Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(23):6949-6963. doi:10.7150/jca.64205 This issue Cite
Review
Advancements in Human Breast Cancer Targeted Therapy and Immunotherapy
Mayassa J. Bou-Dargham1#, Sophia Draughon1#, Vance Cantrell1, Zahraa I. Khamis1,2 ![]() , Qing-Xiang Amy Sang1,3
, Qing-Xiang Amy Sang1,3 ![]()
1. Department of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida, United States of America.
2. Department of Chemistry and Biochemistry, Faculty of Sciences-I, Lebanese University, Beirut, Lebanon.
3. Institute of Molecular Biophysics, Florida State University, Tallahassee, Florida, United States of America.
#These authors contributed equally to this work.
Received 2021-6-23; Accepted 2021-9-16; Published 2021-10-11
Abstract

Human breast cancer treatment regimens have evolved greatly due to the significant advances in understanding the molecular mechanisms and pathways of the common subtypes of breast cancer. In this review, we discuss recent progress in breast cancer targeted therapy and immunotherapy as well as ongoing clinical trials. We also highlight the potential of combination therapies and personalized approaches to improve clinical outcomes. Targeted therapies have surpassed the hormone receptors and the human epidermal growth factor receptor 2 (HER2) to include many other molecules in targetable pathways such as the epidermal growth factor receptor (EGFR), poly (adenosine diphosphate-ribose) polymerase (PARP), and cyclin-dependent kinase 4/6 (CDK4/6). However, resistance to targeted therapy persists, underpinning the need for more efficacious therapies. Immunotherapy is considered a milestone in breast cancer treatments, including the engineered immune cells (CAR-T cell therapy) to better target the tumor cells, vaccines to stimulate the patient's immune system against tumor antigens, and checkpoint inhibitors (PD-1, PD-L1, and CTLA4) to block molecules that mediate immune inhibition. Targeted therapies and immunotherapy tested in breast cancer clinical trials are discussed here, with special emphasis on combinatorial approaches which are believed to maximize treatment efficacy and enhance patient survival.
Keywords: Human breast cancer, targeted therapy, immunotherapy
Introduction
Despite the recent medical advancements, breast cancer remains the most common malignancy and the second deadly cancer among women according to the Centers for Disease Control and Prevention (CDC 2016) [1]. In 2021, it is estimated that breast cancer will affect 2811,550 women and cause the death of 43,600 women in the United States [2]. Like other cancers, breast cancer shows inter- and intra-tumor heterogeneity. Inter-tumor heterogeneity occurs among various individuals and is elucidated by the TNM (tumor-nodes-metastasis) staging system, the histopathologic classification, and the tumor grade [3]. The breast cancer clinical staging system runs from I-IV and is based on tumor size, lymph node involvement, and distant metastases [4]. The morphologic heterogeneity of breast carcinomas is evaluated based on histological type and histological grade that take into consideration the growth pattern and the degree of differentiation of the tumor, respectively [5]. The major histologic types of breast cancer involve invasive ductal carcinoma (50%-75%) and invasive lobular carcinoma (5%-15%) with the remainder showing mixed or other special histology [4]. The other morphologic heterogeneity, the tumor grade, depends on three architectural features, namely the degree of tubule or gland formation, nuclear pleomorphism, and mitotic rate [5]. On the other hand, breast cancer intra-tumor heterogeneity is observed at the morphologic, genomic, transcriptomic, and proteomic levels, and is due to the presence of distinct cellular phenotypes within the same individual tumor. The aforementioned heterogeneities in breast carcinomas have a dire impact on diagnosis, prognosis, and treatment of the disease.
While clinical and morphologic heterogeneities are important [6], expression heterogeneity is one of the main parameters used to classify breast tumors. Expression heterogeneity is mostly observed in the differential expression of the hormone receptors (HR), estrogen and progesterone, and the human epidermal growth factor receptor 2 (HER2/ERBB2). The major subtypes of human breast cancer are luminal A, luminal B, HER2-enriched (HER2+), and triple-negative breast cancers (TNBC). Luminal A and B are the hormone receptor positive (HR+) subtypes characterized by the expression of the estrogen receptor (ER+) and progesterone receptor (PR+) and generally respond to hormone therapy [7]. Unlike the luminal A subtype, luminal B breast cancers show lower expression of hormone receptors and higher expression of proliferation markers and HER2. In general, both luminal A and luminal B may be associated with a good prognosis and long-term survival due to many available treatments [8]. However, the HER2-enriched subtype, characterized by HER2 overexpression, is responsive to anti-HER2 antibodies but is associated with a poor clinical outcome. The efficacy, safety, dosage, and mode of administration of HER2-targeted drugs are still under investigation in clinical settings [9]. The triple-negative breast cancer (TNBC) lacks the expression of the hormone receptors and the amplification of HER2, and commonly overexpresses the epidermal growth factor receptor (EGFR). TNBC is further stratified into basal-like, normal breast-like, and claudin-low molecular subtypes [10,11]. While hormone therapies have shown good results for hormone receptor-positive patients during early stages, the treatment of late-stage and triple-negative breast cancers remains challenging.
Treatment of breast cancer has evolved greatly in recent years due to significant advances in understanding the molecular mechanisms and pathways of the most common subtypes of breast cancer. In this review, we discuss recent progress in targeted therapy and immunotherapy as well as ongoing clinical trials in breast cancer. We also highlight the outcomes of combined therapies and suggest the potential application of combination therapy and personalized approaches to improve clinical outcomes.
Evolution of Breast Cancer Treatments
To determine the optimal therapeutic protocol, breast cancer stage and subtype must be identified. In the United States, 62% of patients suffer from localized breast cancer upon diagnosis, while 31% have tumors spread to sentinel lymph nodes and 6% to distant sites. Treatment strategies for patients with nonmetastatic disease aim to eradicate the tumor from the breast tissue and adjacent lymph nodes, and to prevent relapse [4]. Local treatment of nonmetastatic breast cancer involves surgical resection (lumpectomy or mastectomy) with regional lymph node biopsy or full dissection that might be followed by adjuvant radiotherapy [12]. For early-stage (I or II) tumors, most patients undergo lumpectomy plus adjuvant radiotherapy (49%); whereas 34% of patients choose mastectomy alone or combined with chemotherapy/radiotherapy. The most common treatment among patients diagnosed with stage III breast cancer is mastectomy with adjuvant chemotherapy/radiotherapy (68%). As for metastatic breast cancer (IV), the treatment options tend to be palliative rather than curative with 56% of women receiving radiation therapy/chemotherapy alone [13].
Breast cancer subtype is relied upon to determine the optimal treatment for all stages of the disease and dictates the administered systemic therapy [12]. The standard systemic therapy for metastatic and nonmetastatic breast cancer includes endocrine therapy for HR+ tumors, HER2-targeted agents for HER2+ tumors, and chemotherapy alone for TNBC. Despite its risks, neoadjuvant and adjuvant chemotherapy continues to be an important adjunct for the systemic treatment of patients with HR+ and ERBB2+ breast cancers [4]. Targeted therapy against hormone receptors and HER2 often leads to resistance in clinical settings. Thus, alternative molecular agents in targetable pathways are urgently needed to overcome resistance and ameliorate the clinical outcome. In addition, other treatment modalities are being investigated to obviate the toxicities associated with various therapies. One such approach is immunotherapy that activates the patient's immune system by arming it with the weapons necessary to destroy the tumor cells.
Targeted Therapies
EGFR Targeted Therapies
The majority of TNBC tumors have increased EGFR expression suggesting this receptor as a potential therapeutic target to treat TNBC. Currently, two types of EGFR-targeted therapies - including small-molecule tyrosine kinase inhibitors and monoclonal antibodies, are being examined in ongoing clinical trials for the treatment of TNBC (Figure 1A) [10]. Gefitinib, an EGFR tyrosine kinase inhibitor, was tested for its ability to treat breast cancer [14]. The efficacy of gefitinib alone or in combination with neoadjuvant epirubicin and cyclophosphamide (EC) was evaluated in estrogen receptor-negative invasive breast cancer patients. While patients receiving EC with or without gefitinib showed no significant difference in the overall survival, TNBC patients showed a significantly higher pathologic complete response compared to non-TNBC patients independent of the treatment [14].
Targeted therapies in breast cancer. (a) Tyrosine kinase inhibitors and EGFR monoclonal antibodies against EGFR block the downstream signaling cascade that inhibits cell cycle progression. (b) PARP inhibitors in BRCA mutated patients result in cancer cell death by synthetic lethality due to BRCA mutation or by trapping PARP on the DNA and interfering with its catalytic activity by blocking its auto-PARylation. (c) Cyclin-dependent kinase (CDK) inhibitors such as CDK4/6 inhibitors block the activation of cyclin D and downstream cell cycle progression by preventing the release of Rb and E2F.
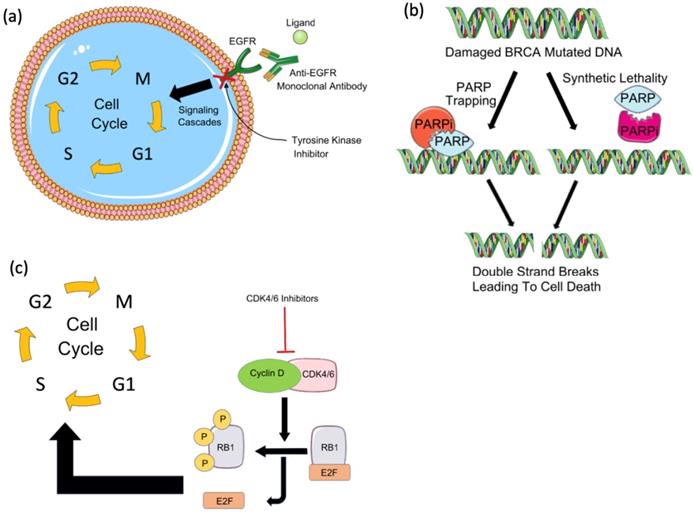
Another trial assessed the effectiveness of cisplatin (an alkylating agent interfering with DNA replication) alone or in combination with cetuximab (anti-EGFR agent) in metastatic TNBC. The objective response rate was higher in the patient group that received cetuximab in combination with cisplatin (20%) compared to the group receiving cisplatin alone (10%), but the results were statistically insignificant [11].
PARP Inhibitors
Poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) are drugs that mainly target breast cancer patients with germline BRCA1 or BRCA2 mutations [15]. Of the PARP enzyme family, PARP-1 plays a key role in signaling DNA damage and mediating base excision repair. Through polymerization of ADP-ribose (PARylation), PARP-1 repairs single-strand breaks (SSBs) through the base excision repair pathway. If PARylation is blocked, SSBs will accumulate and transform into double-stranded breaks (DSBs), which require homologous recombination (HR) for repair. The failure of HR to repair DSBs results in genomic instability and subsequent cell death. Cells deficient in HR, like those with BRCA1 or BRCA2 mutations, are sensitive to PARP inhibitors and usually die by synthetic lethality or PARP trapping. Synthetic lethality depends on the deadly effect of the mutation of two alleles on a cell or organism [15,16]. While in PARP trapping, PARPi traps the PARP-1 enzyme on DNA preventing its autoPARylation and its release from the site of damage; thus, interfering with the catalytic cycle of PARP1 and causing irreparable DNA damage (Figure 1B) [17].
The FDA approved the PARP inhibiting drugs talazoparib and olaparib for use in breast cancer. FDA approval of talazoparib was based on the EMBRACA trial. This trial investigated the effects of talazoparib compared to standard therapy in patients with BRCA-mutation breast cancer [18]. EMBRACA results showed that the patients who received talazoparib had an overall response rate of 62.6% compared to 27.2% in patients receiving chemotherapy. The initial results also showed that the talazoparib group had a significantly higher progression-free survival of 8.6 months compared to 5.6 months in the standard therapy group [19].
Olaparib's FDA approval was based on the OlympiAD trial which compared chemotherapy to olaparib monotherapy in patients with HER2-, BRCA-mutated metastatic breast cancer [19,20]. OlympiAD found that the median progression-free survival and the overall survival were both higher in the patients who received olaparib than in the patients who received chemotherapy. The response rate was 60% in the olaparib group compared to only 28.8% in the chemotherapy group [19]. Collectively, EMBRACA and OlympiAD trials demonstrate the effectiveness of PARP inhibitors as a therapy for breast cancers with BRCA mutations.
CDK4/6 Inhibitors
Cyclin-dependent kinase (CDK)4/6 plays a key role in cellular proliferation. By phosphorylating retinoblastoma (Rb), CDK4/6 induces the release of the transcription factor E2F which activates the expression of genes needed for DNA replication and thereby, promoting the transition of the cell cycle from the G1 to the S phase (Figure 1C) [21]. The CDK4/6-Rb interaction increases cancer cells' survival and CDK4/6 is known to play a role in breast cancer pathogenesis and tumorigenesis [22]. CDK4 was found to be amplified in 14% of luminal A and 25% of luminal B breast cancer patients. Cyclin-D was found to be amplified in 29% of luminal A and 58% of luminal B breast cancer patients [21].
CDK4/6 inhibitors have had a great deal of success in treating hormone receptor-positive (HR+) and HER2- advanced breast cancer patients. Three CDK4/6 inhibitors were recently approved for the treatment of HR+/HER2- breast cancer patients: palbociclib, ribociclib, and abemaciclib [23]. Palbociclib was granted accelerated approval in 2015 with the aromatase inhibitor letrozole for the treatment of ER+/HER2- advanced breast cancer as an initial endocrine-based therapy in postmenopausal women. In 2017, the FDA granted regular approval for palbociclib in combination with another anti-estrogen drug, fulvestrant, for the treatment of HR+/HER2- advanced or metastatic breast cancer in women who have developed disease progression following an endocrine therapy [23]. The approval of palbociclib was based on a series of trials known as the Paloma trials, conducted on HR+/HER2- breast cancer patients. Paloma I, II, and III demonstrated the ability of palbociclib to improve progression-free survival for breast cancer patients [23,24]. In April 2019, the FDA extended the approval of palbociclib to include men [25].
The results of the MONALEESA trials granted ribociclib FDA approval as a first-line treatment for HR+/HER2- advanced breast cancer in postmenopausal women [23]. In 2018, the FDA approved the combination of ribociclib with an aromatase inhibitor as an initial endocrine-based therapy for pre- or perimenopausal women with HR+/HER2- advanced or metastatic breast cancer. A series of clinical trials known as the MONARCH trials in HR+/HER2- patients led to the approval of another CDK4/6 inhibitor known as abemaciclib. Abemaciclib was first approved in 2017 for either second or later-line therapy with fulvestrant. It was also approved for either third or later-line therapy for both men and women as a single agent. In 2018, abemaciclib was later approved in combination with an aromatase inhibitor as initial endocrine-based therapy for postmenopausal women with HR+/HER2- cancer [23]. Novel combinations with CDK4/6 inhibitors and the use of CDK4/6 inhibitors beyond HR+/HER2- breast cancer are both areas of current research (Supplementary file 1) [23].
Anti-HER2 antibodies
As upregulated HER2 expression distinguishes tumor cells from normal breast cells, antibodies against HER2 have emerged as a successful approach for HER2+ breast cancer. Although Trastuzumab, a monoclonal antibody targeting HER2, was approved by the FDA for the treatment of metastatic HER2+ breast cancer patients in 1998, minimizing its cardiac-related side effects has been the aim of several other trials [26,27].
The standard schedule for trastuzumab monotherapy starts with an initial “loading dose” of 4 mg/kg of body weight followed by a weekly administration of 2 mg/kg. However, the administration of higher doses of trastuzumab may provide a more convenient treatment option if it was not for the major adverse effect of symptomatic cardiac dysfunction observed in about 2% to 4.7% of the patients [9]. One phase II trial sought to investigate the efficacy and safety of Trastuzumab administered at a higher dose and longer intervals. In this trial, patients with previously untreated HER2-positive metastatic breast cancer received a median number of five therapy cycles starting with an initial dose of 8 mg/kg and then 6 mg/kg of Trastuzumab intravenously at 3-week intervals until the patients withdrew from the trial or showed disease progression. The study was conducted on 105 HER2+ breast cancer patients and showed an overall response rate of 19% with a clinical benefit rate of 33%. Only one patient experienced a cardiac-related adverse event. This patient had a history of obesity and hypertension and was previously on epirubicin therapy which is associated with cardiac toxicity [9,28].
Trastuzumab is tested in combination with chemotherapy in a randomized phase III clinical trial known as the PANTHER. The node-negative breast cancer patients received cyclophosphamide with dose-dependent epirubicin followed by either docetaxel alone or in combination with standard 5-fluorouracil, cyclophosphamide, and epirubicin every three weeks [29]. Although long-term cardiac complications were relatively low, the efficacy of the treatment was compromised as the results were not statistically significant [29].
Another anti-HER2 monoclonal antibody, pertuzumab, was examined for its potential to treat breast cancer patients in combination with trastuzumab [30]. The CLEOPATRA trial tested the combination of pertuzumab, trastuzumab, and docetaxel against a placebo, trastuzumab, and docetaxel [30]. The trial showed that the addition of pertuzumab significantly increased the overall survival rate when compared to the placebo group while maintaining long-term cardiac safety [30]. The pertuzumab-receiving group had a median overall survival of 56.5 months whereas the placebo group had 40.8 months [30]. Another clinical trial known as NeoSphere showed similar results. Patients treated with pertuzumab, trastuzumab, and docetaxel combination had a significantly improved pathological complete response when compared to patients given only trastuzumab and docetaxel, without significant difference in treatment tolerability [31]. In the combination group, 45.8% of patients experienced complete pathological response; whereas the other group showed notably lower pathological complete response [31]. Overall, these two studies demonstrate that pertuzumab combined with trastuzumab and docetaxel improves clinical outcomes. More studies targeting HER2 in combination with immunotherapy are discussed in the following sections, and other completed clinical trials are summarized in Supplementary file 1.
Immunotherapies
CAR-T Cell Therapy
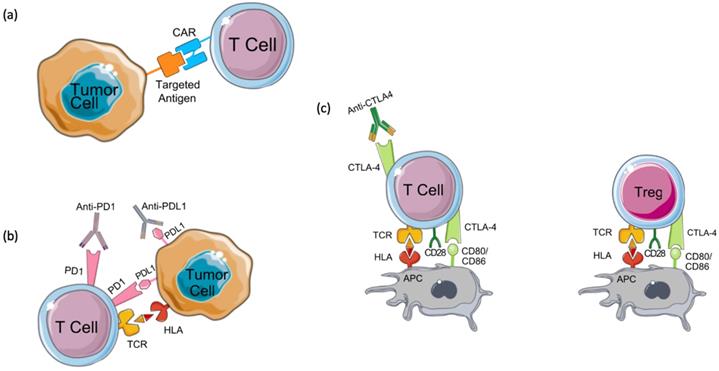
Chimeric antigen receptor-T cell (CAR-T) therapy is a type of T-cell-based adoptive immunotherapy designed to enhance anti-tumor T-cell activity [32]. CAR-T cell therapy relies on modifying T cell receptors (TCR) to express chimeric antigen receptors (CARs) that target a specific tumor antigen (Figure 2A) [32,33]. CARs are composed of four domains: extracellular, intracellular, transmembrane, and spacer. The extracellular domain is most often built from a single-chain variable fragment (ScFV) of a specific antibody, directed against a target antigen [34]. While the transmembrane domain holds the CAR into the cell membrane, the intracellular domain transduces the signals into the cell. Several efforts have been made to enhance the activation and the specificity of CAR-T cells which have resulted in 3 generations of CARs classified depending on the number of costimulatory domains [35,36]. Although a perfect CAR structure does not exist yet, the challenge remains in identifying a tumor-associated antigen that allows for a minimal recognition of normal cells' antigens [34,37]. Several CAR-T cells have been engineered to target different breast cancer antigens, however, they are still at an early stage and require further validation in clinical trials.
Mechanisms of action of immunotherapies. (a) Chimeric antigen receptor (CAR) T cells are engineered to express a chimeric receptor that recognizes an antigen expressed on tumor cells. This recognition results in the activation of cytotoxic T cell immune response targeted against tumor cells. Another class of immunotherapy targets immune checkpoints such as (b) programmed cell death-1 (PD-1), its ligand PD-L1, and (c) CTLA4. These molecules keep the immune response under control and prevent its over-activation and autoimmunity development. Tumor cells seize this mechanism by expressing PD-L1 which binds to PD-1 expressed on T cells and by inhibiting cytotoxic T cell response. CTLA4, the other immune checkpoint is expressed on regulatory T cells (Tregs) and activated T cells. Tumors induce Treg differentiation and chemotaxis to the tumor microenvironment to inhibit immune activation on one hand, and tolerance and overactivation result in T cell's expression of CTLA4, on the other hand, resulting in the inhibition of co-stimulation and activation of T cells by antigen-presenting cells (APCs) which prime and activate T cells against the tumor antigen. By targeting these immune checkpoints with antibodies that block their functions, tolerance can be overcome, and anti-tumor immune response can be re-activated.
AXL-targeted CAR-T
A recent study investigated the expression of AXL, a receptor tyrosine kinase, in breast tumors and cell lines, and the viability of an AXL-targeted CAR-T cell treatment [37]. This study tested patient-derived breast cancer tumors for AXL expression and found that patients with TNBC had the highest expression of AXL. AXL-CAR-T cells were co-cultured with the AXL-positive breast cancer cell lines (MDA-MB-231, 786-0, 769-P, Panc1, and MIAPaca2) and the AXL-negative breast cancer cell line, MCF-7. The co-culture resulted in significant cytotoxicity in the AXL-positive cell lines only [37]. The in-vivo mouse model xenografted with the cell line MDA-MB-231 and treated with AXL-CAR-T cell therapy had the lowest tumor volume compared to the other groups treated with either non-transduced T cells or PBS [37]. Although these results point out the promising effects of AXL-CAR-T cell therapy in TNBC, further validation in clinical settings is necessary.
c-Met-targeted CAR-T
The hepatocyte growth factor receptor, c-Met, is a cell-surface molecule expressed in 50% of breast cancer tumors rendering it a valuable immunotherapeutic target. In this context, a clinical trial evaluating c-Met as a target antigen was conducted on a small set of six breast cancer patients [38]. Each patient was injected with 1ml of mRNA-transfected c-Met-CAR-T cells two days before tumor excision [38]. The tumors were observed post-surgery, and found to experience necrosis, hemorrhage, and inflammatory cell infiltration at the injection site. c-Met expression was also lost in post-injection tumors [38]. The effectiveness of c-Met as a target antigen for CAR-T cell therapy requires further investigation in larger groups of breast cancer patients.
NKG2DL targeted CAR-T
Triple-negative breast cancer cells usually upregulate their expression of stress-induced ligands some of which are recognized by natural-killer group 2, member D (NKG2D) [39,40]. These ligands include MICA/B and ULBP 1-5 in breast cancer [39,41]. Since the adoptive transfer of NK cells has failed to induce tumor regression, CAR constructs containing full-length NKG2D were tested by Stentman's laboratories [40,42,43]. The results of the NKG2D-CAR-T preclinical trial were promising in solid tumors (including breast cancer) and the THINK clinical trial is currently recruiting to test the safety and activity of human NKG2D CAR referred to as NKR-2 [43-45].
HER2 targeted CAR-T
CAR was transduced in CD3+ cells to target HER2 and was found to specifically target and induce apoptosis in the HER2 overexpressing breast cancer cell line [46]. Herceptin-based CARs with modified signaling domains generated to target HER2 resulted in antitumor activity in breast cancer cell lines and a breast cancer xenograft mouse model [47]. Further studies showed that the adoptive transfer of autologous HER2-specific T-lymphocyte clones to a patient with metastatic HER2-overexpressing breast cancer prevented tumor cell dissemination to the bone marrow. However, the T cells were found unable to penetrate solid tumor metastases masses in the liver [48]. Her2-CAR-T therapy requires more optimization as a case study reported multiple organ dysfunction syndrome due to cytokine storm and resulted in the patient's death [49].
Experimentally promising CAR-T
HERV-K targeted CAR-T
Human endogenous retroviruses (HERVs) account for 8% of the human genome [50]. They have been incorporated into the genome millions of years ago after germline infections and are now referred to as “fossil” sequences [50-52]. The transcription of HERVs is controlled epigenetically, such that HERV expression is inhibited in normal adult cells [53,54]. In diseases where epigenetic mechanisms are disrupted, as in cancer, HERV protein expression is upregulated. Specifically, the expression levels of certain genes in the HERV-K group, such as env, gag, and np9 mRNA were elevated in breast cancer cells and were suggested as biomarkers for early breast cancer diagnosis [55,56]. The HERV-K env gene is expressed in 70% of breast cancers and is associated with breast cancer progression [57]. In addition, the overall survival of breast cancer patients with a high HERV-K env was lower compared to patients with low or moderate expression [57]. HERV-K viruses of the HML-2 subtype are the most intact retroviruses in the human genome and can be found in very high titers in the plasma of patients with breast cancer [58,59]. The transcripts of the env gene of HERV-K HML-2 subtype were shown to induce cancer development, and enhance in vitro invasion and migration, and cancer metastasis in MDA-MB 231 xenograft mouse model [58,60]. The env protein of HERV-K was found to be an immunogenic tumor-associated antigen and antibodies targeting it possess anti-tumor activity. Zhou et al injected breast cancer patients and normal female donors with anti-HERV-K monoclonal antibodies and derived chimeric antigen receptor (CAR)-T cells specific for HERV-K env protein (K-CAR) from the peripheral blood mononuclear cells of breast cancer patients and normal donors. The single-chain variable fragment was then introduced to the K-CARs and tested both in vitro on breast cancer cells and in vivo on human tumor xenograft mouse models. A significant decrease in tumor size and weight and cancer cell growth was observed in cells treated with K-CAR compared to control [58,60]. Even so, K-CAR may be a promising immunotherapy, more clinical testing is required to validate its safety and efficacy in humans.
Folate receptor-alpha targeted CAR-T
Folate receptor-alpha (FR-α) is upregulated in non-mucinous tumors of epithelial origins such as breast cancer; however, its overexpression is mostly detected in the TNBC subtype [61,62]. FR-α overexpression was found to be associated with poor outcomes in breast cancer and worse clinical outcomes in TNBC [63,64]. Estrogens control FR-α expression in breast cancer, and 17-β-estradiol downregulates its expression by a direct action on the estrogen receptor at the FR-α promoter [62,65]. This suggests a negative correlation between ER and FR-α expression, which explains why ER-negative breast cancers express more FR-α [66]. FR-α targeted CAR-T showed cytotoxic activity in breast cancer in vitro [66,67], and resulted in reduced tumor progression in a TNBC xenograft mouse model [66]. More experiments and trials are required and the variability in FR-α expression may demand further patient stratification in clinical studies [68].
MUC1 and ErbB2-targeted CAR-T
CAR-T cells engineered to target two antigens MUC1 and ErbB2 have shown successful results in breast cancer in vitro [69]. Dual targeting is an appealing approach for CAR-T cell therapy because it may reduce the toxic potential of the treatment and promote T-cell survival within the tumor. The reduced toxic potential results from optimized T-cell homing and tumor specificity. Nevertheless, enhanced T-cell survival in the tumor is attributed to the synergistic signals primarily delivered to T cells within the tumor microenvironment [69]. In vivo studies are still underway to compare the potency, efficacy, and safety of dual targeting.
Vaccines
Peptide-based Vaccines
Peptide-based vaccines focus mainly on eliciting a cellular antigen-specific T-cell response against antigens highly expressed on tumor cells such as HER2 and MUC1 in breast cancer [70-72]. Since the response to HER2 and MUC1 antibodies is low in breast cancer, peptide vaccines are mostly used with GM-CSF as a cytokine adjuvant to increase efficacy [73].
HER2
Vaccines made from peptides that may help the body build an effective immune response against HER2/neu expressing tumor cells are currently in clinical trials. HER2/neu immunogenic peptides vaccines are stretches of peptides from the HER2/neu protein. HER2 immunogenic peptides include G89 (HER-2/neu: 777-789), F7 (HER-2/neu: 776-788), p776 (HER-2/neu: 774-788), AE36 (HER-2/neu:776-790), GP2 (HER2/neu: 654-662), and E75 (HER2/neu: 366-37) [74-77]. Fusion proteins with the Ii-key (amino acids 77-80 of the immune-regulatory Ii protein), LRMK sequence, and ε-aminovaleric acid (Ava) were tested for increased antigenicity and T cell response [76]. AE37 is a Ii-Key hybrid of AE36 that induces a generalized immune response without the use of an adjuvant [78,79].
The trials that tested AE37, GP2, and E75 with GM-CSF showed that all three peptide vaccines were safe and well-tolerated [80]. In phase II clinical trial, the overall intention-to-treat analysis demonstrated no benefit to vaccination. However, the results did confirm the safety of the vaccine and suggested that vaccination may have clinical benefits in patients with low HER2-expressing tumors [81]. As for GP2 combination with GM-CSF, the phase II clinical trial did not demonstrate clinical benefit when administered in the adjuvant setting to node-positive and high-risk node-negative breast cancer patients with tumors expressing any degree of HER2 [82].
A recently completed clinical trial investigated the safety and clinical efficacy of GP2 and AE37 combined with the immunoadjuvant GM-CSF in high-risk breast cancer patients with any level of HER2 expression in adjuvant settings. Since HLA-A2 status is thought to affect peptide vaccine outcomes, this trial sought to investigate HLA-A2 and HER2 statuses [83]. While the results of the HLA-A2 status did not significantly influence outcomes, the HER2 expression in patients highly treated with adjuvant trastuzumab had a significantly better disease-free survival than patients with low HER2 expression [83].
E75 peptide (Nelipepimut-S) was evaluated in phase I/II clinical trial on node-positive and high-risk node-negative breast cancer patients with tumors expressing any degree of HER2 in the adjuvant setting. The five-year disease-free survival rate was 94.6% in optimally dosed patients (P = 0.05 versus the CG) and 87.1% in suboptimally dosed patients [84]. A phase III clinical trial tested the efficacy of Nelipepimut-S with GM-CSF in preventing breast cancer metastasis. The results showed that the treatment could not significantly decrease recurrence in HER2 low node-positive breast cancer patients in the adjuvant settings [85]. Synergistic effects have been observed when passive immunotherapy (monoclonal antibodies) is combined with active immunotherapy (cancer vaccines). Preclinical studies in murine models have shown that both cellular and humoral anti-neu immune responses are necessary to eliminate HER2/neu-expressing tumors [86-89]. This mechanism occurs through antibody-dependent cellular cytotoxicity mediated by natural killer cells, such that the antibody-induced cytotoxicity causes the tumor cells to lyse and release antibody-coated tumor antigens [90-92]. Further studies are required on HER2/neu vaccines to know their efficacy compared to trastuzumab.
MUC1
Due to their low antibody immune response, MUC1 peptide vaccines have been used in various combinatorial approaches. One such promising combination uses Bacille Calmette-Guérin (BCG) conjugated with the MUC1 antigen and human interleukin-2 (IL-2) vaccine. The preclinical study showed an increased CD8+ T cell response and inhibited breast cancer growth [73,93]. Further clinical trials are required to test its safety and efficacy in humans. Another trial targeted MUC1 using liposomal BLP25 (L-BLP25; a homolog of the protein backbone of MUC1) vaccination therapy, Tecemotide. The results of this study have been recently published and showed no significant increase in the residual cancer burden or pathologic complete response between patients taking Tecemotide and the control group [94].
Gene-based vaccine
Gene-based vaccines can be either (i) recombinant viral vectors modified to express tumor-associated antigens or (ii) bacterial plasmids constructed to function as a shuttle system that delivers and expresses a tumor antigen to help activate targeted cellular and humoral immunity [93,95].
Viral vaccines
Viral vaccines have been tested in breast cancer, such as the Modified Vaccinia Ankara (MVA) which consists of the Twist transgene and a TRIad of COstimulatory Molecules (B7-1, ICAM-1, LFA-3; TRICOM) (MVA-TWIST/TRICOM). This vaccine was shown to induce both CD8+ and CD4+ T cell responses against the transcription factor Twist, thereby reducing tumor growth and metastasis in a metastatic breast cancer model [96]. Viral vaccines, however, pose a clinical risk as they can induce strong immunity to the viral constructs and the production of neutralizing antibodies [97].
DNA vaccines
In breast cancer, DNA vaccines are combined with immunostimulatory molecules, such as toll-like receptors (TLR). The combination of DNA vaccines targeting HER2 with the agonist TLR9 showed potent anti-tumor activity and antibody-dependent cytotoxicity in mice [98]. Another DNA vaccine was designed by fusing the extracellular domain of CTLA-4 to HER-2/Neu to facilitate the detection of tumor antigens by APCs. This DNA vaccine-induced protective humoral and cellular immune responses, which delayed the onset of spontaneous Neu-driven mammary carcinomas and improved tumor-free survival of HER-2/Neu-driven mammary carcinoma in mice [99]. Similar to viral vaccines, DNA vaccines are not translated successfully in clinical settings due to efficacy rather than toxicity, and thus more improvements on the constructs and methods of administration are under investigation to enhance their utility in patients.
Whole-cell vaccine
Instead of using a specific antigen, whole tumor lysates were investigated for their immunogenicity. Whole-cell tumor vaccines, prepared by irradiating autogenic or allogenic cancer cells, increased cancer recognition by CTL and antigen-presenting cells [100]. Whole cell-based vaccines may thus induce a broader immune response given the broader load of antigenic components with a high yet rare risk of developing autoimmune diseases [101,102].
However, current research has shown that they are poorly immunogenic [103]. Modifications to these vaccines have been evaluated, such as modifying the irradiated cancer cells to secrete GM-CSF, in an attempt to have a better effect when combined with chemotherapy [104]. Other modifications included the addition of IL2, VEGFR2, and BCG in adjuvant settings to improve the immunogenicity and efficacy of whole-cell vaccines [105,106]. The whole-cell vaccine with BCG adjuvant showed improved survival in 60% of breast cancer patients after a 5-year follow-up [106]. The vaccination of a murine mammary cancer model with whole-cancer cells infected with irradiated adenovirus encoding VEGFR2 inhibited subsequent tumor growth, angiogenesis, and pulmonary metastasis. In addition, the number of CD8+ T lymphocytes was increased within the tumors of vaccinated mice [107]. This modified whole-cell lysate vaccine may be a potentially effective strategy for breast cancer treatment.
Dendritic cell-based vaccine
Dendritic cells (DC) are antigen-presenting cells that help in activating the anti-tumor immune response. DCs recognize the antigen on tumor cells, phagocytose and process the antigen, and present it on their cell surface to prime and activate cytotoxic T lymphocytes (CTL) [108]. DCs can be activated against tumor antigens in vitro, modified genetically using recombinant viral vectors, or fused with tumor cells using polyethylene glycol (PEG) or electrofusion to maintain antigen presence [109,110].
Lapuleucel-T vaccine is an example of in vitro activated DCs. It is derived from autologous peripheral blood mononuclear cells, including antigen-presenting cells overexpressing HER-2/neu-GM-CSF. This vaccine was tested in HER2+ metastatic patients and resulted in induced T cell response and tolerability [111].
Genetically modified DCs generated via recombinant adenoviral transduction of bone marrow-derived DCs to express a truncated HER2 protein were developed and tested in vivo. The vaccine increased the production of anti-HER2 antibodies, enhanced T cell response, and delayed the onset of mammary carcinomas in mice [110].
The fusion of DCs with tumor cells was tested in vitro and in vivo. This DC vaccine showed a high CTL response and resulted in the eradication of the tumor within 90 days [112]. However, these results were not further corroborated as other studies on PEG fusion-based DC vaccines were conducted on a mammary dog model using autologous and allogenic cancer cells and did not show an effective response as in the mouse model [113,114].
A more recent study sought to test the anti-tumor ability of DC administration in combination with chemotherapy in a mouse breast cancer model [115]. After adjusting the dosage of paclitaxel to minimize T cell inhibition, the combined treatment of paclitaxel and DC was found to induce antigen-specific CD8-mediated response in all 9 tested mice and CD4-mediated response in 6 out of the 9 treated mice. Furthermore, the group of mice treated with DC and paclitaxel had a significantly longer survival (45 days) compared to the untreated control group (29 days) and the tumor size was diminished compared to paclitaxel or DC administration alone. Therefore, the combination of paclitaxel and dendritic cells may be a potentially successful treatment in breast cancer [115].
Another clinical trial examined the effect of DC in combination with adoptive cell transfer, cytokine-induced killer T-cell therapy, and high dose chemotherapy on 166 metastatic breast cancer patients. This combination aims to eliminate the chemotherapeutic-resistant cancer stem cells to improve response to chemotherapy. The control group received a standard dose of chemotherapy (75 mg/m2 docetaxel and 75 mg/m2 thiotepa); while the treatment group received high-dose chemotherapy (120 mg/m2) in combination with DC and autologous cytokine-induced killer immunotherapy (DC/CIK) [116]. The results of the study showed that the treatment group had a significantly improved progression-free survival and overall survival compared to the control group [116]. Another study investigated the role of DC vaccines in targeting breast cancer stem cells in vitro and in vivo. DCs loaded with 4T1 tumor antigens improved CTL response against breast cancer stem cells in vitro and decreased tumor size in vivo [117]. More clinical trials including combination therapies with DC are listed in Table 1.
Current breast cancer immunotherapy trials
| Therapy | Condition | Trial | Trial ID | Phase |
|---|---|---|---|---|
| DC | Stage II and III | Chemotherapy followed by Autologous DC and surgery with/without radiation and/or hormone Therapy | NCT00499083 | II |
| Locally recurrent or metastatic | Vaccine therapy, trastuzumab, and vinorelbine | NCT00266110 | II | |
| TNBC and ER+/HER2- | Safety trial for chemotherapy and DC vaccine | NCT02018458 | I, II | |
| P53 overexpression and Stage III | Vaccine therapy + adjuvant/neoadjuvant chemotherapy + adjuvant radiation therapy | NCT00082641 | I, II | |
| Metastatic BC | Vaccine therapy + 1-MT | NCT01042535 | I, II | |
| Stage IV | DC/tumor fusion + IL12 | NCT00622401 | I, II | |
| HER-2 driven invasive breast cancer at least Stage IIIA | HER-2 pulsed Dendritic Cell Vaccine | NCT02063724 | I | |
| Locally recurrent/metastatic BC | Vaccine therapy + with trastuzumab + vinorelbine | NCT00088985 | II | |
| Adoptive T-cell therapy | Stage IV | Ex vivo-expanded HER2-specific T cells | NCT00791037 | I, II |
| Anti-PD-1/PD-L1 | Advanced, trastuzumab-resistant, HER2+ BC | Ant-PD-1 (Pembrolizumab; MK-3475) | NCT02129556 | I, II |
| Metastatic TNBC | Pembrolizumab + radiotherapy | NCT02730130 | II | |
| TNBC | Pembrolizumab (MK-3475) + chemotherapy as neoadjuvant treatment | NCT02622074 | I | |
| Advanced/metastatic TNBC and OC | Niraparib + pembrolizumab | NCT02657889 | I, II | |
| HER2 overexpressing metastatic BC | Pembrolizumab + monoclonal antibody therapy | NCT02318901 | ||
| TNBC | Pembrolizumab + epacadostat | NCT02178722 | I, II | |
| Trastuzumab emtansine + atezolizumab/atezolizumab-placebo | NCT02924883 | II | ||
| Metastatic TNBC | Cobimetinib (MEK inhibitor) + paclitaxel, Cobimetinib + atezolizumab Plus Paclitaxel, or Cobimetinib + atezolizumab + Nab-Paclitaxel | NCT02322814 | II | |
| Relapsed/refractory BC | Ibrutinib + durvalumab (MEDI4736) | NCT02403271 | I, II | |
| Stage IV HER2- BC | Ipilimumab and Nivolumab | NCT02892734 | II | |
| Metastatic TNBC | SYK inhibitor (TAK-659) + nivolumab | NCT02834247 | I | |
| TNBC | Anti-PDL1 (MEDI4736) monotherapy or MEDI4736 + tremelimumab (anti-CTLA4) | NCT02527434 | II | |
| Anti-CTLA4 | Recurrent Stage IV HER2- BC | Ipilimumab and Nivolumab | NCT02892734 | II |
BC: Breast cancer; 1MT: 1-methyl-d-tryptophan; OC: Ovarian cancer; TNBC: triple-negative breast cancer; HER2: Human epidermal growth factor receptor 2; ER: estrogen receptor, IL: interleukin.
Checkpoint inhibitors
Anti-PD1/PDL1 inhibitors
Expressed on T-cells, B cells, and NK cells, programmed cell death 1 (PD-1) is a checkpoint that keeps the immune response in check to prevent autoimmune diseases and immune overactivation [118,119]. The inhibition of PD-1 occurs when it binds to its ligands, PD-L1 and PD-L2, which are commonly expressed on myeloid and tumor cells [119-121]. A meta-analysis of five studies (2,546 patients) has shown that PD-L1 positive expression in breast cancer ranges between 21.7% to 56.6% [122]. The expression levels of PD-1 and its ligand, PD-L1, vary depending on the subtype of breast cancer. For example, basal-like breast cancer has the highest overall PD-1 expression on Tumor-Infiltrating Lymphocytes (TILs) (27.4%) while the luminal A subtype has the lowest overall expression (4.7%). Furthermore, PD-L1 expression has been associated with positive lymph node metastasis, higher histological grade, larger tumor size, triple-negative subtype, and HER2 positivity [123].
Drugs targeting the PD-1/PD-L1/PD-L2 signaling pathway block the interaction between PD-1 and its ligands by targeting either PD-1 (Pembrolizumab and Nivolumab) or PD-L1 (Atezolizumab, Avelumab, and Durvalumab) (Figure 2B). While it is thought that patients with a high PD-1 expression would respond well to anti-PD-1 treatments, the response is highly variable among patients. This is mainly due to the lack of well-defined cutoff criteria, technical discrepancies, and T cell infiltration to the tumor [124,125]. Furthermore, a variety of genetic aberrations can cause a constitutive PD-L1 expression, which may arbitrarily induce PD-L1 positivity regardless of the presence or absence of T-cell infiltration. Recently, we have identified a list of biomarkers that classify the subsets of breast cancer patients that would respond best to checkpoint inhibitors or other combinations of immunotherapies [126].
Several studies have shown that PD-1/PD-L1 signaling antagonists induce clinical response durability in some metastatic TNBC patients. This has led to the FDA approval of the first immunotherapy in breast cancer, anti-PD-L1 (atezolizumab) in combination with chemotherapy (nab-paclitaxel) for metastatic TNBC [127,128]. The phase III trial showed prolonged progression-free survival from 5.5 months in the placebo group plus nab-paclitaxel to about 7.2 months for the atezolizumab plus nab-paclitaxel combination group. The PD-L1 positive subgroup also had a prolonged progression-free survival of 7.5 months in the combination group compared to 5 months in the control group [127]. However, several questions have been raised including how to enrich the responsive TNBC population, how to assess PD-L1 positivity and thus patients' response to atezolizumab, whether PD-L1 expression should be tested in the tumor or immune cells, if nab-paclitaxel is the best chemotherapy to use and if atezolizumab monotherapy could have been beneficial for a certain subset of patients without chemotherapy [129]. Several studies have shown the relevance of these questions. We have classified the TNBC patients into 2 clusters, one that would benefit from chemotherapy and the other would benefit from anti-PD-L1 treatment in combination with other immunotherapeutic drugs, such as anti-CTLA4 [126,130]. A concise review on how to enrich the TNBC population is addressed by Marra et al. [129].
Multiple completed trials have tested the effects of PD-1 inhibitors as monotherapies in breast cancer patients. The Keynote-086 phase II trial evaluated pembrolizumab as first- or later-line treatment for PDL-1 positive patients with metastatic TNBC untreated or previously treated with chemotherapy [131,132]. In the previously treated cohort, the overall response rate (5.3%) was lower than single-agent chemotherapy. However, pembrolizumab abolished the common toxicities associated with chemotherapy and resulted in durable responses. In addition, 75.0% and 62.5% of responders had a response duration of ≥6 and ≥12 months, respectively compared to the typical duration of response (1-3 months) seen in standard chemotherapy in the metastatic TNBC setting. Thus, pembrolizumab showed a durable effect in patients who achieved a response [132]. In the previously untreated cohort, pembrolizumab monotherapy demonstrated an acceptable safety profile despite the common adverse effects occurring in 63% of patients. The antitumor activity was robust and durable with a median time to response of 2.0 months, a median duration of response of 10.4 months, median overall survival of 18 months, and an overall survival rate of 48.7% at 18 months. Thus, pembrolizumab demonstrated antitumor durability as a first line of treatment.
One combinatorial approach involved the blockade of PD-1/PD-L1 signaling together with CTLA4. A pilot study evaluated durvalumab (anti-PD-1 drug) and tremelimumab (CTLA4 blocking drug) in refractory metastatic breast cancer patients [133]. The overall response rate was 17% which was mostly seen in TNBC patients (43%) with no response to treatment detected in the ER-positive patients. This study also found that TNBC patients who did respond to treatments had a higher non-synonymous mutation load along with a higher prevalence of neoantigens; both of which increased the number of activated T cells [133-135]. Despite the limitations attributed to this study, it provides important observations to plan future trials. Other ongoing clinical trials are summarized in Table 1.
Anti-CTLA-4 inhibitors
Cytotoxic T lymphocyte antigen 4 (CTLA4) is an immune checkpoint molecule typically expressed on the surface of regulatory T-cells and cytotoxic T cells shortly after activation [121,136]. CTLA4 limits T-cell activation by interacting with its two ligands CD80 and CD86 (Figure 2C) [137]. A recent study found that high CTLA4 expression is associated with poor prognosis in breast cancer [136].
Only a few studies investigated the effects of CTLA4 blockade on breast cancer, most of which combined CTLA4 blocking drugs with other treatments such as aromatase inhibitors, radiotherapy, and chemotherapy.
Anti-CTLA4 + aromatase inhibitor
The effect of tremelimumab (anti-CTLA-4 antibody) in combination with exemestane - a steroidal aromatase inhibitor - was evaluated in breast cancer [2]. The study was done on postmenopausal women with life expectancies greater than six months who had estrogen receptor-positive breast cancer except for one [137]. Forty-two percent of the patients developed a stable disease; however, none of the patients experienced partial or objective responses. It should be noted that 36% of the patients who developed a stable disease had previously experienced tumor progression while on exemestane [137].
Anti-CTLA4 + radiotherapy
Anti-CTLA4 was tested in combination with single-dose and fractionated radiotherapy in a mouse breast cancer model to determine if it can produce what is known as an “abscopal effect” on secondary and primary tumors. The abscopal effect is tumor regression that occurs outside of the field of radiation [138]. It was found that CTLA4 blockade had no effect on its own but caused tumor regression when combined with radiotherapy. However, the abscopal effect to the secondary tumor only occurred when radiotherapy doses were fragmented and not given in single-dose form [138]. Furthermore, the higher radiation regimen (8Gy) showed a more efficient abscopal effect than a lower dose (6Gy) regimen [138].
Anti-CTLA4 + chemotherapy
Anti-CTLA-4 combination with metronomic chemotherapy has also been tested in breast cancer [139]. Metronomic chemotherapy is a type of chemotherapy that involves the administration of a chemotherapeutic drug in relatively low doses over a long period to prevent the tumor from becoming drug-resistant [140]. This combination was investigated in preclinical settings in breast cancer and metronomic cyclophosphamide was shown to enhance the outcome of anti-CTLA4 therapy. CTLA4 antibody administration alone decreased tumor size in mice but resulted in tumor relapse [139]. More impressive results were seen using a sequential regimen of CTLA4 blockade followed by metronomic gemcitabine chemotherapy [139,141]. Despite these results, resistance was still observed as well as spontaneous metastases.
Conclusion
Several advances have been made to increase breast cancer patient's survival and mitigate tumor growth. Approvals have been granted for several targeted therapies and combination therapies and 2019 highlights the first approved immunotherapy in combination with chemotherapy for metastatic TNBC. More improvements, however, can be made by taking a more personalized approach to tackle the intra- and inter-tumor heterogeneities, and by identifying better biomarkers to make the personalization more clinically feasible. Thus, more basic, pre-clinical, translational, and clinical investigations are required to enhance patients' response by further categorizing patients based on their tumor phenotypic and genotypic characteristics.
Abbreviations
APC: antigen-presenting cell; BCG: Bacille Calmette-Guérin; BRCA: Breast cancer; CAR: Chimeric antigen receptor; CD: Cluster of differentiation; CDK: Cyclin-dependent kinase; CIK: Cytokine-induced killer; c-Met: tyrosine-protein kinase Met or hepatocyte growth factor receptor (HGFR); CTL: Cytotoxic T-lymphocyte; CTLA4: Cytotoxic T-lymphocyte-associated protein 4; DC: Dendritic cell; DSB: Double-stranded break; E2F: E2 transcription factor; EGFR: Epidermal Growth Factor Receptor; ER: Estrogen receptor; ErbB2: Human epidermal growth factor receptor 2 (HER2); FDA: Food and drug administration; FRα: Folate receptor-alpha; GM-CSF: Granulocyte monocyte colony-stimulating factor; HER2: Human epidermal growth factor receptor 2; HERV-K: Human endogenous retrovirus-K; HLA: Human leukocyte antigen; HML-2: Human endogenous retrovirus-K; HR: Hormone receptor; IL2: Interleukin 2; MUC1: Mucin-1; MVA: Modified vaccinia Ankara; Nab: Nanoparticle albumin-bound; NKG2D: Natural killer group 2 member D; PARP: Poly(ADP-ribose) polymerase; PD1: Programmed death 1; PDL1: Programmed death-ligand 1; PEG: Polyethylene glycol; PR: Progesterone receptor; RB: Retinoblastoma; ScFV: Single-chain variable fragment; SSB: Single-stranded break; TCR: T cell receptor; TLR: Toll-like receptor; TNBC: Triple-negative breast cancer; VEGFR2: Vascular endothelial growth factor receptor 2.
Supplementary Material
Supplementary data.
Acknowledgements
The authors wish to thank Gail Lopez and Jasmine Colangelo for their help with literature search on some of the topics covered in this paper.
Funding information
This work was supported by the Florida State University Council on Research & Creativity grants, the Pfeiffer Professorship for Cancer Research in Chemistry and Biochemistry, and an Endowed Chair Professorship in Cancer Research from anonymous donors (to QXS), and a Lebanese grant (to ZIK).
Competing Interests
The authors have declared that no competing interest exists.
References
1. U.S. Cancer Statistics Working Group. U.S. Cancer Statistics Data Visualizations Tool, based on 2019 submission data (1999-2017). US Dep Heal Hum Serv Centers Dis Control Prev Natl Cancer Inst. 2017 https://gis.cdc.gov/Cancer/USCS/DataViz.html
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7-33
3. Turashvili G, Brogi E. Tumor heterogeneity in breast cancer. Front Med. 2017;4:227
4. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA - J Am Med Assoc. 2019;321:288-300
5. Rakha EA, Reis-Filho JS, Baehner F. et al. Breast cancer prognostic classification in the molecular era: The role of histological grade. Breast Cancer Res. 2010;12:1-12
6. Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403-410
7. El Sayed R, El Jamal L, El Iskandarani S, Kort J, Abdel Salam M, Assi H. Endocrine and Targeted Therapy for Hormone-Receptor-Positive, HER2-Negative Advanced Breast Cancer: Insights to Sequencing Treatment and Overcoming Resistance Based on Clinical Trials. Front Oncol. 2019;9:510
8. Rivenbark AG, O'Connor SM, Coleman WB. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am J Pathol. 2013;183:1113-1124
9. Baselga J, Carbonell X, Castañeda-Soto NJ. et al. Phase II study of efficacy, safety, and pharmacokinetics of trastuzumab monotherapy administered on a 3-weekly schedule. J Clin Oncol. 2005;23:2162-2171
10. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6:1609-1623
11. Tomao F, Papa A, Zaccarelli E. et al. Triple-negative breast cancer: New perspectives for targeted therapies. Onco Targets Ther. 2015;8:177-193
12. Ruddy KJ, Ganz PA. Treatment of Nonmetastatic Breast Cancer. JAMA - J Am Med Assoc. 2019;320:1716-1717
13. Miller KD, Nogueira L, Mariotto AB. et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363-385
14. Bernsdorf M, Ingvar C, Jörgensen L. et al. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res Treat. 2011;126:463-470
15. Zimmer AS, Gillard M, Lipkowitz S, Lee JM. Update on PARP Inhibitors in Breast Cancer. Curr Treat Options Oncol. 2018;19:21
16. D'Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018;71:172-176
17. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152-1158
18. Exman P, Barroso-Sousa R, Tolaney SM. Evidence to Date: Talazoparib in the treatment of breast cancer. Onco Targets Ther. 2019;12:5177-5187
19. Turk AA, Wisinski KB. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer. 2018;124:2498-2506
20. Caulfield SE, Davis CC, Byers KF. Olaparib: A Novel Therapy for Metastatic Breast Cancer in Patients With a BRCA1/2 Mutation. J Adv Pract Oncol. 2019;10:167-174
21. Nur Husna SM, Tan HT, Mohamud R, Dyhl-Polk A, Wong KK. Inhibitors targeting CDK4/6, PARP and PI3K in breast cancer: a review. Ther Adv Med Oncol. 2018;10:1758835918808509
22. Saleh L, Wilson C, Holen I. CDK4/6 inhibitors in breast cancer - from in vitro models to clinical trials. Acta Oncol. 2020;59:219-232
23. Bunney PE, Zink AN, Holm AA, Billington CJ, Kotz CM. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol Behav. 2017;176:139-148
24. Vasiliki TC, Aristeidis GT, Christos AK, Timotheadou ET. The Pharmacological Profile of Cyclin-dependent Kinase (CDK) 4/6 Inhibitors: Clinical Management of Toxicity and Drug Interactions Related to CDK 4/6 Inhibitor-based Treatment in Advanced/Metastatic Breast Cancer. Forum Clin Oncol. 2019;10:2-14
25. Wedam S, Fashoyin-Aje L, Bloomquist E. et al. FDA approval summary: Palbociclib for Male patients with metastatic breast cancer. Clin Cancer Res. 2020;26:1208-1212
26. Mohan N, Jiang J, Dokmanovic M, Wu WJ. Trastuzumab-mediated cardiotoxicity: Current understanding, challenges, and frontiers. Antib Ther. 2018;1:13-17
27. Huszno J, Leś D, Sarzyczny-Słota D, Nowara E. Cardiac side effects of trastuzumab in breast cancer patients - Single centere experiences. Wspolczesna Onkol. 2013;17:190-195
28. Ryberg M, Nielsen D, Cortese G, Nielsen G, Skovsgaard T, Andersen PK. New insight into epirubicin cardiac toxicity: Competing risks analysis of 1097 breast cancer patients. J Natl Cancer Inst. 2008;100:1058-1067
29. Papakonstantinou A, Matikas A, Bengtsson NO. et al. Efficacy and safety of tailored and dose-dense adjuvant chemotherapy and trastuzumab for resected HER2-positive breast cancer: Results from the phase 3 PANTHER trial. Cancer. 2020;126:1175-1182
30. Swain SM, Baselga J, Kim S-B. et al. Pertuzumab, Trastuzumab, and Docetaxel in HER2-Positive Metastatic Breast Cancer. N Engl J Med. 2015;372:724-734
31. Swain SM, Miles D, Kim SB. et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21:519-530
32. Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M. The application of CAR-T cell therapy in hematological malignancies: advantages and challenges. Acta Pharm Sin B. 2018;8:539-551
33. Bajgain P, Tawinwung S, D'Elia L. et al. CAR T cell therapy for breast cancer: Harnessing the tumor milieu to drive T cell activation. J Immunother Cancer. 2018;6:1-13
34. Elahi R, Khosh E, Tahmasebi S, Esmaeilzadeh A. Immune Cell Hacking: Challenges and Clinical Approaches to Create Smarter Generations of Chimeric Antigen Receptor T Cells. Front Immunol. 2018;9:1717
35. Petersen CT, Krenciute G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front Oncol. 2019;9:69
36. Guedan S, Calderon H, Posey AD, Maus M V. Engineering and Design of Chimeric Antigen Receptors. Mol Ther - Methods Clin Dev. 2019;12:145-156
37. Wei J, Sun H, Zhang A. et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol. 2018;331:49-58
38. Tchou J, Zhao Y, Levine BL. et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017;5:1152-1161
39. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55-61
40. Roberti MP, Mordoh J, Levy EM. Biological role of NK cells and immunotherapeutic approaches in breast cancer. Front Immunol. 2012;3:375
41. Demoulin B, Cook WJ, Murad J. et al. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Futur Oncol. 2017;13:1593-1605
42. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287-6297
43. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544-1551
44. Han Y, Xie W, Song DG, Powell DJ. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol. 2018;11:92
45. Lonez C, Verma B, Hendlisz A. et al. Study protocol for THINK: A multinational open-label phase i study to assess the safety and clinical activity of multiple administrations of NKR-2 in patients with different metastatic tumour types. BMJ Open. 2017;7:e017075
46. Munisvaradass R, Kumar S, Govindasamy C, Alnumair KS, Mok PL. Human CD3+ T-cells with the anti-ERBB2 chimeric antigen receptor exhibit efficient targeting and induce apoptosis in ERBB2 overexpressing breast cancer cells. Int J Mol Sci. 2017;18:1797
47. Zhao Y, Wang QJ, Yang S. et al. A Herceptin-Based Chimeric Antigen Receptor with Modified Signaling Domains Leads to Enhanced Survival of Transduced T Lymphocytes and Antitumor Activity. J Immunol. 2009;183:5563-5574
48. Bernhard H, Neudorfer J, Gebhard K. et al. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother. 2008;57:271-280
49. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843-851
50. Bannert N, Kurth R. Retroelements and the human genome: New perspectives on an old relation. Proc Natl Acad Sci U S A. 2004;101:14572-14579
51. Grandi N, Tramontano E. Human endogenous retroviruses are ancient acquired elements still shaping innate immune responses. Front Immunol. 2018;9:2039
52. Hohn O, Hanke K, Bannert N. HERV-K(HML-2), the Best Preserved Family of HERVs: Endogenization, Expression, and Implications in Health and Disease. Front Oncol. 2013;3:246
53. Misiak B, Ricceri L, Sasiadek MM. Transposable elements and their epigenetic regulation in mental disorders: Current evidence in the field. Front Genet. 2019;10:580
54. Hurst TP, Magiorkinis G. Epigenetic control of human endogenous retrovirus expression: Focus on regulation of long-terminal repeats (LTRs). Viruses. 2017;9:130
55. Tavakolian S, Goudarzi H, Faghihloo E. Evaluating the expression level of HERV-K env, np9, rec and gag in breast tissue. Infect Agent Cancer. 2019;14:1-5
56. Curty G, Marston JL, De Mulder Rougvie M, Leal FE, Nixon DF, Soares MA. Human Endogenous Retrovirus K in Cancer: A Potential Biomarker and Immunotherapeutic Target. Viruses. 2020;12:726
57. Zhao J, Rycaj K, Geng S. et al. Expression of Human Endogenous Retrovirus Type K Envelope Protein is a Novel Candidate Prognostic Marker for Human Breast Cancer. Genes and Cancer. 2011;2:914-922
58. Zhou F, Krishnamurthy J, Wei Y. et al. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology. 2015;4:e1047582
59. Contreras-Galindo R, Kaplan MH, Leissner P. et al. Human Endogenous Retrovirus K (HML-2) Elements in the Plasma of People with Lymphoma and Breast Cancer. J Virol. 2008;82:9329-9336
60. Zhou F, Li M, Wei Y. et al. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget. 2016;7:84093-84117
61. Kelley KMM, Rowan BG, Ratnam M. Modulation of the folate receptor α gene by the estrogen receptor: Mechanism and implications in tumor targeting. Cancer Res. 2003;63:2820-2828
62. Rochman H, Selhub J, Karrison T. Folate binding protein and the estrogen receptor in breast cancer. Cancer Detect Prev. 1985;8:71-75
63. Zhang Z, Wang J, Tacha DE. et al. Folate receptor α associated with triple-negative breast cancer and poor prognosis. Arch Pathol Lab Med. 2014;138:890-895
64. Hartmann LC, Keeney GL, Lingle WL. et al. Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer. 2007;121:938-942
65. O'Shannessy DJ, Somers EB, Maltzman J, Smale R, Fu YS. Folate receptor alpha (FRA) expression in breast cancer: identification of a new molecular subtype and association with triple negative disease. Springerplus. 2012;1:22
66. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol. 2016;9:56
67. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119:696-706
68. Norton N, Youssef B, Hillman DW. et al. Folate receptor alpha expression associates with improved disease-free survival in triple negative breast cancer patients. NPJ Breast Cancer. 2020;6:4
69. Wilkie S, Van Schalkwyk MCI, Hobbs S. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059-1070
70. Weber J. Peptide vaccines for cancer. Cancer Invest. 2002;20:208-221
71. Lazoura E, Apostolopoulos V. Rational Peptide-Based Vaccine Design for Cancer Immunotherapeutic Applications. Curr Med Chem. 2010;12:629-639
72. Kaumaya PTP, Foy KC. Peptide vaccines and targeting HER and VEGF proteins may offer a potentially new paradigm in cancer immunotherapy. Futur Oncol. 2012;8:961-987
73. Chung MA, Luo Y, O'Donnell M. et al. Development and preclinical evaluation of a Bacillus Calmette-Guérin-MUC1-based novel breast cancer vaccine. Cancer Res. 2003;63:1280-1287
74. Tuttle TM, Anderson BW, Thompson WE. et al. Proliferative and cytokine responses to class II HER-2/neu associated peptides in breast cancer patients. Clin Cancer Res. 1998;4:2015-2024
75. Clifton GT, Gall V, Peoples GE, Mittendorf EA. Clinical Development of the E75 Vaccine in Breast Cancer. Breast Care. 2016;11:116-121
76. Li Y, Ishiyama S, Matsueda S, Tsuda N, Ioannides CG. HER-2 peptides p776 and F7, N-terminal-linked with Ii-Key tetramer (LRMK) help the proliferation of E75-TCR+ cells: The dependency of help on the side chains of LRMK-extended peptide pointed towards the T cell receptor. Oncol Rep. 2008;19:1445-1452
77. Razazan A, Behravan J, Arab A. et al. Conjugated nanoliposome with the HER2/ neu-derived peptide GP2 as an effective vaccine against breast cancer in mice xenograft model. Pappalardo F, ed. PLoS One. 2017;12:e0185099
78. Holmes JP, Benavides LC, Gates JD. et al. Results of the first phase I clinical trial of the novel Ii-key hybrid preventive HER-2/neu peptide (AE37) vaccine. J Clin Oncol. 2008;26:3426-3433
79. Sears AK, Perez SA, Clifton GT. et al. AE37: A novel T-cell-eliciting vaccine for breast cancer. Expert Opin Biol Ther. 2011;11:1543-1550
80. Benavides LC, Sears AK, Gates JD. et al. Comparison of different HER2/neu vaccines in adjuvant breast cancer trials: Implications for dosing of peptide vaccines. Expert Rev Vaccines. 2011;10:201-210
81. Mittendorf EA, Ardavanis A, Symanowski J. et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann Oncol. 2016;27:1241-1248
82. Mittendorf EA, Ardavanis A, Litton JK. et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide GP2 vaccine in breast cancer patients to prevent recurrence. Oncotarget. 2016;7:66192-66201
83. Jackson Doreen O, Francois TA, Travis CG. et al. Effects of HLA status and HER2 status on outcomes in breast cancer patients at risk for recurrence - Implications for vaccine trial design. Clin Immunol. 2018;195:28-35
84. Mittendorf EA, Clifton GT, Holmes JP. et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25:1735-1742
85. Mittendorf EA, Lu B, Melisko M. et al. Efficacy and safety analysis of nelipepimut-S vaccine to prevent breast cancer recurrence: A randomized, multicenter, phase III clinical trial. Clin Cancer Res. 2019;25:4248-4254
86. Wolpoe ME, Lutz ER, Ercolini AM. et al. HER-2/neu-Specific Monoclonal Antibodies Collaborate with HER-2/neu-Targeted Granulocyte Macrophage Colony-Stimulating Factor Secreting Whole Cell Vaccination to Augment CD8 + T Cell Effector Function and Tumor-Free Survival in Her-2/ neu -Transgenic Mice. J Immunol. 2003;171:2161-2169
87. Reilly RT, Machiels JPH, Emens LA. et al. The collaboration of both humoral and cellular HER-2/neu-targeted immune responses is required for the complete eradication of HER-2/neu-expressing tumors. Cancer Res. 2001;61:880-883
88. Reilly RT, Gottlieb MBC, Ercolini AM. et al. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569-3576
89. Park SG, Jiang Z, Mortenson ED. et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010;18:160-170
90. Arab A, Yazdian-Robati R, Behravan J. HER2-Positive Breast Cancer Immunotherapy: A Focus on Vaccine Development. Arch Immunol Ther Exp (Warsz). 2020;68:2
91. Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: Clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28:4390-4399
92. Clifton GT, Peoples GE, Mittendorf EA. The development and use of the E75 (HER2 369-377) peptide vaccine. Futur Oncol. 2016;12:1321-1329
93. Allahverdiyev A, Tari G, Bagirova M, Abamo ES. Current approaches in development of immunotherapeutic vaccines for breast cancer. J Breast Cancer. 2018;21:343-353
94. Singer CF, Pfeiler G, Hubalek M. et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur J Cancer. 2020;132:43-52
95. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines. Past, present, and future. In: Advances in Cancer Research. Vol 119. Academic Press Inc. 2013:421-475
96. Kwilas AR, Ardiani A, Dirmeier U, Wottawah C, Schlom J, Hodge JW. A poxviral-based cancer vaccine targeting the transcription factor twist inhibits primary tumor growth and metastases in a model of metastatic breast cancer and improves survival in a spontaneous prostate cancer model. Oncotarget. 2015;6:28194-28210
97. Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17:359-371
98. Aurisicchio L, Peruzzi D, Conforti A. et al. Treatment of mammary carcinomas in HER-2 transgenic mice through combination of genetic vaccine and an agonist of toll-like receptor 9. Clin Cancer Res. 2009;15:1575-1584
99. Sloots A, Mastini C, Rohrbach F. et al. DNA vaccines targeting tumor antigens to B7 molecules on antigen-presenting cells induce protective antitumor immunity and delay onset of HER-2/Neu-driven mammary carcinoma. Clin Cancer Res. 2008;14:6933-6943
100. Sharma A, Bode B, Wenger RH, Lehmann K, Sartori AA, Moch H, Knuth A, Boehmer Lv, Broek Mv. γ-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS One. 2011;6:e28217
101. Srivatsan S, Patel JM, Bozeman EN. et al. Allogeneic tumor cell vaccines: The promise and limitations in clinical trials. Hum Vaccines Immunother. 2014;10:52-63
102. Dols A, Meijer SL, Smith JW 2nd, Fox BA, Urba WJ. Allogeneic breast cancer cell vaccines. Clin Breast Cancer. 2003;3(Suppl 4):S173-180
103. Srivatsan S, Patel JM, Bozeman EN. et al. Allogeneic tumor cell vaccines: The promise and limitations in clinical trials. Hum Vaccines Immunother. 2014;10:52-63
104. Simons JW, Sacks N. Granulocyte-macrophage colony-stimulating factor-transduced allogeneic cancer cellular immunotherapy: the GVAX vaccine for prostate cancer. Urol Oncol. 2006;24:419-424
105. Huang X, Ye D, Thorpe PE. Enhancing the potency of a whole-cell breast cancer vaccine in mice with an antibody-IL-2 immunocytokine that targets exposed phosphatidylserine. Vaccine. 2011;29:4785-4793
106. Convit J, Montesinos H, Oviedo H. et al. Autologous tumor lysate/Bacillus Calmette-Guérin immunotherapy as an adjuvant to conventional breast cancer therapy. Clin Transl Oncol. 2015;17:884-887
107. Yan HX, Cheng P, Wei HY. et al. Active immunotherapy for mouse breast cancer with irradiated whole-cell vaccine expressing VEGFR2. Oncol Rep. 2013;29:1510-1516
108. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. 2013;39:1-10
109. Gelao L, Criscitiello C, Esposito A. et al. Dendritic cell-based vaccines: Clinical applications in breast cancer. Immunotherapy. 2014;6:349-360
110. Sakai Y, Morrison BJ, Burke JD. et al. Vaccination by genetically modified dendritic cells expressing a truncated neu oncogene prevents development of breast cancer in transgenic mice. Cancer Res. 2004;64:8022-8028
111. Peethambaram PP, Melisko ME, Rinn KJ. et al. A phase I trial of immunotherapy with lapuleucel-T (APC8024) in patients with refractory metastatic tumors that express HER-2/neu. Clin Cancer Res. 2009;15:5937-5944
112. Koido S, Tanaka Y, Tajiri H, Gong J. Generation and functional assessment of antigen-specific T cells stimulated by fusions of dendritic cells and allogeneic breast cancer cells. Vaccine. 2007;25:2610-2619
113. Bird RC, DeInnocentes P, Church Bird AE, Van Ginkel FW, Lindquist J, Smith BF. An autologous dendritic cell canine mammary tumor hybrid-cell fusion vaccine. Cancer Immunol Immunother. 2011;60:87-97
114. Bird RC, DeInnocentes P, Lenz S, Thacker EE, Curiel DT, Smith BF. An allogeneic hybrid-cell fusion vaccine against canine mammary cancer. Vet Immunol Immunopathol. 2008;123:289-304
115. Bin Y, Kusmartsev S, Fengdong C. et al. Effective combination of chemotherapy and dendritic cell administration for the treatment of advanced-stage experimental breast cancer. Clin Cancer Res. 2003;9:285-294
116. Ren J, Di L, Song G. et al. Selections of appropriate regimen of high-dose chemotherapy combined with adoptive cellular therapy with dendritic and cytokine-induced killer cells improved progression-free and overall survival in patients with metastatic breast cancer: Reargument of such contentious therapeutic preferences. Clin Transl Oncol. 2013;15:780-788
117. Nguyen ST, Nguyen HL, Pham VQ. et al. Targeting specificity of dendritic cells on breast cancer stem cells: In vitro and in vivo evaluations. Onco Targets Ther. 2015;8:323-334
118. Nanda R, Chow LQM, Dees EC. et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib keynote-012 study. J Clin Oncol. 2016;34:2460-2467
119. Yu LY, Li MP, Kuang D Bin, Zhang CM, Chen XP. New immunotherapy strategies in breast cancer. Chinese Pharmacol Bull. 2016;32:1037-1040
120. Ansell SM, Lesokhin AM, Borrello I. et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin's Lymphoma. N Engl J Med. 2015;372:311-319
121. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264
122. Zhang M, Sun H, Zhao S, Wang Y, Pu H, Zhang Q. Expression of PD-L1 and prognosis in breast cancer: A metaanalysis. Oncotarget. 2017;8:31347-31354
123. Kim HM, Lee J, Koo JS. Clinicopathological and prognostic significance of programmed death ligand-1 expression in breast cancer: A meta-analysis. BMC Cancer. 2017;17:690
124. Grigg C, Rizvi NA. PD-L1 biomarker testing for non-small cell lung cancer: Truth or fiction? J Immunother Cancer. 2016;4:48
125. Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med. 2016;213:2835-2840
126. Bou-Dargham MJ, Liu Y, Sang QXA, Zhang J. Subgrouping breast cancer patients based on immune evasion mechanisms unravels a high involvement of transforming growth factor-beta and decoy receptor 3. PLoS One. 2018;13:e0207799
127. Schmid P, Adams S, Rugo HS. et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379:2108-2121
128. FDA Approves Atezolizumab for Triple-Negative Breast Cancer - National Cancer Institute. https://www.cancer.gov/news-events/cancer-currents-blog/2019/atezolizumab-triple-negative-breast-cancer-fda-approval
129. Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019;17:90
130. Kagihara JA, Andress M, Diamond JR. Nab-paclitaxel and atezolizumab for the treatment of PD-L1-positive, metastatic triple-negative breast cancer: review and future directions. Expert Rev Precis Med Drug Dev. 2020;5:59-65
131. Adams S, Loi S, Toppmeyer D. et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:405-411
132. Adams S, Schmid P, Rugo HS. et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:397-404
133. Santa-Maria CA, Kato T, Park JH. et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget. 2018;9:18985-18996
134. Tumeh PC, Harview CL, Yearley JH. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-571
135. Le DT, Durham JN, Smith KN. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-413
136. Yu H, Yang J, Jiao S, Li Y, Zhang W, Wang J. Cytotoxic T lymphocyte antigen 4 expression in human breast cancer: implications for prognosis. Cancer Immunol Immunother. 2015;64:853-860
137. Vonderheide RH, Lorusso PM, Khalil M. et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010;16:3485-3494
138. Dewan MZ, Galloway AE, Kawashima N. et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379-5388
139. Parra K, Valenzuela P, Lerma N. et al. Impact of CTLA-4 blockade in conjunction with metronomic chemotherapy on preclinical breast cancer growth. Br J Cancer. 2017;116:324-334
140. Banys-Paluchowski M, Schtz F, Ruckhberle E, Krawczyk N, Fehm T. Metronomic Chemotherapy for Metastatic Breast Cancer a Systematic Review of the Literature. Geburtshilfe Frauenheilkd. 2016;76:525-534
141. Francia G, Shaked Y, Hashimoto K. et al. Low-dose metronomic oral dosing of a prodrug of gemcitabine (LY2334737) causes antitumor effects in the absence of inhibition of systemic vasculogenesis. Mol Cancer Ther. 2012;11:680-689
Author contact
![]() Corresponding authors: E-mail: qxsangfsu.edu (QXS) and zahraakhamiscom (ZIK).
Corresponding authors: E-mail: qxsangfsu.edu (QXS) and zahraakhamiscom (ZIK).