosome 19 had the highest density of differentially expressed EccDNA genes, followed by chromosome 17. Among the six samples, only T3 was del(17p), according to fluorescence in situ hybridization. Among all the upregulated differentially expressed EccDNA genes from chromosome 17 (137) in the six samples, approximately 92% were present in the T3 sample. We further integrated Circle-seq and mRNA-seq data and obtained several potentially functional candidate protein-coding genes and miRNAs from chromosome 17. Further analysis revealed that these candidate genes were located on three EccDNA molecules, with two being EcDNA and one being microDNA. However, EcDNA is too long to be synthesized. To further explore the function of EccDNA, we first assessed the prognostic value of the three-candidate protein-coding genes in the MMRF-COMPASS clinical trial and then selected CNP for WB, CCK8, and Annexin V-FITC/PI assays. We found that all three-candidate protein-coding genes were poor prognostic indicators for MM and that the overexpression of CNP increased bortezomib resistance in MM cells.
Materials and Methods
Patient selection and sample collection for sequencing
Due to the large volume of bone marrow samples required during remission and the fact that samples taken at complete remission and at relapse were obtained from the same patient, the samples were not purified for plasma cells. The effect evaluation of MM patients hospitalized at Xiangya Third Hospital of Central South University was performed according to the International Myeloma Working Group (IMWG) response criteria [27]. When patients achieve complete remission and when they relapse, we collect bone marrow samples, further collect bone marrow mononuclear cells, and immediately transfer them to a -80°C freezer. The study was conducted with the patients' written informed consent and the approval of the Xiangya Third Hospital of Central South University's Ethics Committee.
Cell lines
Four human MM cell lines—NCI-H929, RPMI 8226, ARP1, and U266—were procured from ATCC (Manassas, VA, USA). These cells were cultivated in RPMI 1640 medium (Basal Media, Shanghai, China), which was supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA), 100 U/mL penicillin-streptomycin (100X) (New Cell & Molecular, Suzhou, China). The cells were cultivated in a humidified incubator with 5% CO2 at 37 °C. Both cell lines were authenticated via short tandem repeat (STR) profiling within the last three years, and cells free from mycoplasma were utilized in all experiments.
EccDNA purification, circle-seq, data analysis
Previous reports have described how to purify, roll-circle amplify, and sequence EccDNA [28,29]. Briefly, we used the QIAamp DNA Mini Kit (USA) to extract genomic DNA from bone marrow samples, followed by DNA quality control using a Nanodrop 1000 instrument and agarose gel electrophoresis. PacI restriction enzyme (New England Biolabs, USA) was used to linearize mitochondrial DNA (mtDNA). phenol/chloroform/isoamyl alcohol (PCI) solution (25:24:1) (Solarbio, Beijing, China) was used for DNA extraction. Phi29 DNA polymerase (New England Biolabs, USA) was used for rolling circle amplification (RCA). The Bioruptor® apparatus (Diagenode, Belgium) was utilized to shear amplified EccDNA to an average fragment size of 400 base pairs (bp). The NEBNext® Ultra™ II DNA Library Prep Kit (New England Biolabs, USA) was employed for library preparation for the EccDNA-seq. The Illumina cBot system and NovaSeq 6000 S4 kit (300 cycles) (USA) were used for library clustering. The Illumina NovaSeq 6000 platform was utilized in the generation of raw sequencing data. In order to trim the QC-filtered raw reads, the fastp software, version 0.23.2, was utilized. The Bioinformatics Workflow Advisor (BWA) software version 0.7.17 was utilized to perform alignment with the human genome (UCSC HG38). The detection of EccDNA was facilitated by the utilization of the Circle-Map V1.1.4 software, which was employed to analyze the sequencing data. A comparison of the differences in EccDNA genes between sample groups was conducted using total base pair evidence normalization (fragments per million: FPM). The cut-off criteria for identifying differentially expressed EccDNAs was established as follows: a |log2FoldChange| value of at least 1 and a p-value less than 0.05. In the event that a valid p-value was not obtained, the criteria were adjusted to |log2FoldChange| ≥ 3.
mRNA enrichment, mRNA-seq, and data analysis
TRIzol reagent (Invitrogen, Carlsbad, California, USA) was utilized to extract total RNA from bone marrow samples. The quality of the extracted RNA was subsequently assessed using a Nanodrop 1000 instrument and agarose gel electrophoresis. The Next® Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, USA) was utilized for the purpose of enriching mRNA. The preparation of mRNA sequencing libraries was undertaken with the utilization of the KAPA Stranded RNA-Seq Library Prep Kit (Roche, Switzerland). The Agilent 2100 Bioanalyzer (USA) was utilized in the evaluation of the quality of the constructed libraries. The NovaSeq 6000 S4 Kit (300 cycles) (Illumina, USA) was employed for in situ dilution and amplification of the prepared libraries. The Illumina NovaSeq 6000 (USA) was used for sequencing. The fastp software (V0.23.2) was used for quality control of the raw sequence data. BWA software (V0.7.17) was used for alignment to the human genome (UCSC HG38). The quantification of sample expression levels, as well as the differential expression analysis of genes and transcripts between groups, were performed using the Ballgown R package. The screening thresholds for differentially expressed genes and transcripts were established as follows: a fold change of at least 1.5, a p-value of less than 0.05%, and an FPKM mean of at least zero for each group.
Transfection
Sangon Biotechnology (Shanghai, China) supplied the CNP-overexpressing pcDNA3.1 plasmids and negative controls, and we used Lipo8000™ (Beyotime, Shanghai, China) for cell transfection in 12-well plates.
RNA isolation, real-time quantitative reverse transcription‒PCR (RT‒qPCR)
RNAiso Plus (TaKaRa, Tokyo, Japan) was used to extract total RNA from MM cells. CNP and β-actin were quantified via HiScript II Q RT Super Mix for qPCR and ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China). The experiment was conducted on three separate occasions. The primers utilized were as follows: β-actin, forward: 5ʹ-GAGACCTTCAACACCCCAGC-3ʹ; reverse: 5ʹ-ATGTCACGCACGATTTCCC-3ʹ; and CNP, forward: 5ʹ-GAGTACGCTCAACAAGATGTGT-3ʹ; reverse: 5ʹ-AGCTTGTCCACATCACTCGG-3ʹ.
Cell viability assay
The half-maximal inhibitory concentration (IC50) was determined via a Cell Counting Kit-8 (Abiowell, Changsha, China). 200 μL cell suspension (1×104 cells/well) were seeded in a 96-well plate and treated with various doses of bortezomib (4-32 nmol/L) (MedChemExpress, USA). After 24 hours, each well was added with 20 µL of CCK-8 solution. After incubating for two hours at 37°C, the absorbance was measured at 450 nm. The experiment was replicated thrice.
Annexin V-FITC/PI assay
Annexin V-FITC/PI apoptosis detection kit (Vazyme, Nanjing, China) was used to analyze apoptosis (Vazyme, Nanjing, China). The cells were exposed to bortezomib (Med Chem Express, USA) at a concentration of 10 nmol/L for a duration of 24 hours. The evaluation of apoptotic cells was subsequently conducted and the subsequent analysis of the data was facilitated. We did the experiment thrice.
Fluorescence in situ hybridization (FISH)
The analysis of CD138-purified plasma cells was conducted using a fluorescence in situ hybridization (FISH) approach, employing the p53/D13S319 probe (provided by HealthCare Biotechnology, Wuhan, China). In each analysis, a minimum of 200 interphase nuclei were considered, and an abnormal threshold of 10% was established.
Western blotting (WB)
The cell lysis buffer (Beyotime, Shanghai, China) was used to lyse the cells after collection for WB analysis, with the buffer containing 10 nmol/L protease and phosphatase inhibitors. The bands were visualized via enhanced chemiluminescence (ECL) reagent (Absin, Shanghai, China). An anti-caspase-3 antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). The anti-β-actin and HRP-conjugated goat anti-rabbit IgG antibodies were obtained from ABclonal in Wuhan, China.
Statistical analysis
The data are expressed as the means ± SDs of at least three independent experiments. The statistical significance of the differences was evaluated via a student's t test using GraphPad Prism software (Prism 10). Statistical significance was set at P < 0.05.
Results
Characterization of MM patients and EccDNA expression in MM
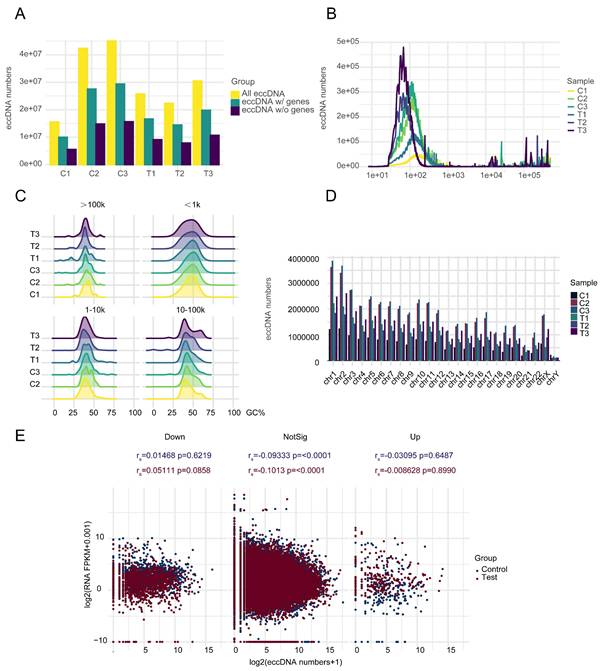
Basic information about three MM patients, including age, sex, monoclonal gammopathy type, treatment, and bone marrow at the time of recurrence has been reported in our previous article [30]. After RNA and DNA samples pass quality control, the mRNA-seq and circle-seq processes are conducted. A large number of EccDNA molecules (341217-1148828) were detected in six samples, with more EccDNA-carrying genes than those not carrying genes (Figure 1A). Most of the EccDNAs were a few hundred bp in length, with lengths ranging from tens to millions of bp, as shown in Figure 1B. The GC content of the EccDNA molecules was approximately 50%. The larger the molecule is, the higher the GC content (Figure 1C). The EccDNA sequences were all aligned to 46 chromosomes, and the results revealed that the longer the chromosome was, the more EccDNAs there were (Figure 1D). Circle-seq combined with mRNA-seq analysis revealed that there is no linear relationship between mRNA expression and EccDNA quantity in either the mRNA upregulation group or the mRNA downregulation group, and in either the control group or the test group (Figure 1E).
EccDNA profile in MM. A Numbers of total EccDNA molecules, EccDNA molecules with genes, and EccDNA molecules without genes detected in six samples. B Length distribution of EccDNA in six samples. C GC content distribution of EccDNA of different sizes in six samples. D EccDNA distribution on chromosomes in six samples. E Correlation analysis of mRNA expression and the number of EccDNA carrying the same gene in the subgroups in which mRNA was upregulated, not significantly different, or downregulated.
Differentially expressed EccDNA genes between the two groups
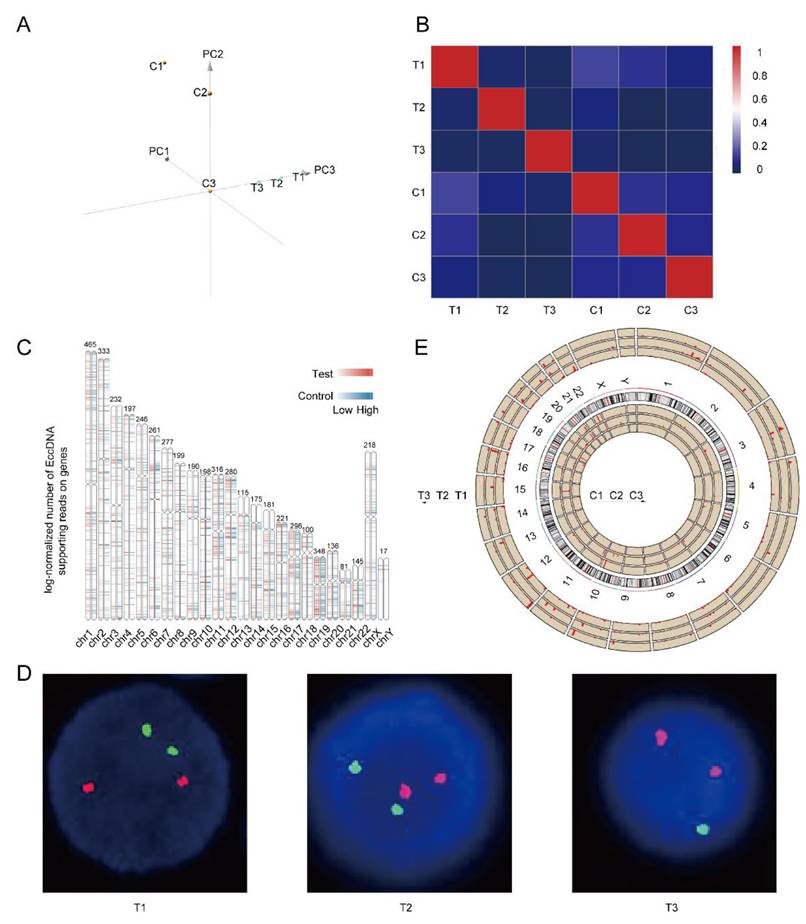
Both principal component analysis (PCA) and correlation heatmaps revealed obvious heterogeneity, not only between the completely relieved group and the relapsed group but also within the six samples, as shown in Figure 2A-B. As shown in Figure 2C, the differentially expressed EccDNA genes originated from all the chromosomes, with the Y chromosome having the fewest. Chromosome 19 had the highest density of differentially expressed genes, followed by chromosome 17. The clinical data of three patients were analyzed, and we found that the T3 sample was del(17p) by fluorescence in situ hybridization (FISH) (Figure 2D). However, no 17p deletion was detected in any of the other five samples (FISH is performed on purified myeloma cells. Because there are no myeloma cells in the bone marrow of patients in complete remission, there is no significance to perform FISH. As myeloma cells were present in the bone marrow of patients in relapse, FISH testing was performed to detect 17p deletion conventionally). The chromosomal distribution of upregulated differentially expressed genes in all six samples is shown in Figure 2E (& Supplementary Table 1). Approximately 91% of the upregulated differentially expressed EccDNA genes (2359 in total) were present in the T3 sample. Only 5% of the differentially expressed EccDNA genes in T3 originated from chromosome 17. However, approximately 92% of all upregulated differentially expressed EccDNA genes (137 in total) in all six samples were from chromosome 17 in T3.
Heterogeneity in the EccDNAs of six samples and differentially expressed EccDNA genes between the two groups. A Principal component analysis (PCA) of six samples. B Correlation heatmap of six samples. C Differentially expressed EccDNA genes distribution on chromosomes. D p53/D13S319 gene deletion probe FISH images of three samples in the relapse group (FISH was not performed in the complete response group), with the D13S319 probe red and the p53 probe green. E Distribution of upregulated differentially expressed EccDNA genes on all chromosomes of all six samples, with the length of the red line indicating the number of upregulated differentially expressed EccDNA genes.
Upregulated differentially expressed EccDNA genes from chromosome 17
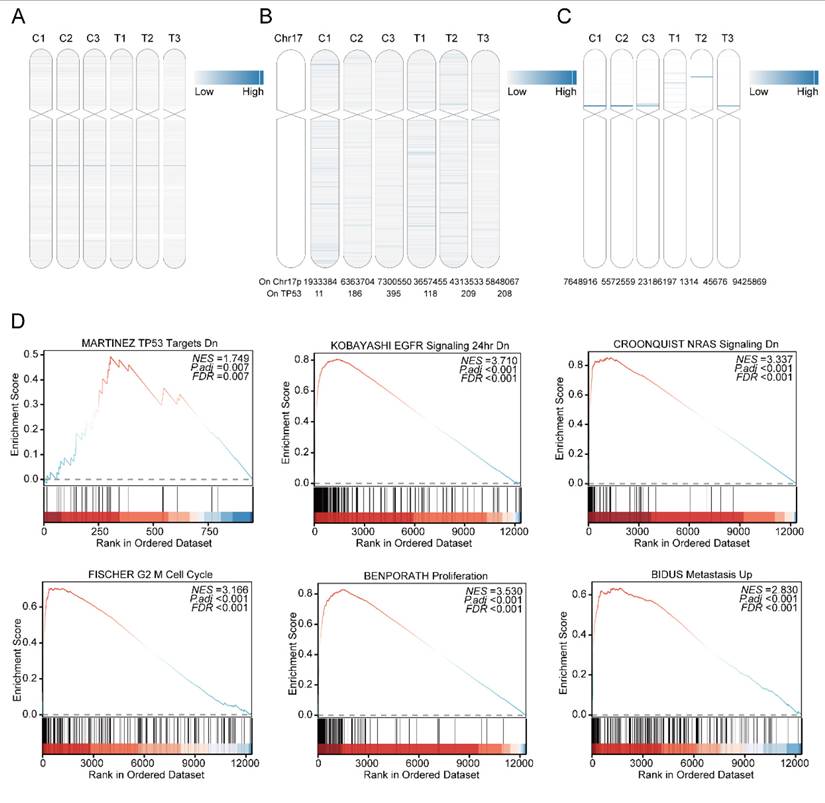
As a recognized high-risk factor in multiple myeloma, del(17p) is present in approximately 10% of patients at first diagnosis and relapse [31]. The gene expression of EccDNA on chromosome 17 in all six samples was determined via mRNA-seq and is shown in Figure 3A. All six samples had EccDNA genes from chromosome 17, and the relieved MM samples did not have fewer EccDNA genes than the relapsed MM samples did. The extremely poor prognosis of MM patients with del(17p) may be due to the deletion of the TP53 gene [32]. The number of EccDNA molecules whose junction site was located on chromosome 17, as well as whose junction site was located on the TP53 gene, in all six samples is shown in Figure 3B, clearly demonstrating that no significant predominance was observed in the T3 sample. As shown in Figure 3C, many super enhancer-harboring EccDNAs from chromosome 17 were detected in all six samples. As shown in Figure 3D, Upregulated differentially expressed EccDNA genes on chromosome 17 are enriched in TP53 target genes, and are enriched in the EGFR and NRAS pathways, which are closely related to tumorigenesis and development, as well as in cell cycle checkpoints, proliferation, and metastasis pathways. This is consistent with the poor prognosis associated with multiple myeloma with del(17p).
Upregulated differentially expressed EccDNA genes from chromosome 17. A mRNA expression levels of genes carried by EccDNA on chromosome 17 in six samples, with the depth of the blue line indicating the expression level of the mRNA. B Number of EccDNA junction sites located at 17p and TP53 in six samples. TP53 is located at the red line, with the depth of the blue line indicating the number of junction sites. The numbers below the picture are specific quantities. C The number of super enhancers carried by EccDNA located at 17p, with the depth of the blue line indicating the number of super enhancers. D GSEA enrichment analysis of upregulated differentially expressed EccDNA genes on chromosome 17 (test group vs control group).
EccDNA carries genes with potential functions
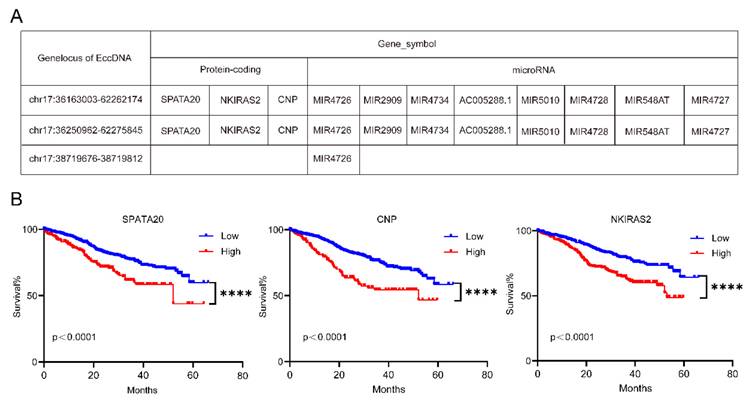
Some candidate genes were obtained after taking the intersection of upregulated differentially expressed EccDNA-protein coding genes and upregulated differentially expressed mRNAs from chromosome 17 and after taking the intersection of upregulated differentially expressed EccDNA-small RNA genes and downregulated differentially expressed mRNAs from chromosome 17 (Supplementary Table 2-3). These candidate genes were localized to three EccDNA molecules, namely, EccDNAchr17:36163003--62262174, EccDNAchr17:36250962--62275845, and EccDNAchr17:38719676--38719812, with the first two being EcDNA and the latter being microDNA. The three EccDNAs and the genes they carry are shown in Figure 4A. We further focused on the protein-coding genes encoded by the EcDNA. We further tested the associations of these three protein-coding genes with clinical outcomes in the MMRF-COMMPASS dataset from the TCGA. We found that all three protein-coding genes were poor prognostic indicators of MM (Figure 4B).
EccDNAs carry genes with potential functions. A Three EccDNA molecules and the candidate genes they carry. B Overall survival (OS) associated with the SPATA20, CNP and NKIRAS2 levels estimated in patients included in the MMRF-COMMPASS dataset. P values were calculated with a log-rank test.
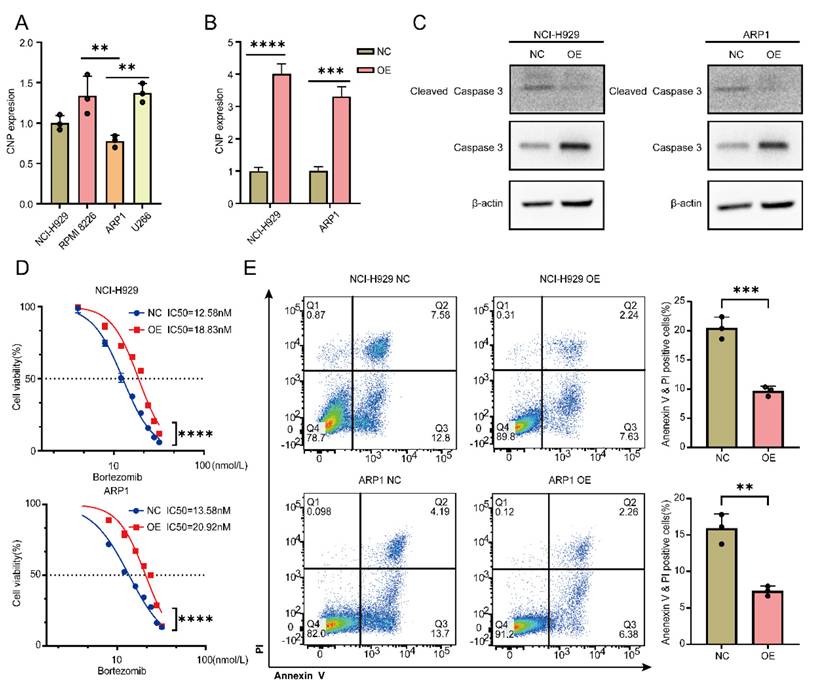
Overexpression of CNP increases bortezomib tolerance in MM cells
We wanted to synthesize EccDNA molecules for further experiments but failed because EcDNA is too long to loop. Moreover, EcDNA carry several genes that are not suitable for subsequent studies. To further verify that the genes carried on EccDNA were functional, we chose CNP for further studies. Four MM cell lines were found to express CNP, with the lowest levels of CNP being detected in NCI-H929 and ARP1 cells (Figure 5A). After successful transfection of the OE-CNP plasmids (Figure 5B), bortezomib was administered to NCI-H929 and ARP1 cells for a period of 24 hours. WB experiments were performed to detect the apoptotic pathway. As shown in Figure 5C, compared with control cells, both NCI-H929 and ARP1 cells overexpressing CNP presented a decrease in activated caspase 3. Furthermore, after 24 hours of bortezomib treatment, the IC50 increased in both NCI-H929 cells and ARP1 cells when CNP was overexpressed, in comparison to the negative control group (Figure 5D). Additionally, CNP overexpression decreased the proportion of apoptotic NCI-H929 and ARP1 cells (Figure 5E).
The overexpression of CNP increases the tolerance of MM cells to bortezomib. A qPCR analysis of CNP expression in 4 MM cell lines. B The expression of CNP in NCI-H929 cells and ARP1 cells was analyzed using qPCR after the cells were infected with OE-CNP plasmids or negative controls for 48 hours. C WB analysis of caspase 3 and cleaved caspase 3 expression was performed in NCI-H929 cells and ARP1 cells that had been transfected with OE-CNP plasmids or negative controls for 24 hours. The cells were then treated with bortezomib for an additional 24 hours. D The cell viability of the NCI-H929 and ARP1 cell lines was examined after they were transfected with OE-CNP plasmids or negative controls and then treated with different concentrations of bortezomib for 24 hours. E Apoptosis in NCI-H929 and ARP1 cells, as detected by the Annexin V-FITC/PI assay, after 24 hours of bortezomib treatment at 10 nmol/L, with OE-CNP plasmids or negative controls. The statistical significance of the test was determined using a student's t-test. *p<0.05, **p<0.01, ***p<0.001. **** p < 0.0001.
Discussion
Many EccDNA molecules have been detected in MM, and our results are in agreement with earlier findings [18-23]. The length distribution, chromosomal distribution, and high GC content of EccDNA molecules in MM are similar to those in other tumor types [18-23]. There are more EccDNA molecules that carry genes (including longer protein coding genes and shorter non-coding genes such as lincRNA, microRNA) than not, suggesting that EccDNA molecules are produced to regulate gene expression. However, we further analyzed the mRNA expression and number of EccDNAs carrying concordant genes and found no correlation between mRNA expression and EccDNA, suggesting that only a few key EccDNA molecules are generated during the disease process and are involved in disease progression and drug resistance.
MM is a highly heterogeneous malignant plasma tumor with a survival period ranging from a few months to 10 years [33]. Numerous studies have reported extensive inter- and intra-patient heterogeneity in patients with multiple myeloma at the genomic level [34-38], including single-cell RNA sequencing, whole-genome sequencing, and bisulfite sequencing. In this study, circle-seq analysis was performed on bone marrow samples from three MM patients when they achieve complete remission or when they relapse. The results also revealed high heterogeneity in all six samples. Importantly, the heterogeneity among the three samples in the relapsed group could be attributed to their different genetic abnormalities. However, there was still high heterogeneity among samples in the complete response group without any genetic abnormalities due to the tumor burden. The high heterogeneity of multiple myeloma is due to extremely frequent clonal evolution and highly unstable genomes [39,40]. Quantitative multigene fluorescence in situ hybridization (QM-FISH) was performed in paired newly diagnosed and relapsed MM patients, and the clonal evolution of multiple myeloma was divided into four models [41], demonstrating that the clonal evolution of multiple myeloma is variable. We speculate that the evolution of MM clones was accompanied by the production of a large amount of EccDNA and that the noncoding RNA and protein-coding genes carried on these EccDNAs are functional, thereby mediating the high heterogeneity of MM. Genes on autosomal chromosomes can be transcribed only after the double helix structure of chromosomes is opened and the chromatin is depolymerized. However, EccDNA can be transcribed directly without restricting the opening of the double helix or depolymerization of the chromatin. Compared with autosomal genes, EccDNA can function more promptly and efficiently in response to environmental changes. Moreover, several studies have shown that large amounts of EccDNA are detected in normal human tissues and body fluids [42-44]. In view of the above findings, we hypothesized that the EccDNA genome, like the autosomal genome, has individualized characteristics. “Good EccDNA” are essential for the survival of cells, and once “bad EccDNA” are produced, they may lead to various disorders.
All 46 chromosomes produced differentially expressed EccDNA genes; the shorter the chromosome was, the fewer differentially expressed EccDNA genes. The density of differentially expressed EccDNA genes was the highest on chromosome 19, followed by chromosome 17. Among the six samples, only T3 was del (17p). Considering the extremely poor prognosis of MM patients with del(17p), we further analyzed the differentially expressed EccDNA genes on chromosome 17. We noticed that only 5% of the differentially expressed EccDNA genes in the T3 sample originated from chromosome 17 and that the T3 sample accounted for 92% of all upregulated differentially expressed EccDNA genes originating from chromosome 17 in all six samples, suggesting that the upregulated differentially expressed EccDNA genes originating from chromosome 17 in the T3 sample may be involved in disease progression and disease resistance. Del(17p) is a high-risk factor for MM [45]. Newly diagnosed MM patients with del(17p) are associated with shorter overall survival (OS) [46], as are patients who acquire del(17p) later during the disease course [47]. Neither chemotherapy nor autologous hematopoietic stem cell transplantation has been shown to completely overcome the adverse prognosis of MM with del(17p) [32,48], even in the era of new drug therapies. Del (17p) has been demonstrated to be a significant predictor of disease progression and drug resistance [31,49,50]. For a long time, TP53 loss of heterozygosity was believed to be responsible for the poor prognosis of MM patients with del (17p). However, there are approximately 500 genes on chromosome 17p, among which both new oncogenes and tumor suppressor genes have been discovered [51-54]. Here, we speculated that del(17p) may have formed a large amount of EccDNA, so we wondered whether other genes from chromosome 17 are involved in the very poor prognosis of del(17p). It is also possible that many EccDNA junction sites are located in the TP53 gene, which leads to its deletion. Therefore, we focused mainly on the upregulated differentially expressed EccDNA genes on chromosome 17p. We further analyzed the junction sites on chromosome 17 and TP53 of the upregulated differentially expressed EccDNA in all six samples. Among them, neither junction sites on chromosome 17 nor TP53 in the T3 sample had the highest frequency of upregulated differentially expressed EccDNA. EccDNA functions as a mobile enhancer [55]. Hence, we analyzed EccDNA carrying super enhancers derived from 17p and found that the T3 sample had a large number of super enhancer-carrying EccDNA but not most of the other six samples.
EccDNA downregulates MUC20 via KIF3C amplification, thereby mediating resistance to bortezomib in MM [24]. In this investigation, some candidate genes were screened after the intersection of upregulated differentially expressed EccDNA-protein coding genes and upregulated differentially expressed mRNAs from chromosome 17 was identified and after the intersection of upregulated differentially expressed EccDNA-small RNA genes and downregulated differentially expressed mRNAs from chromosome 17 was identified. These results indicate that these potential genes might be linked to bortezomib resistance in MM patients with del(17p). These candidate genes are located on three EccDNA molecules: two EcDNA and one microDNA. As mentioned above, EcDNA are sufficiently large to carry complete genes and thus participate in oncogene amplification and genomic heterogeneity [5]. Therefore, we wanted to synthesize EcDNA to further confirm its function in the cell; however, we failed because it was too long to form a loop. EcDNA carry several genes and are not ideal targets for research. More evidence is needed to support the involvement of EccDNA in bortezomib resistance. We focused on three candidate protein-coding genes and found that all were poor prognostic indicators for MM by analyzing the MMRF-COMPASS dataset. We further selected CNP for cell function experiments. The results revealed that the overexpression of CNP inhibited the apoptosis pathway, leading to an increase in the IC50 and a decrease in the apoptosis of MM cells after treatment with bortezomib. All the above results suggest that some of the genes carried by EccDNA may be highly expressed and thus involved in bortezomib resistance. Outward PCR and Sanger sequencing have been reported to identify the circular characteristics of EccDNA molecules in studies on EccDNA[19-21,29,44]. In the future, we will identify specific EccDNA on chromosome 17, which will facilitate further research.
Our study revealed that a small number of key EccDNA molecules were generated during disease resistance, progression, and relapse, rather than a large number, for the following reasons. First, many EccDNA molecules and differentially expressed EccDNA genes remained in the complete remission group. Second, in both the complete remission group and the relapse group, whether in the mRNA upregulation group or the downregulation group, there is no correlation between mRNA expression and EccDNA quantity. Third, the samples without del (17p) still had many EccDNA with junction sites at 17p, TP53 and EccDNA carrying super enhancers originating from 17p.
Conclusion
In conclusion, we first investigated and characterized the role of EccDNA in complete response MM and relapsed MM via circle-seq analysis. Our findings highlight that the heterogeneity of EccDNA is consistent with the heterogeneity of MM and may be a driver of the heterogeneity of MM. Moreover, we found that only some key EccDNA molecules may be involved in disease resistance, progression, and relapse by analyzing circle-seq data in combination with mRNA-seq data. Analyses of MM datasets in public databases, as well as functional experiments, have revealed that some key EccDNA molecules may be involved in bortezomib resistance. However, this investigation was conducted on three samples, and the sample volumes were limited. A larger sample size would pave the way for further confirmation and discovery of novel biomarkers. Moreover, we failed to synthesize EccDNA, and more direct evidence of EccDNA function is needed.
Abbreviations
EccDNA: Extrachromosomal circular DNA; MM: Multiple myeloma; CNP: 2',3'-cyclic nucleotide 3' phosphodiesterase; BFB: Breakage-fusion-bridge; spcDNA: Small polydispersed DNA; IMWG: International Myeloma Working Group; FBS: Fetal bovine serum; STR: Short tandem repeat; RCA: Rolling Circle Amplification; mtDNA: Mitochondrial DNA; PCI: Phenol/chloroform/isoamyl alcohol; IC50: Half maximal inhibitory concentration; FPM: Fragments per million; FISH: Fluorescence in situ hybridization; WB: Western blotting; PCA: Principal component analysis; QM-FISH: Quantitative multigene fluorescence in situ hybridization; OS: Overall survival; BM: Bone marrow morphology.
Supplementary Material
Supplementary table 1: sig_diff_TvsC_ecc.
Supplementary table 2: Overlapping_Result 1.
Supplementary table 3: Overlapping_Result 2.
Acknowledgements
The authors would like to express their gratitude to the patients who participated in this study. The authors would like to thank Shanghai Kangcheng BIO-TECH Co., Ltd., for sequencing technique support; Shanghai Dache Biotechnology Co., Ltd., for statistical analysis technique support; and medical writers, proofreaders, and editors for their assistance.
Funding
This project was funded by the Natural Science Foundation of Hunan Province [2025JJ50706]; the "co-PI" initiative from The Third Xiangya Hospital of Central South University [202408]; the Natural Science Foundation of Changsha City [kq2502026]; and the General Project of the Hunan Health Commission [20253585].
Data availability
All the data are available in the main text or the supplementary material.
Author contributions
F.L.: Conceptualization, methodology, software, validation, formal analysis, data curation, writing—original draft preparation, and visualization. X.L.: methodology and validation. S.T.: Methodology and validation. J.L.: Conceptualization, investigation, resources, writing—review and editing, supervision, and project administration. Y.F.: Conceptualization, investigation, resources, writing—review and editing, supervision, project administration, and funding acquisition.
Ethics approval and consent to participate
Informed consent in written form was obtained from all patients. The study was approved by the ethics committee of The Third Xiangya Hospital, Central South University, Changsha. All experimental procedures were performed in accordance with the principles of the Declaration of Helsinki.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu W, Liu J, Song Y, Wang X, Zhou M, Wang L. et al. Mortality of lymphoma and myeloma in China, 2004-2017: an observational study. J Hematol Oncol. 2019;12(1):22
2. Wang S, Xu L, Feng J, Liu Y, Liu L, Wang J. et al. Prevalence and Incidence of Multiple Myeloma in Urban Area in China: A National Population-Based Analysis. Front Oncol. 2020;9:1513
3. Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S. et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122-8
4. Van De Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. The Lancet. 2021;397(10272):410-27
5. Yang L, Jia R, Ge T, Ge S, Zhuang A, Chai P. et al. Extrachromosomal circular DNA: biogenesis, structure, functions and diseases. Sig Transduct Target Ther. 2022;7(1):342
6. Ling X, Han Y, Meng J, Zhong B, Chen J, Zhang H. et al. Small extrachromosomal circular DNA (eccDNA): major functions in evolution and cancer. Mol Cancer. 2021;20(1):113
7. Lanciano S, Carpentier MC, Llauro C, Jobet E, Robakowska-Hyzorek D, Lasserre E. et al. Sequencing the extrachromosomal circular mobilome reveals retrotransposon activity in plants. PLoS Genet. 2017;13(2):e1006630
8. Cohen S, Houben A, Segal D. Extrachromosomal circular DNA derived from tandemly repeated genomic sequences in plants. The Plant Journal. 2008;53(6):1027-34
9. Wang K, Tian H, Wang L, Wang L, Tan Y, Zhang Z. et al. Deciphering extrachromosomal circular DNA in Arabidopsis. Computational and Structural Biotechnology Journal. 2021;19:1176-83
10. Yerlici VT, Lu MW, Hoge CR, Miller RV, Neme R, Khurana JS. et al. Programmed genome rearrangements in Oxytricha produce transcriptionally active extrachromosomal circular DNA. Nucleic Acids Research. 2019;47(18):9741-60
11. Møller HD, Parsons L, Jørgensen TS, Botstein D, Regenberg B. Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci USA. 2015;112(24):E3114-22
12. Cohen S, Méchali M. Formation of extrachromosomal circles from telomeric DNA in Xenopus laevis. EMBO Reports. 2002;3(12):1168-74
13. Cohen S, Yacobi K, Segal D. Extrachromosomal Circular DNA of Tandemly Repeated Genomic Sequences in Drosophila. Genome Res. 2003;13(6a):1133-45
14. Cohen Z, Bacharach E, Lavi S. Mouse major satellite DNA is prone to eccDNA formation via DNA Ligase IV-dependent pathway. Oncogene. 2006;25(33):4515-24
15. Møller HD, Ramos-Madrigal J, Prada-Luengo I, Gilbert MTP, Regenberg B. Near-Random Distribution of Chromosome-Derived Circular DNA in the Condensed Genome of Pigeons and the Larger, More Repeat-Rich Human Genome. Genome Biology and Evolution. 2020;12(2):3762-77
16. Van Leen E, Brückner L, Henssen AG. The genomic and spatial mobility of extrachromosomal DNA and its implications for cancer therapy. Nat Genet. 2022;54(2):107-14
17. Yi E, Gujar AD, Guthrie M, Kim H, Zhao D, Johnson KC. et al. Live-Cell Imaging Shows Uneven Segregation of Extrachromosomal DNA Elements and Transcriptionally Active Extrachromosomal DNA Hubs in Cancer. Cancer Discov. 2022;12(2):468-83
18. Kumar P, Kiran S, Saha S, Su Z, Paulsen T, Chatrath A. et al. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci Adv. 2020;6(20):eaba2489
19. Lin C, Chen Y, Zhang F, Liu B, Xie C, Song Y. Encoding gene RAB3B exists in linear chromosomal and circular extrachromosomal DNA and contributes to cisplatin resistance of hypopharyngeal squamous cell carcinoma via inducing autophagy. Cell Death Dis. 2022;13(2):171
20. Jiang X, Pan X, Li W, Han P, Yu J, Li J. et al. Genome-wide characterization of extrachromosomal circular DNA in gastric cancer and its potential role in carcinogenesis and cancer progression. Cell Mol Life Sci. 2023;80(7):191
21. Cen Y, Fang Y, Ren Y, Hong S, Lu W, Xu J. Global characterization of extrachromosomal circular DNAs in advanced high grade serous ovarian cancer. Cell Death Dis. 2022;13(4):342
22. Yang Y, Yang Y, Huang H, Song T, Mao S, Liu D. et al. PLCG2 can exist in eccDNA and contribute to the metastasis of non-small cell lung cancer by regulating mitochondrial respiration. Cell Death Dis. 2023;14(4):257
23. Zeng T, Huang W, Cui L, Zhu P, Lin Q, Zhang W. et al. The landscape of extrachromosomal circular DNA (eccDNA) in the normal hematopoiesis and leukemia evolution. Cell Death Discov. 2022;8(1):400
24. Wang X, Shi Y, Shi H, Liu X, Liao A, Liu Z. et al. MUC20 regulated by extrachromosomal circular DNA attenuates proteasome inhibitor resistance of multiple myeloma by modulating cuproptosis. J Exp Clin Cancer Res. 2024;43(1):68
25. Zou S, Chen S, Rao G, Zhang G, Ma M, Peng B. et al. Extrachromosomal circular MiR-17-92 amplicon promotes HCC. Hepatology. 2024;79(1):79-95
26. Paulsen T, Shibata Y, Kumar P, Dillon L, Dutta A. Small extrachromosomal circular DNAs, microDNA, produce short regulatory RNAs that suppress gene expression independent of canonical promoters. Nucleic Acids Research. 2019;47(9):4586-96
27. Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P. et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. The Lancet Oncology. 2016;17(8):e328-46
28. Henriksen RA, Jenjaroenpun P, Sjøstrøm IB, Jensen KR, Prada-Luengo I, Wongsurawat T. et al. Circular DNA in the human germline and its association with recombination. Molecular Cell. 2022;82(1):209-217.e7
29. Møller HD, Mohiyuddin M, Prada-Luengo I, Sailani MR, Halling JF, Plomgaard P. et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun. 2018;9(1):1069
30. Li F, Long X, Tang S, Yan J, Jing L, Fu Y. MIR4726EccDNA drives bortezomib resistance in multiple myeloma by enhancing MIR4726-5p/NXF1/NKIRAS2 axis dependent autophagy. 2025;23(1):346.
31. Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA. et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569-75
32. Corre J, Perrot A, Caillot D, Belhadj K, Hulin C, Leleu X. et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood. 2021;137(9):1192-5
33. Joseph NS, Kaufman JL, Dhodapkar MV, Hofmeister CC, Almaula DK, Heffner LT. et al. Long-Term Follow-Up Results of Lenalidomide, Bortezomib, and Dexamethasone Induction Therapy and Risk-Adapted Maintenance Approach in Newly Diagnosed Multiple Myeloma. JCO. 2020;38(17):1928-37
34. Merz M, Merz AMA, Wang J, Wei L, Hu Q, Hutson N. et al. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat Commun. 2022;13(1):807
35. Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5(1):2997
36. Derrien J, Guérin-Charbonnel C, Gaborit V, Campion L, Devic M, Douillard E. et al. The DNA methylation landscape of multiple myeloma shows extensive inter- and intrapatient heterogeneity that fuels transcriptomic variability. Genome Med. 2021;13(1):127
37. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D. et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell. 2014;25(1):91-101
38. Vo JN, Wu YM, Mishler J, Hall S, Mannan R, Wang L. et al. The genetic heterogeneity and drug resistance mechanisms of relapsed refractory multiple myeloma. Nat Commun. 2022;13(1):3750
39. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100-13
40. Dutta AK, Alberge JB, Sklavenitis-Pistofidis R, Lightbody ED, Getz G, Ghobrial IM. Single-cell profiling of tumour evolution in multiple myeloma — opportunities for precision medicine. Nat Rev Clin Oncol. 2022;19(4):223-36
41. Yan Y, Qin X, Liu J, Fan H, Yan W, Liu L. et al. Clonal phylogeny and evolution of critical cytogenetic aberrations in multiple myeloma at single-cell level by QM-FISH. Blood Advances. 2022;6(2):441-51
42. Lv W, Pan X, Han P, Wang Z, Feng W, Xing X. et al. Circle-Seq reveals genomic and disease-specific hallmarks in urinary cell-free. Clin Transl Med. 2022;12(4):e817
43. Gerovska D, Araúzo-Bravo MJ. Systemic Lupus Erythematosus Patients with DNASE1L3·Deficiency Have a Distinctive and Specific Genic Circular DNA Profile in Plasma. Cells. 2023;12(7):1061
44. Yang H, He J, Huang S, Yang H, Yi Q, Tao Y. et al. Identification and Characterization of Extrachromosomal Circular DNA in Human Placentas With Fetal Growth Restriction. Front Immunol. 2021;12:780779
45. Sonneveld P, Avet-Loiseau H, Lonial S, Usmani S, Siegel D, Anderson KC. et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955-62
46. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD. et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15):e56-65
47. Lakshman A, Painuly U, Rajkumar SV, Ketterling RP, Kapoor P, Greipp PT. et al. Impact of acquired del(17p) in multiple myeloma. Blood Advances. 2019;3(13):1930-8
48. Gaballa S, Saliba RM, Srour S, Lu G, Brammer JE, Shah N. et al. Outcomes in patients with multiple myeloma with TP53 deletion after autologous hematopoietic stem cell transplant. American J Hematol. 2016;91(10):E442-7
49. Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM. et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6(1):6997
50. Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C. et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489-95
51. Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J. et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531(7595):471-5
52. Tavtigian SV, Simard J, Teng DHF, Abtin V, Baumgard M, Beck A. et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27(2):172-80
53. Chen M, Chen X, Li S, Pan X, Gong Y, Zheng J. et al. An Epigenetic Mechanism Underlying Chromosome 17p Deletion-Driven Tumorigenesis. Cancer Discovery. 2021;11(1):194-207
54. Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I. et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature. 2012;487(7406):244-8
55. Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, Gong L. et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell. 2021;39(5):694-707.e7
Author contact
![]() Corresponding authors: Jing Liu, Email: jingliu0318com; Yunfeng Fu, Email: fuyunfengedu.cn.
Corresponding authors: Jing Liu, Email: jingliu0318com; Yunfeng Fu, Email: fuyunfengedu.cn.