Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(6):1675-1686. doi:10.7150/jca.90457 This issue Cite
Review
Inside anticancer therapy resistance and metastasis. Focus on CD36
Ioana M. Lambrescu1,2, Gisela F. Gaina1,2, Laura C. Ceafalan1,2 ![]() , Mihail E. Hinescu2,3
, Mihail E. Hinescu2,3
1. Cell Biology, Neurosciences, and Experimental Myology Laboratory, Victor Babeș Institute of Pathology, 050096 Bucharest, Romania.
2. Department of Cellular and Molecular Biology and Histology, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania.
3. National Institute of Pathology "Victor Babes," 050096 Bucharest, Romania.
Received 2023-9-23; Accepted 2023-11-28; Published 2024-1-27
Abstract

Despite recent advances in targeted cancer therapies, drug resistance remains an important setback in tumor control. Understanding the complex mechanisms involved in both innate and acquired drug resistance represents the first step in discovering novel therapeutic agents. Because of its importance in tumorigenesis, progression, and metastasis, lipid metabolism is increasingly garnering attention. CD36 is a membrane receptor at the top of the signaling cascade that transports lipids. Its expression has been repeatedly presented as an unfavorable prognostic factor for various tumor types, raising the question: could CD36 be a critical factor in cancer treatment resistance? In our review, we set out to explore the most prominent studies on the implication of CD36 in resistance to platinum-based drugs and other adjuvant cancer therapies in solid and haematological neoplasia. Moreover, we provide an overview of the latest anti-CD36 cancer therapies, thus opening new perspectives for future personalized medicine.
Keywords: CD36, cancer treatment, drug resistance, chemotherapy, lipid metabolism
Introduction
The importance of lipid metabolism in tumor initiation, growth, and metastasis is receiving much attention in cancer research. To fulfill the demands for rapid proliferation and development, tumor cells can potentially increase lipid accumulation and metabolism (1). CD36 is a transmembrane glycoprotein of the class B scavenger receptor family, also known as Fatty Acid translocase (FAT), platelet GPIV, or GP88, that interacts with a variety of ligands such as apoptotic cells, thrombospondin-1 (TSP-1) and fatty acids (FAs) (2,3).
The history of CD36 goes back to 1977 when it was first described by Clemstone and colleagues (4). The past four decades have been extremely generous in terms of scientific research into the influence of CD36 on FA transport, insulin resistance, and tumorigenesis, as stated in Figure 1. Created with BioRender.com.
The most significant historical milestones of CD36. Created with BioRender.com
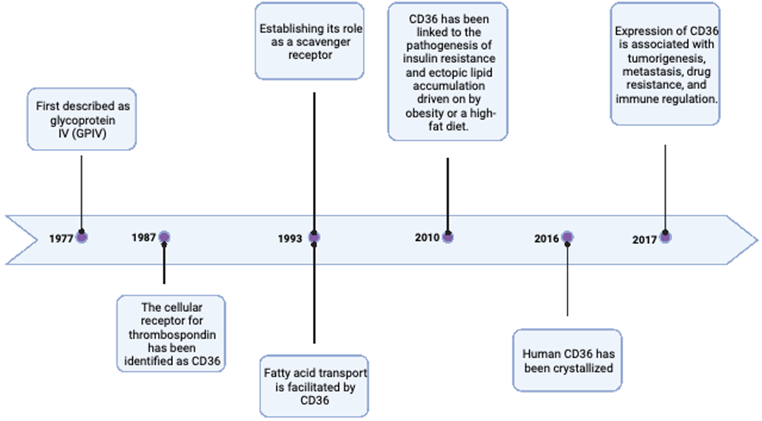
Due to the extensive glycosylation, the 472 amino acid protein has an apparent molecular mass of 88 kDa with two membrane-spanning domains and many palmitoylation sites (5). Numerous non-immune cells, such as platelets, immature erythrocytes, adipocytes, myocytes, some specialized epithelial cells, and microvascular endothelial cells, as well as a wide range of innate and adaptive immune cells, including macrophages, monocytes, dendritic cells, and subsets of T and B cells, express CD36 on their surfaces (6).
According to a thorough evaluation of several omics data from The Cancer Genome Atlas (TCGA), fatty acid (FA) metabolism is one of the most commonly altered pathways in lipid metabolism in pan-cancer (7). Furthermore, tumor immunological tolerance and carcinogenesis can be linked to CD36-driven lipid metabolic reprogramming and the function of tumor-associated immune cells (8).
Drug resistance is a significant drawback for cancer patients regarding disease control. Tumor heterogeneity and the diversity of their milieu could explain therapy resistance. Furthermore, 90% of chemotherapy failures occur during the invasion and metastatic spread of the malignancy due to drug resistance (9). Several mechanisms were suggested through which cancer cells are either innately resistant to medication treatments or develop resistance to drug therapies after exposure. Molecular alterations in drug efflux pumps, drug-metabolizing enzymes, and apoptotic regulating pathways have been found to protect tumor cells against chemotherapy (10).
Consequently, these processes may influence the drug molecule itself or its target (11). Many solid tumors, including ovarian, gastric, and oral cancers, have CD36 levels that are significantly upregulated (3,12,13). However, tumoral stroma and microvessels show downregulation in CD36 expression compared to the surrounding tissues (1), as demonstrated for breast cancer (14).
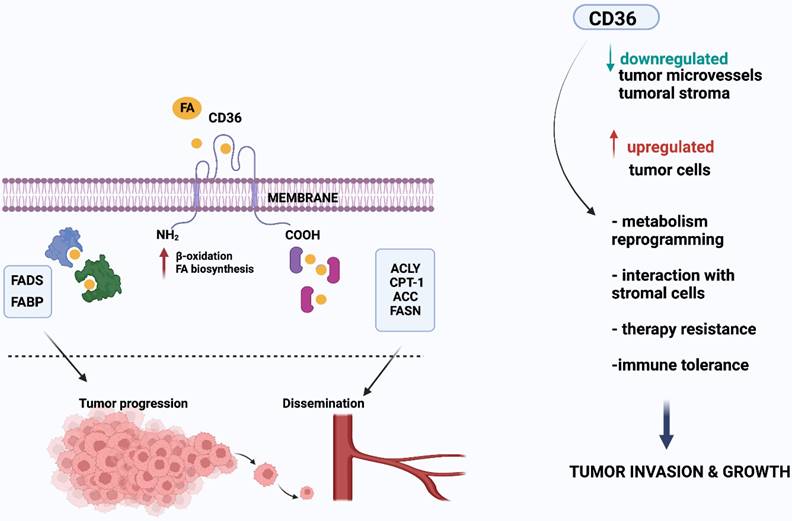
CD36 expression has been repeatedly presented as an unfavorable prognostic factor for various tumor types, such as ovarian and colon cancer (3,15,16). Recently, further studies have linked CD36 to tumor progression and treatment resistance via increased lipid uptake and FA oxidation (FAO) for ATP production (1,17-20) (Figure 2).
CD36 contributes to the growth and spread of tumors by boosting lipid absorption and fatty acid (FA) oxidation. Several proteins, notably fatty acid binding protein (FABP) and fatty acyl-CoA desaturases (FADS), coordinate tumor cell development. As seen on the right side of the picture, lipid metabolism alterations may also affect cell motility, which may ultimately lead to metastasis. Several enzymes, including ATP citrate lyase (ACLY), carnitine palmitoyl transferase 1 (CTP-1), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), are involved in this process. In addition, FA are synthesized from scratch, and FASN is overexpressed in a range of cancer types. Created with BioRender.com
In 2017, Dong and colleagues published an exciting study that linked the progression of estrogen receptor (ER) positive breast cancer in female mice to diet-induced obesity (DIO) and the lysophosphatidic acid/protein kinase D1 (LPA/PKD-1)-CD36 signaling axis (21). According to this study, tumor angiogenesis may be driven by the downregulation of CD36 in the tumor endothelium. This is critical because targeting this pathway might represent a future therapeutic approach (21).
This review aims to highlight the impact of CD36 on cancer therapies with an emphasis on drug resistance and the direct targeting of CD36 for improving cancer control.
The crosstalk between CD36, metastasis and drug resistance
The study of tumor biology has shown that malignancies are not produced by a single cluster of cells but rather by various cellular populations (22). The diversity of cells in the tumor microenvironment (TME) and the genetic variability could help explain this tumor heterogeneity. A tumor comprised of distinct cell populations in terms of behavior and response to treatment defines the concept of intratumoral heterogeneity (23). Furthermore, various cellular subpopulations are responsible for building the groundwork for inter- and intrametastatic heterogeneity. This is a significant deterrent to therapy and a potentiator of acquired resistance (22,24).
Cancer cells that have been exposed to chemotherapy or radiation over an extended period may become resistant to therapy, resulting in recurrence and a poor prognosis. Resistance to cancer drugs can be intrinsic. However, acquired resistance in tumor cells is more common (25).
Increased expression of proteins involved in the epithelial-mesenchymal transition (EMT) has been linked to drug resistance, thus enhancing cell migration and metastasis. In 2015, Deng and colleagues reported an interplay between CD36 and TGF-β to drive EMT in cervical cancer cells (26). Additionally, breast cancer cells acquire FAs from adipocytes via CD36 trafficking, inducing EMT (27). Finally, CD36 was shown to promote EMT through the PI3K/AKT/mTOR pathway in gastric cancer with peritoneal metastasis (PM) (28).
CD36 and metastatic potential
CD36 seems to be more involved in the metastatic process rather than promoting the development of the primary tumor (29). Furthermore, the CD36 gene was discovered to be often amplified in metastatic cancers, with the worst survival rates in the high-copy number group (15).
The metastatic potential of CD36 was also evaluated by Pheiler and colleagues in 2018. The authors reported that CD36 is a key modulator of immune cell engulfment of tumor microvesicles, stressing the role of CD36 in transporting tumor microvesicles to the perivascular region, resulting in premetastatic cell clusters (30).
FAT/CD36 is upregulated and increases FA uptake under tumor-derived cytokines signaling in infiltrating polymorphonuclear myeloid-derived suppressor cells (MDSC). MDSC block anti-tumor T-cell responses, thus promoting tumor growth (31). The genetic depletion of CD36 limits oxidative metabolism and the induction of the immunosuppressive mechanisms, resulting in a CD8+ T cell-dependent delay in tumor development (31). These findings highlight the importance of CD36 in immune suppression and tumor development, which further recommends CD36 as a potential therapeutic target, especially for metastatic cancers.
Metastasis-initiating cells respond to dietary lipids. High-fat diets or high quantities of palmitic acid (PA) significantly increase the metastatic potential. In vivo, studies on both immunocompetent and immunodeficient mouse strains revealed that PA or a high-fat diet enhances the ability of CD36+ metastasis-initiating cells to proliferate (32). Moreover, Pascual et al. proved that the number and size of metastases were reduced by CD36 inhibition or knockdown, as well as by employing cancer cells expressing mutant CD36 (32).
Finding novel treatment strategies for patients with metastases is now of utmost importance. The liver is among the most typical sites for metastatic illness, and patients who progress to this stage have a poor prognosis and reduced therapy response. Although immunotherapy for cancer is an influential and rapidly expanding therapeutic option, approximately 15% to 20% of patients with liver metastases benefit from it (33,34).
One of the reasons immunotherapy is ineffective in metastatic liver disease is the accumulation of macrophages at this level. Recent evidence suggests that one of the factors promoting tumor growth could be CD36-mediated lipid metabolism of the tumor-associated immune cells' (35).
Thus, one potential therapeutic strategy could focus on CD36 inhibition to target the tumor-macrophage metabolic interface. In this regard, it was established in preclinical animal models that blocking CD36 in macrophages restores CD8+ T cell immunity and reduces liver metastases (34).
Dysregulation of lipid metabolism and anticancer drug resistance
It has been hypothesized that the metabolic switch that enables cancer cells to adapt to treatment-induced cellular stress could be the abnormal lipid metabolism. Several intricate mechanisms are responsible for the dysregulation of lipid metabolism in cancer. De novo lipid synthesis or lipogenesis, FAO in cancer cell mitochondria, and the coexistence of lipolysis and lipogenesis in cancer cells come together to meet the needs of tumor cell proliferation and growth (36). FAs are coupled to fatty acid binding proteins (FABPs) in the cytoplasm and subsequently metabolized to acyl-CoA. Carnitine palmitoyltransferase 1 (CPT1) is required to transport acyl-CoA into the mitochondria and represents the major rate-limiting enzyme for FAO (36,37).
Numerous posttranslational modification sites on CD36 have been identified. These modifications, which control CD36 stability, protein folding, and translocation, can be glycosylated, phosphorylated, palmitoylated, acetylated, or ubiquitylated. This causes CD36 to become involved in several signaling pathways once ligands bind at various subcellular sites (1). These mechanisms are particularly essential since they serve as the basis for developing novel therapeutic lines that may be used to combat resistance to already approved drugs.
The increased FA uptake by breast cancer cells due to the up-regulation of CD36 expression can be demonstrated by co-culturing these cells with adipocytes (27). Thus, the uptake of FA released by adipocytes provides the energetic requirements for cancer cell survival (38-42). Moreover, the adipocyte-derived interleukin-6 (IL-6) and leptin regulate epithelial-mesenchymal transition (EMT) in cancer cells and supports stem cell renewal and chemoresistance (43-47).
Anticancer therapy may lead to an enrichment of a pre-existing cellular subpopulation exhibiting an abnormal lipid metabolism, which is thought to represent the cancer stem cells (CSCs). It has been observed that depending on cancer cell type, CSCs can undergo various changes in their lipid metabolism, such as an increase in de novo lipogenesis or lipid uptake (36). According to Yasumoto et al., de novo lipogenesis appears more active in glioma stem cells (GSCs) than in differentiated bulk cells. The authors discovered that GSCs have higher levels of 14[C]-glucose and 14[C]-acetate incorporation into lipids in comparison with non-GSCs (48). Additionally, in various cancer cell types, including glioma, pancreatic tumors, and breast cancer, fatty acid synthase (FASN) was identified as an essential factor in CSC survival (49).
A substantial increase in lipid droplet concentration and stearoyl-CoA desaturase-1 expression was seen in non-small cell lung cancer (NSCLC) tumor samples from patients before and after Gefitinib therapy (50). Furthermore, cytarabine (AraC)-treated acute myeloid leukemia cells have altered lipid metabolism, as demonstrated by an increased CD36 expression and mitochondrial FAO (51). Thus, another critical point in maintaining the CSC pool could be represented by FAO. Two significant publications highlighted this concept focusing on the pharmacological inhibition of FAO by etomoxir (irreversible inhibitor of CTP-1) that either decreased the number of leukemic stem and progenitor cells or sensitized the CSCs to sorafenib, a tyrosine kinase inhibitor (52,53).
Another mechanism through which lipid metabolism can be influenced is via HIF-2α. This member of the hypoxia-inducible factor family is responsible for the proliferation, angiogenesis, and pharmacologic resistance in various types of cancer (54). One of the proposed mechanisms suggests that the increased expression of HIF-2α in tumor cells may increase the expression of PLIN2 - a cytosolic lipid droplet coat protein frequently used as a marker of intracellular lipid accumulation (55,56). Overexpression of PLIN2 is sufficient to enhance lipid production and storage in murine fibroblast in vitro and in the liver in vivo (55,57). Interestingly, in mouse hepatocytes, in vivo, PLIN2 expression is associated with HIF-2 activation.
Moreover, in another study, microarray data indicated that HIF-2 enhances PLIN2 mRNA expression in vitro in clear cell renal carcinoma (58,59). Thus, PLIN2 expression driven by HIF-2α can promote tumorigenesis and pharmacologic resistance to endoplasmic reticulum stress (58). Consequently, it could be interesting to investigate further if CD36 expression is influenced by hypoxia in the tumor environment.
The modulation of CD36 in various cancer therapies with emphasis on drug resistance
Changes in lipid metabolism have long been linked to resistance to traditional chemotherapies and targeted treatments for various malignancies. Thus, in the context of personalized medicine, many studies have focused over the last two decades on CD36, as it is one of the most reputable FA transporters.
Most studies suggest that the lipid metabolism of cancer-resistant cells varies in response to therapy and environmental and cellular contexts (36). In the following section, we present the most prominent studies of the past decade focusing on the modulation of CD36 in different cancer settings emphasizing drug resistance (Table 1).
The most significant research of the last decade focuses on regulating CD36 in various cancer contexts, with a particular emphasis on treatment resistance.
| Compound | Target /Mechanism | Type of cancer | Result | Source | Ref |
|---|---|---|---|---|---|
| Quercetin | miR-1254/CD36 signaling pathway | Oral squamous cell carcinoma | Anti-tumor effects on proliferation and invasion | CAL-27 cell line | (59) |
| Quercetin | Regulate TSP1 by increasing the expression of CD36 | Pancreatic | • Enhancing cell adhesion • Stimulating the immune response • May reduce the death rate | Drug Bank String and TCGA databases | (60) |
| Gemcitabine siRNA | Downregulation of CD36 | Pancreatic | siCD36 suppressed Bcl-2 in Gemcitabine-resistant (GR) PDAC cell lines | Resected specimen from pancreatic ductal adenocarcinoma (GR) PDAC cell lines | (20) |
| Lapatinib | CD36-mediated metabolic rewiring | Breast | • CD36 has a key role in HER2-targeted treatment resistance • Re-sensitizing rBT474 cells to lapatinib with SSO | Experimental model: MMTV-neu, MMTV-Cre, NSG mice Human breast cancer cells: BT474, SKBR3, HCC202 | (61) |
| Tamoxifen siRNA | Regulation of CD36 expression | Breast | • Inhibition of CD36 expression • Restoring Tamoxifen′s capacity to inhibit cell growth | ERα- positive MCF-7 cells | (62) |
| Nanoparticle load-CD36 siRNA Genistein | CD36/phospho-p38 MAPK axis | Breast | • Silencing the expression of CD36 • Suppressing the proliferation of MDA-MB-231 cells • Cellular apoptosis | MDA-MB-231 cell line | (63) |
| Nobiletin | CD36/ (STAT3)/NF-κB signaling axis | Breast | • Suppressing CD36 expression • Inhibiting CD36-dependent breast cancer cell migration • Inhibiting angiogenesis • Decreasing the expression of TSP-1 and TGF-β1 | MCF-7, and MDA-MB-231 cell lines | (64) |
| Decitabine Chidamide | Methylation of CD36 | Lung | De-methylation and re-expression of silenced CD36 | Lung cancer tissue Cell line: A549, NCI-H520, Calu-1 | (65) |
| Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) | Upregulation of TRAIL receptors through engagement of CD36 by three type-1 repeat (3TSR) domain of TSP1 | Glioblastoma (GBM) | Inhibits tumor growth Prolonged survival of mice bearing intracranial TRAIL-resistant GBM | GBM tumor cells and GBM-associated endothelial cells, mouse models of TRAIL-resistant GBMs | (66) |
| Cisplatin Pt(IV) pro-drugs | Exploiting the upregulation of CD36 | Ovarian | Mitochondria-damaging FA-like drugs | Cisplatin-sensitive ovarian cancer (A2780), Cisplatin-resistant (A2780cis) and (HEK293) non-cancerous cell lines | (67) |
Breast cancer
Adipocytes influence breast cancer behavior, as these cells release FAs into the tumor microenvironment. When human adipocytes and breast cancer cells were co-cultured, the expression of CD36 was increased (27). Furthermore, the hormone leptin, released by adipocytes, promotes cancer stem cell renewal and chemoresistance (46,47).
In 2007, the group conducted by Vasquez-Martin showed that due to the bidirectional crosstalk between the FASN and the human epidermal growth factor receptor-2 (HER2), cancer cells undergo apoptosis as the endogenous FA lipogenic pathway is inhibited (68). According to this study, FASN-driven cellular signaling is implicated in HER2-induced malignant transformation, and FASN blockage suppresses HER2 transcription (68).
More than ten years later, Feng and colleagues discovered that breast cancer cells could escape lapatinib (an oral dual tyrosine kinase inhibitor that targets epidermal growth factor receptor-EGFR and HER2) and consequently survive by displaying a change toward CD36-mediated FA uptake (61,69). This study aimed to determine whether lapatinib-resistant cells developed resistance over time or were formed from an innately resistant subpopulation expressing high levels of CD36. The authors concluded that after continuous lapatinib therapy, drug-naïve cells significantly increased CD36 expression rather than a selection process of resistant cells, which already exhibited a CD36 overexpression (61).
Although Tamoxifen is a standard treatment used in patients with ER-positive breast cancer, oncologists must be aware that therapy resistance can develop in some instances. Liang and colleagues published an interesting study demonstrating that Tamoxifen's influence on the proliferation of ER-positive MCF-7 cells was connected to regulating CD36 expression. Additionally, the authors showed that Tamoxifen's growth inhibitory impact was decreased by activating CD36 expression, whereas siRNA-mediated CD36 inhibition caused a synergistic suppression of cell growth (62).
Due to its poor prognosis and aggressive nature, triple-negative breast cancer (TNBC), which lacks estrogen, progesterone, and human epidermal growth factor receptor 2, represents a therapeutic challenge (70). Thus, finding appropriate therapy combinations with the lowest risk of toxicity is critical in this scenario.
Genistein is a classic example of a phytoestrogen molecule, similar to other plant components like lignans, with estrogenic action (71). Furthermore, this compound is crucial for the phospholipid breakdown of MDA-MB-231 cells (TNBC cell line that is highly aggressive, invasive, and poorly differentiated) and tyrosine kinase signal transduction pathways (63). A crucial metabolic mechanism for the development of TNBC is FA oxidation. Consequently, CD36 targeting may be a possible TNBC treatment approach. Exploring this pathway, using nanomaterials such as siRNA carriers in combination with Genistein in MDA-MB-231 cells revealed a significant reduction of CD36 expression through phosphorylation of the p38 MAPK pathway, resulting in cell growth inhibition (63). Nobilet is another flavonoid whose effectiveness has been tested in the context of ER-positive breast cancer cells. This compound reduced tumor angiogenesis, modulating Src, FAK, and STAT3 signaling through PXN (72). In 2017, Sp and colleagues hypothesized that Nobiletin's antiangiogenic effect included regulation of CD36 through signal transducer and activator of transcription 3 (STAT3) rather than via TSP-1 since CD36 also interacts with the oncogene STAT3. The authors demonstrated that CD36 siRNA decreased cell invasion by over 65% in the MDA-MB-231 cell line, indicating that CD36 plays a role in tumor metastasis (64).
Pulmonary cancer
Lung cancer is the most common cause of cancer death worldwide; Non-Small Cell Lung Cancer (NSCLC) accounts for the majority of cases (73,74). Lipid metabolic reprogramming is one of the leading mechanisms for cancer development, progression, and resistance to therapies (73). The targeting of HMG-box transcription factor 1 (HBP1) by miR-21 has previously been demonstrated to promote the invasion and migration of drug-resistant lung adenocarcinoma cancer cells (75). In 2019, Ni and colleagues published a study that examines the relationship between miR-21 and CD36-regulated FA metabolism in NSCLC cell lines (76). The authors demonstrated that miRNA-21 mimic treatment stimulated CD36 expression and cell proliferation and migration. Moreover, CD36 silencing decreased intracellular lipid content and hindered the development of tumor cells mediated by miRNA-21(76).
Using weighted gene co-expression network analysis (WGCNA), Sun and colleagues identified CD36 as a hub gene with a low expression in lung cancer (65). However, they outline a new link between increased CD36 gene methylation and tumor development. The authors concluded a decrease in CD36 methylation after administering Decitabine, a DNA methylation inhibitor, and Chidamide, a particular class I histone deacetylase inhibitor, alone and in combination, ultimately inhibited tumor growth (65).
Haematological malignancies
Many hypotheses have been proposed to elucidate chemoresistance in acute myeloid leukemia (AML). However, this phenomenon continues to represent an issue regarding survival rates. An interesting study from 2019 analyzed AraC resistance in AML patient-derived xenografts and targeted mitochondrial metabolism via the CD36-mitochondrial FA β-oxidation and oxidative phosphorylation (51). The authors identified the CD36 receptor as one of the most differentially expressed genes associated with FA and lipid metabolism in chemoresistant leukemic cells. This may open up new treatment options for AML patients with chemoresistance as inhibiting oxidative phosphorylation via CD36, might boost AraC's antileukemic impact (51).
Interestingly, the gonadal adipose tissue (GAT) can be used as a niche by a subpopulation of leukemic stem cells. This interaction increases FA metabolism, as observed in a murine model of blast crisis chronic myeloid leukemia (CML). A high level of FAO and a drug-resistant phenotype was demonstrated in CD36+ cells together with the capacity of GAT to protect the cells against chemotherapy (77).
Reducing drug resistance and simultaneously reducing tumor growth and dissemination can sometimes be obtained by switching from one cancer drug to another. According to this principle, Landberg and colleagues observed in CML cells that the second-generation tyrosine kinase inhibitor (TKI) nilotinib can overcome the decreased in vitro sensitivity of CD36-expressing cells to imatinib (78).
Pancreatic cancer
With a 5-year survival rate of barely 6%, pancreatic ductal adenocarcinoma (PDAC) is the fourth most significant cause of cancer-related deaths globally (79). Jia and colleagues showed that in pancreatic cancer cell lines and tumor tissue, CD36 expression is considerably reduced (80). Moreover, lower TNM staging and CA19-9 levels were predicted by a low expression of CD36. However, the latter also predicted larger tumors and poor survival. This might be because CD36-negative tissue had fewer lymph node metastases and infiltrated surrounding tissue, decreasing TNM staging (80).
In gemcitabine-treated patients, CD36 expression in resected pancreatic ductal adenocarcinoma (PDAC) specimens was also correlated to cancer prognosis (20). The group led by Kubo revealed that, consequently, to CD36 down-regulation after siRNA transduction, PDAC cell resistance to gemcitabine was reduced, and anti-apoptosis proteins were inhibited, supporting the idea that chemoresistance can be fought with anti-CD36 treatments (20). The heterogeneity in patient samples may cause inconsistency between these studies regarding CD36 expression. A larger and more homogeneous cohort of samples would benefit the continuing study of CD36 expression in connection to pancreatic cancer with an emphasis on treatment resistance.
Ovarian cancer
There is no proof yet of the involvement of CD36 in ovarian cancer development and progression. However, some studies used CD36 as a therapeutic escape for Cisplatin-resistant ovarian cancer. Resistance to platinum-based therapies and toxicity are two crucial issues that must be addressed to obtain remission without affecting the quality of life. In this regard, Pt(IV) pro-drugs that resemble the fatty acid structure (FALPs) have been developed to upregulate CD36 receptors, allowing them to enter the ovarian cancer cell. The difference between FAs (necessary for lipid metabolism) and Pt(IV) pro-drugs consists in the fact that the latter can cause mitochondrial damage, rendering them effective in fighting cisplatin resistance in ovarian cancer (67). Using a platinum-resistant ovarian cancer PDX model, a prosaposin-derived therapeutic cyclic peptide was expected to promote cancer regression (81). The authors of this study demonstrated that the prosaposin-based therapeutic agent inhibits the progression of ovarian cancer through TSP-1 and downstream CD36 signaling, which further highlights the importance of CD36 in the tumor microenvironment (81).
Melanoma
The use of targeted treatments and immunotherapies for metastatic melanoma has evolved in the past decade, yet the vast majority of patients are still left uncured. It remains a major clinical issue to overcome resistance to treatment. A TCGA melanoma cohort research reveals that melanoma patients with high CD36 expression have a worse clinical prognosis (82). A possible explanation could be the up-regulation of CD36 in response to mitogen-activated protein kinase inhibitors (MAPKi) in BRAF-mutated melanomas (83). Both in vitro and in vivo testing demonstrated that during both adaptation and drug tolerance states, MAPKi promotes and sustains the expression of CD36 in BRAF-mutated melanomas (83). Martini and colleagues identified CD36 as a regulator of vascular mimicry (VM) in melanoma cells. The authors concluded that, as part of the tumor microenvironment, CD36 works in tandem with adhesion molecules like integrin-3 and different microenvironment components such as laminin for organizing VM channels (82).
Glioblastoma
Glioblastoma (GBM) is the most prevalent high-grade primary malignant brain tumor with a meager survival rate. Consequently, novel therapeutic strategies are highly needed due to the poor prognosis of individuals receiving currently authorized GBM treatments (84). Highly vascularized tumors like GBMs depend on developing new tumor-associated blood vessels (85). Thus, in 2015, Choi and colleagues investigated in vitro and in vivo the antiangiogenic potential of three type-1 repeat (3TSR) domain of TSP-1 on GBM cells (66,86,87). The authors demonstrated that 3TSR sensitized GBM lines to caspase-3/7-mediated apoptosis by upregulating tumor-necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor DR4/5 expression in a CD36-dependent manner. Furthermore, the results showed a decrease in tumor growth, with prolonged survival of mice bearing TRAIL-resistant GBM using the combination of engineered human mesenchymal stem cells (MSC) with 3TSR/TRAIL (66).
A crucial method by which cells identify, phagocytose, and eliminate what is unnecessary is via scavenger receptors (88). Multiple cell types in the brain, including microglia, endothelial cells, astrocytes, and neurons, use CD36 as a the scavenger receptor (89). It was hypothesized that CSCs might recognize and react to an unfavorable environment by expressing scavenger receptors as an adaptive strategy (90). In 2015, the group conducted by Hale published an exciting paper that reports the crosstalk between patient-derived and in vivo xenograft models of CSCs and CD36 (90). The authors demonstrated the presence of oxidized phospholipids in GBM cells, which are CD36 ligands. Additionally, exposure to oxidized low-density lipoprotein increased the proliferation of CSCs, rendering this molecular mechanism an important target for future therapeutic approaches (90).
Different settings of anti-CD36 cancer therapies - future perspectives
In terms of FA uptake, normal cells preferentially choose the exogenous sources. In cancer cells, it was established that more than 90% of FA are de novo synthesized and FASN is overexpressed in a variety of cancer types (61). Cancer cells usually display a "lipogenic phenotype" defined by both FASN overexpression and high FA biogenesis, even in the presence of circulating exogenous FA. However, CD36 is closely connected to lipid homeostasis, angiogenesis, and tumor dissemination. Therefore it was often regarded as a potential cancer therapeutic target (1). To support this idea, addressing CD36 lipid transport activity is important as the development of resistance to HER2 inhibitors is influenced by CD36 up-regulation (61). In vitro studies on lapatinib-resistant breast cancer cells (rBT474) revealed that the small-molecule inhibitor sulfosuccinimidyl oleate (SSO) provides pharmacological inhibition of CD36, re-sensitizing rBT474 cells to lapatinib (61). Other studies that target CD36 in the context of breast cancer (63,64) are summarised in Table 1.
In mice, the administration of neutralizing monoclonal antibodies (JC63.1) to block CD36 has been shown to inhibit gastric cancer metastases (91). An impediment to anti-tumor immunity and cancer treatment could be explained by a large number of regulatory T cells (Treg cells), which is why depleting them is a promising cancer treatment. However, it must be taken into account that this therapeutic strategy is hampered by autoimmunity (92). In 2020, the group led by Wang explored the regulation of CD36 in intratumoral Treg cells. YUMMI.7 melanoma-engrafted mice were treated with an anti-CD36 monoclonal antibody, which resulted in a decrease in tumor growth with a reduction of intratumoral Treg cells, whereas Treg cells in the spleen and draining lymph nodes remained stable (92).
The role of natural bioactive compounds in the treatment and prevention of different types of cancer has received considerable attention in the literature (59,60). Among them is the flavonoid quercetin, which is thought to be an antioxidant that scavenges free radicals (93). It has been shown that quercetin activates the miR-1254/CD36 signaling pathway, which inhibits oral squamous cell carcinoma cells' ability to survive and invade (59).
Hypercholesterolemia is a common metabolic disease that has been linked to the development of malignancies that are steroid-targeted. However, in 2021, Yang and colleagues published a study that investigates the link between hypercholesterolemia and the aggressiveness of bladder cancer, a non-steroid-cancer (94). The authors concluded that the CD36/STAT3 signaling pathway increases cancer progression on the background of hypercholesterolemia-induced oxidized LDL (ox-LDL). Considering these mechanisms, targeting the CD36/STAT3 axis could represent a future therapeutic promise for patients who associate hypercholesterolemia and urinary bladder cancer (94).
In high-risk localized disease patient-derived xenografts (PDXs), the CD36 monoclonal antibody inhibited prostate cancer development. Thus, targeting FA absorption by blocking CD36 might also be a potential treatment strategy for patients diagnosed with prostate cancer (95). Another recent study on human prostate cancer cell lines analyzed the potency of C75, a radiosensitizing FASN inhibitor (96). The authors reported an increased C75 sensitivity in cells by CD36 neutralizing antibody, which suggests that the availability of FAs enhances the effect of the radiosensitizer (96).
A founding member of the MADS-transcriptional regulator factor that may influence the pathways of certain malignant tumors is the myocyte enhancer factor 2C-genecards (MEF2C) (97). The MEF2C-CD36 pathway is mentioned in a recent study published in 2023 as a possible approach to comprehending the mechanism of tumor regulation in colorectal cancer. Thus, MEF2C inhibits tumor growth through modulation of CD36 transcription, rendering this pathway a promising therapeutic target (97).
CD36 antibody targeting was also explored as a potential therapy in the CML setting. Thus, the group conducted by Landberg demonstrated that directing human NK cells to eliminate CML cells specifically can be obtained by CD36 targeting antibodies considering the antibody-dependent cellular toxicity (78).
The future perspective of personalized medicine resides in perfecting new therapeutic strategies that combine the strengths of peptides and monoclonal antibodies. Anticancer clinical trials exploring modified peptides with a TSP-1 mimetic activity targeting CD36, such as ABT-510 in haematological and solid neoplasia, were terminated due to severe side effects and ineffective performance (98). A problem with peptides is the rapid degradation and, therefore, the need for frequent compound administration (99). To overcome this problem and extend the half-life of the TSP-1 peptide, an antibody scaffold was added, resulting in a TSR peptide mimetic-antibody fusion molecule with favorable pharmacokinetics (CVX-045, CVX-22). CVX-045 was administered in advanced solid tumors, causing a decrease in tumor microvasculature, while CVX-22 was administered in melanoma. Even though both therapies had encouraging preclinical results, CVX-045 failed a phase 1 clinical trial due to severe adverse effects (100).
Conclusions
Despite current therapeutic advances in treating cancer patients, drug resistance continues to be one of the limiting factors. Consequently, novel treatment approaches have been created based on a better knowledge of the molecular mechanisms disrupted during the transition of a normal cell into a malignant one. CD36 is a critical participant in cancer development and progression via lipid uptake, immunological recognition, apoptosis, and anti-angiogenesis. Additionally, its prognostic value has been extensively discussed in the literature, making this FA-transporter a potential target for cancer patients. Numerous inhibitors targeting different aspects of lipid metabolism, including CD36, have been discovered and have already proved potential effectiveness.
In this review, we have highlighted the most relevant research of the past decade investigating the role of CD36 in treatment resistance in various neoplasms, either solid or haematological. Understanding the signaling pathways, cell dependencies, and the variety of molecules involved in tumor growth and metastasis opens new avenues for cancer patients once drug resistance occurs. Nevertheless, aiming at a particular molecule or mechanism involved in lipid metabolism may not be enough to induce long-term cancer cell growth inhibition and, thus, disease control.
Acknowledgements
Funding
This work was supported by the Ministry of Research, Innovation, and Digitalization in Romania, under Program 1—The Improvement of the National System of Research and Development, Subprogram 1.2—Institutional Excellence-Projects of Excellence Funding in RDI, Grant No. 31PFE/30.12.2021 and the National Recovery and Resilience Plan—Creation, Operational and Development of the National Center of Competence in the field of Cancer (CNCC), Grant No. 760009/30.12.2022.
Author contributions
Ioana Lambrescu and Mihail Hinescu had the idea for the article. Literature search and data analysis were performed by [Ioana Lambrescu], [Gisela Gaina] and [Laura Ceafalan]. The first draft of the manuscript was written by [Ioana Lambrescu] and [Laura Ceafalan] and [Mihail Hinescu] critically revised the work. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang J, Li Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics. 2019;9(17):4893-908
2. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009May;2(72):re3
3. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CSO. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature [Internet]. 2017;541(7635):41-5 Available from: http://dx.doi.org/10.1038/nature20791
4. Clemetson KJ, Pfueller SL, Luscher EF, Jenkins CSP. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. BBA - Biomembr. 1977;464(3):493-508
5. Lehner R, Quiroga AD. Fatty Acid Handling in Mammalian Cells. In: Biochemistry of Lipids, Lipoproteins and Membranes [Internet]. Elsevier. 2016 p. 149-84. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780444634382000055
6. Chen Y, Zhang J, Cui W, Silverstein RL. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J Exp Med [Internet]. 2022 Jun 6;219(6). Available from: https://rupress.org/jem/article/219/6/e20211314/213166/CD36-a-signaling-receptor-and-fatty-acid
7. Hao Y, Li D, Xu Y, Ouyang J, Wang Y, Zhang Y. et al. Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data. BMC Bioinformatics [Internet]. 2019May1;20(S7):195 Available from: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2734-4
8. Chen Y-J, Liao W-X, Huang S-Z, Yu Y-F, Wen J-Y, Chen J. et al. Prognostic and immunological role of CD36: A pan-cancer analysis. J Cancer [Internet]. 2021;12(16):4762-73 Available from: https://www.jcancer.org/v12p4762.htm
9. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull [Internet]. 2017Sep25;7(3):339-48 Available from: http://apb.tbzmed.ac.ir/Abstract/APB_386_20170226104729
10. Luanpitpong S, Janan M, Thumanu K, Poohadsuan J, Rodboon N, Klaihmon P. et al. Deciphering the Elevated Lipid via CD36 in Mantle Cell Lymphoma with Bortezomib Resistance Using Synchrotron-Based Fourier Transform Infrared Spectroscopy of Single Cells. Cancers (Basel) [Internet]. 2019Apr24;11(4):576 Available from: https://www.mdpi.com/2072-6694/11/4/576
11. Ward RA, Fawell S, Floc'h N, Flemington V, McKerrecher D, Smith PD. Challenges and Opportunities in Cancer Drug Resistance. Chem Rev [Internet]. 2021Mar24;121(6):3297-351 Available from: https://pubs.acs.org/doi/10.1021/acs.chemrev.0c00383
12. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S. et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene [Internet]. 2018;37(17):2285-301 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29398710
13. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F. et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J Exp Clin Cancer Res [Internet]. 2019Dec4;38(1):52 Available from: https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1049-7
14. DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen Y-Y, Fontenay G V. et al. CD36 Repression Activates a Multicellular Stromal Program Shared by High Mammographic Density and Tumor Tissues. Cancer Discov. 2012Sep;2(9):826-39
15. Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep [Internet]. 2016;6(November 2015):1-13 Available from: http://dx.doi.org/10.1038/srep18669
16. Ji Z, Shen Y, Feng X, Kong Y, Shao Y, Meng J. et al. Deregulation of Lipid Metabolism: The Critical Factors in Ovarian Cancer. Front Oncol [Internet]. 2020 Oct 19;10. Available from: https://www.frontiersin.org/article/10.3389/fonc. 2020 593017/full
17. Coburn CT, Knapp FF, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective Uptake and Utilization of Long Chain Fatty Acids in Muscle and Adipose Tissues of CD36 Knockout Mice. J Biol Chem [Internet]. 2000Oct;275(42):32523-9 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925820891285
18. Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K. et al. Muscle-specific Overexpression of FAT/CD36 Enhances Fatty Acid Oxidation by Contracting Muscle, Reduces Plasma Triglycerides and Fatty Acids, and Increases Plasma Glucose and Insulin. J Biol Chem [Internet]. 1999Sep;274(38):26761-6 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925819550876
19. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu Rev Nutr [Internet]. 2014Jul17;34(1):281-303 Available from: http://www.ajnr.org/cgi/doi/10.3174/ajnr.A1256
20. Kubo M, Gotoh K, Eguchi H, Kobayashi S, Iwagami Y, Tomimaru Y. et al. Impact of CD36 on Chemoresistance in Pancreatic Ductal Adenocarcinoma. Ann Surg Oncol [Internet]. 2020Feb;27(2):610-9 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31605325
21. Dong L, Yuan Y, Opansky C, Chen Y, Aguilera-Barrantes I, Wu S. et al. Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling. Oncotarget [Internet]. 2017;8(14):22550-62 Available from: http://www.oncotarget.com/abstract/15123
22. El-Sayes N, Vito A, Mossman K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers (Basel) [Internet]. 2021Feb15;13(4):806 Available from: https://www.mdpi.com/2072-6694/13/4/806
23. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science (80- ) [Internet]. 2013Mar29;339(6127):1546-58 Available from: https://www.sciencemag.org/lookup/doi/10.1126/science.1235122
24. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell [Internet]. 2017Feb;168(4):707-23 Available from: https://linkinghub.elsevier.com/retrieve/pii/S009286741730065X
25. Norouzi S, Gorgi Valokala M, Mosaffa F, Zirak MR, Zamani P, Behravan J. Crosstalk in cancer resistance and metastasis. Crit Rev Oncol Hematol [Internet]. 2018Dec;132:145-53 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1040842818301999
26. Deng M, Cai X, Long L, Xie L, Ma H, Zhou Y. et al. CD36 promotes the epithelial-mesenchymal transition and metastasis in cervical cancer by interacting with TGF-β. J Transl Med [Internet]. 2019;17(1):352 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31655604
27. Gyamfi J, Yeo JH, Kwon D, Min BS, Cha YJ, Koo JS. et al. Interaction between CD36 and FABP4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. npj Breast Cancer [Internet]. 2021Dec24;7(1):129 Available from: https://www.nature.com/articles/s41523-021-00324-7
28. Wang C, Yang Z, Xu E, Shen X, Wang X, Li Z. et al. Apolipoprotein C-II induces EMT to promote gastric cancer peritoneal metastasis via PI3K/AKT/mTOR pathway. Clin Transl Med [Internet]. 2021 Aug 9;11(8). Available from: https://onlinelibrary.wiley.com/doi/10.1002/ctm2.522
29. Tanase C, Gheorghisan-Galateanu A-A, Popescu ID, Mihai S, Codrici E, Albulescu R. et al. CD36 and CD97 in Pancreatic Cancer versus Other Malignancies. Int J Mol Sci [Internet]. 2020 Aug 6;21(16). Available from: http://www.ncbi.nlm.nih.gov/pubmed/32781778
30. Pfeiler S, Thakur M, Grünauer P, Megens RTA, Joshi U, Coletti R. et al. CD36-triggered cell invasion and persistent tissue colonization by tumor microvesicles during metastasis. FASEB J [Internet]. 2019Feb12;33(2):1860-72 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1096/fj.201800985R
31. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J. et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology [Internet]. 2017Oct3;6(10):e1344804 Available from: https://www.tandfonline.com/doi/full/10.1080/2162402X.2017.1344804
32. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature [Internet]. 2017;541(7635):41-5 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27974793
33. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med [Internet]. 2021Jan4;27(1):152-64 Available from: https://www.nature.com/articles/s41591-020-1131-x
34. Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng H. et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun [Internet]. 2022Oct2;13(1):5782 Available from: https://www.nature.com/articles/s41467-022-33349-y
35. Liao X, Yan S, Li J, Jiang C, Huang S, Liu S. et al. CD36 and Its Role in Regulating the Tumor Microenvironment. Curr Oncol [Internet]. 2022Oct27;29(11):8133-45 Available from: https://www.mdpi.com/1718-7729/29/11/642
36. Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology (Basel) [Internet]. 2020Dec16;9(12):474 Available from: https://www.mdpi.com/2079-7737/9/12/474
37. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J. et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev [Internet]. 2011May15;25(10):1041-51 Available from: http://genesdev.cshlp.org/lookup/doi/10.1101/gad.1987211
38. Balaban S, Lee LS, Schreuder M, Hoy AJ. Obesity and Cancer Progression: Is There a Role of Fatty Acid Metabolism? Biomed Res Int [Internet]. 2015;2015:1-17 Available from: http://www.hindawi.com/journals/bmri/2015/274585/
39. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer [Internet]. 2013Apr28;13(4):227-32 Available from: http://www.nature.com/articles/nrc3483
40. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T. et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov [Internet]. 2018Aug;8(8):1006-25 Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-17-1371
41. Kuo C-Y, Ann DK. When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun [Internet]. 2018Dec;38(1):47 Available from: http://doi.wiley.com/10.1186/s40880-018-0317-9
42. Nieman KM, Kenny HA, Penicka C V, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med [Internet]. 2011Nov30;17(11):1498-503 Available from: http://www.nature.com/articles/nm.2492
43. Gyamfi J, Lee Y-H, Eom M, Choi J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci Rep [Internet]. 2018Dec11;8(1):8859 Available from: http://www.nature.com/articles/s41598-018-27184-9
44. Gyamfi J, Lee Y-H, Min BS, Choi J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Sci Rep [Internet]. 2019Dec5;9(1):11336 Available from: http://www.nature.com/articles/s41598-019-47707-2
45. Lee Y, Jung WH, Koo JS. Adipocytes can induce epithelial-mesenchymal transition in breast cancer cells. Breast Cancer Res Treat [Internet]. 2015Sep19;153(2):323-35 Available from: http://link.springer.com/10.1007/s10549-015-3550-9
46. Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C. et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab [Internet]. 2018Jun;27(6):1357 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413118303024
47. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci [Internet]. 2018Dec6;25(1):20 Available from: https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-018-0426-4
48. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y. et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. Rishi A, editor. PLoS One [Internet]. 2016Jan25;11(1):e0147717 Available from: https://dx.plos.org/10.1371/journal.pone.0147717
49. Begicevic R-R, Arfuso F, Falasca M. Bioactive lipids in cancer stem cells. World J Stem Cells [Internet]. 2019Sep26;11(9):693-704 Available from: https://www.wjgnet.com/1948-0210/full/v11/i9/693.htm
50. Huang Q, Wang Q, Li D, Wei X, Jia Y, Zhang Z. et al. Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J Exp Clin Cancer Res [Internet]. 2019Dec15;38(1):129 Available from: https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1120-4
51. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R. et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov [Internet]. 2017Jul;7(7):716-35 Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-16-0441
52. Chen C-L, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM. et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab [Internet]. 2016Jan;23(1):206-19 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413115006191
53. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B. et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest [Internet]. 2010Jan4;120(1):142-56 Available from: http://www.jci.org/articles/view/38942
54. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol Cell [Internet]. 2010Oct;40(2):294-309 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097276510007501
55. Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H. et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med [Internet]. 2012Jun6;18(6):934-42 Available from: http://www.nature.com/articles/nm.2744
56. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C. et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm [Internet]. 2021Mar24;2(1):27-59 Available from: https://onlinelibrary.wiley.com/doi/10.1002/mco2.27
57. Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N. et al. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Metab [Internet]. 2002Oct1;283(4):E775-83 Available from: https://www.physiology.org/doi/10.1152/ajpendo.00040.2002
58. Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A. et al. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov [Internet]. 2015Jun;5(6):652-67 Available from: http://cancerdiscovery.aacrjournals.org/lookup/doi/10.1158/2159-8290.CD-14-1507
59. Chen L, Xia J-S, Wu J-H, Chen Y-G, Qiu C-J. Quercetin suppresses cell survival and invasion in oral squamous cell carcinoma via the miR-1254/CD36 cascade in vitro. Hum Exp Toxicol [Internet]. 2021Sep9;40(9):1413-21 Available from: http://journals.sagepub.com/doi/10.1177/0960327121991912
60. Pang B, Xu X, Lu Y, Jin H, Yang R, Jiang C. et al. Prediction of new targets and mechanisms for quercetin in the treatment of pancreatic cancer, colon cancer, and rectal cancer. Food Funct [Internet]. 2019;10(9):5339-49 Available from: http://xlink.rsc.org/?DOI=C9FO01168D
61. Feng WW, Wilkins O, Bang S, Ung M, Li J, An J. et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep [Internet]. 2019Dec;29(11):3405-3420.e5 Available from: https://linkinghub.elsevier.com/retrieve/pii/S2211124719314792
62. Liang Y, Han H, Liu L, Duan Y, Yang X, Ma C. et al. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis [Internet]. 2018Dec21;7(12):98 Available from: http://www.nature.com/articles/s41389-018-0107-x
63. Wang B, Yan N, Wu D, Dou Y, Liu Z, Hu X. et al. Combination inhibition of triple-negative breast cancer cell growth with CD36 siRNA-loaded DNA nanoprism and genistein. Nanotechnology [Internet]. 2021Sep24;32(39):395101 Available from: https://iopscience.iop.org/article/10.1088/1361-6528/ac0d1e
64. Sp N, Kang D, Kim D, Park J, Lee H, Kim H. et al. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Κb Signaling Axis. Nutrients [Internet]. 2018Jun15;10(6):772 Available from: http://www.mdpi.com/2072-6643/10/6/772
65. Sun Q, Zhang W, Wang L, Guo F, Song D, Zhang Q. et al. Hypermethylated CD36 gene affected the progression of lung cancer. Gene [Internet]. 2018Dec;678:395-406 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0378111918307649
66. Choi SH, Tamura K, Khajuria RK, Bhere D, Nesterenko I, Lawler J. et al. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol Ther. 2015Feb;23(2):235-43
67. Jayawardhana AMDS, Stilgenbauer M, Datta P, Qiu Z, Mckenzie S, Wang H. et al. Fatty acid-like Pt(iv) prodrugs overcome cisplatin resistance in ovarian cancer by harnessing CD36. Chem Commun [Internet]. 2020;56(73):10706-9 Available from: http://xlink.rsc.org/?DOI=D0CC02174A
68. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting “HER2 super-expression” occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol [Internet]. 2007Oct;31(4):769-76 Available from: http://www.ncbi.nlm.nih.gov/pubmed/17786307
69. Johnston SRD, Leary A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs of Today [Internet]. 2006;42(7):441 Available from: http://journals.prous.com/journals/servlet/xmlxsl/pk_journals.xml_summary_pr?p_JournalId=4&p_RefId=985637&p_IsPs=N
70. Almansour NM. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front Mol Biosci [Internet]. 2022 Jan 25;9. Available from: https://www.frontiersin.org/articles/10.3389/fmolb. 2022 836417/full
71. Spagnuolo C, Russo GL, Orhan IE, Habtemariam S, Daglia M, Sureda A. et al. Genistein and Cancer: Current Status, Challenges, and Future Directions. Adv Nutr [Internet]. 2015Jul1;6(4):408-19 Available from: https://academic.oup.com/advances/article/6/4/408/4568643
72. Sp N, Kang D, Joung Y, Park J, Kim W, Lee H. et al. Nobiletin Inhibits Angiogenesis by Regulating Src/FAK/STAT3-Mediated Signaling through PXN in ER+ Breast Cancer Cells. Int J Mol Sci [Internet]. 2017Apr30;18(5):935 Available from: http://www.mdpi.com/1422-0067/18/5/935
73. Eltayeb K, La Monica S, Tiseo M, Alfieri R, Fumarola C. Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer. Cells [Internet]. 2022Jan25;11(3):413 Available from: https://www.mdpi.com/2073-4409/11/3/413
74. de Sousa VML, Carvalho L. Heterogeneity in Lung Cancer. Pathobiology [Internet]. 2018;85(1-2):96-107 Available from: https://www.karger.com/Article/FullText/487440
75. Su C, Cheng X, Li Y, Han Y, Song X, Yu D. et al. MiR-21 improves invasion and migration of drug-resistant lung adenocarcinoma cancer cell and transformation of EMT through targeting HBP1. Cancer Med [Internet]. 2018Jun;7(6):2485-503 Available from: https://onlinelibrary.wiley.com/doi/10.1002/cam4.1294
76. Ni K, Wang D, Xu H, Mei F, Wu C, Liu Z. et al. miR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int [Internet]. 2019Dec23;19(1):219 Available from: https://cancerci.biomedcentral.com/articles/10.1186/s12935-019-0941-8
77. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M. et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell [Internet]. 2016Jul;19(1):23-37 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1934590916301515
78. Landberg N, von Palffy S, Askmyr M, Lilljebjörn H, Sandén C, Rissler M. et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica [Internet]. 2018Mar;103(3):447-55 Available from: http://www.haematologica.org/lookup/doi/10.3324/haematol.2017.169946
79. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin [Internet]. 2014Jan;64(1):9-29 Available from: http://doi.wiley.com/10.3322/caac.21208
80. Jia S, Zhou L, Shen T, Zhou S, Ding G, Cao L. Down-expression of CD36 in pancreatic adenocarcinoma and its correlation with clinicopathological features and prognosis. J Cancer [Internet]. 2018;9(3):578-83 Available from: http://www.jcancer.org/v09p0578.htm
81. Wang S, Blois A, El Rayes T, Liu JF, Hirsch MS, Gravdal K. et al. Development of a prosaposin-derived therapeutic cyclic peptide that targets ovarian cancer via the tumor microenvironment. Sci Transl Med [Internet]. 2016 Mar 9;8(329). Available from: https://www.science.org/doi/10.1126/scitranslmed.aad5653
82. Martini C, DeNichilo M, King DP, Cockshell MP, Ebert B, Dale B. et al. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer [Internet]. 2021Dec2;21(1):765 Available from: https://bmccancer.biomedcentral.com/articles/10.1186/s12885-021-08482-4
83. Aloia A, Müllhaupt D, Chabbert CD, Eberhart T, Flückiger-Mangual S, Vukolic A. et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF -mutated Melanoma to MAPK Inhibitors. Clin Cancer Res [Internet]. 2019Nov15;25(22):6852-67 Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-19-0253
84. Rong L, Li N, Zhang Z. Emerging therapies for glioblastoma: current state and future directions. J Exp Clin Cancer Res [Internet]. 2022Dec15;41(1):142 Available from: https://jeccr.biomedcentral.com/articles/10.1186/s13046-022-02349-7
85. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci [Internet]. 2007Aug;8(8):610-22 Available from: http://www.nature.com/articles/nrn2175
86. Jiménez B, Volpert O V, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med [Internet]. 2000Jan;6(1):41-8 Available from: http://www.nature.com/articles/nm0100_41
87. Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res [Internet]. 2007Sep;74(2-3):90-9 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0026286207000532
88. Greenberg ME, Li X-M, Gugiu BG, Gu X, Qin J, Salomon RG. et al. The Lipid Whisker Model of the Structure of Oxidized Cell Membranes. J Biol Chem [Internet]. 2008Jan;283(4):2385-96 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925820776714
89. Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia [Internet]. 2002Nov4;40(2):195-205 Available from: https://onlinelibrary.wiley.com/doi/10.1002/glia.10148
90. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K. et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem Cells [Internet]. 2014Jul;32(7):1746-58 Available from: http://doi.wiley.com/10.1002/stem.1716
91. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S. et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics [Internet]. 2019;9(18):5359-73 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31410220
92. Wang H, Franco F, Tsui Y-C, Xie X, Trefny MP, Zappasodi R. et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol [Internet]. 2020Mar17;21(3):298-308 Available from: http://www.nature.com/articles/s41590-019-0589-5
93. Gibellini L, Pinti M, Nasi M, Montagna JP, De Biasi S, Roat E. et al. Quercetin and Cancer Chemoprevention. Evidence-Based Complement Altern Med [Internet]. 2011;2011:1-15 Available from: http://www.hindawi.com/journals/ecam/2011/591356/
94. Yang L, Sun J, Li M, Long Y, Zhang D, Guo H. et al. Oxidized Low-Density Lipoprotein Links Hypercholesterolemia and Bladder Cancer Aggressiveness by Promoting Cancer Stemness. Cancer Res [Internet]. 2021Nov15;81(22):5720-32 Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-21-0646
95. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R. et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med [Internet]. 2019 Feb 6;11(478). Available from: https://www.science.org/doi/10.1126/scitranslmed.aau5758
96. Rae C, Fragkoulis GI, Chalmers AJ. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv Radiat Oncol [Internet]. 2020Sep;5(5):994-1005 Available from: https://linkinghub.elsevier.com/retrieve/pii/S2452109420301718
97. Wang M, Jiang Y, Fan Z-L, Zhang N, Liang J, Zheng Z. Exosomal MEF2C's Association With the Progression of CRC in Regulating CD36 Transcription. Altern Ther Health Med [Internet]. 2023Jan;29(1):198-209 Available from: http://www.ncbi.nlm.nih.gov/pubmed/36074971
98. Jeanne A, Schneider C, Martiny L, Dedieu S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front Pharmacol. 2015;6(OCT):1-13
99. Coronella J, Li L, Johnson K, Pirie-Shepherd S, Roxas G, Levin N. Selective activity against proliferating tumor endothelial cells by CVX-22, a thrombospondin-1 mimetic CovX-Body. Anticancer Res [Internet]. 2009Jun;29(6):2243-52 Available from: http://www.ncbi.nlm.nih.gov/pubmed/19528489
100. Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: Highlights of the 2008 American Society of Clinical Oncology meeting. J Hematol Oncol [Internet]. 2008Dec29;1(1):20 Available from: https://jhoonline.biomedcentral.com/articles/10.1186/1756-8722-1-20
Author contact
![]() Corresponding author: Laura C. Ceafalan; laura.ceafalanro.
Corresponding author: Laura C. Ceafalan; laura.ceafalanro.