Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(21):6553-6562. doi:10.7150/jca.61996 This issue Cite
Research Paper
The E3 Ubiquitin Ligase HOIP inhibits Cancer Cell Apoptosis via modulating PTEN stability
Zhiguo Niu1,2*, Xin Li2*, Shuxiao Dong3*, Jianhui Gao2, Qingsong Huang2, Huijie Yang2, Hui Qian1, Shu Zhuo4, Ting Zhuang2, Jian Zhu2,5 ![]() , Yinlu Ding5
, Yinlu Ding5 ![]() , Wenrong Xu1
, Wenrong Xu1 ![]()
1. Jiangsu Key Laboratory of Medical Science and Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, 212000, China.
2. Henan Key Laboratory of Immunology and Targeted Drugs, School of Laboratory Medicine, Xinxiang Medical University, 453000, China.
3. Department of Gastroenterology surgery, Shandong Provincial Third Hospital, Jinan, 250000, China.
4. Signet Therapeutics Inc, Shenzhen, China. Research Institute of Tsinghua University in Shenzhen, Shenzhen, 518000, China.
5. Department of general surgery, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250033, China.
*These authors contributed equally to this work.
Received 2021-4-24; Accepted 2021-8-30; Published 2021-9-9
Abstract

Chemotherapy is widely used in a variety of solid tumors, such as lung cancer, gastric cancer and breast cancer. The genotoxic drugs, such as cisplatin, suppress cancer progression either by inhibition cell proliferation or facilitating apoptosis. However, the chemotherapy resistance remains an urgent challenge in cancer therapy, especially in advanced stages. Several studies showed that the activation of pro-survival pathways, such as PI3K-AKT, participated in mediating chemotherapy resistance. The insights into the molecular mechanisms for underlying chemotherapy resistance are of great importance to improve cancer patient survival in advanced stages. The HOIP protein belongs to the RING family E3 ubiquitin ligases and modulates several atypical ubiquitination processes in cellular signaling. Previous studies showed that HOIP might be an important effector in modulating cancer cell death under genotoxic drugs. Here, we report that HOIP associates with PTEN and facilitates PTEN degradation in cancer cells. Depletion of HOIP causes cell cycle arrest and apoptosis, which effects could be rescued by PTEN silencing. Besides, the survival data from public available database show that HOIP expression correlates with poor survival in several types of chemotherapy-treated cancer patients. In conclusion, our study establishes a novel mechanism by which HOIP modulates PTEN stability and facilitates chemotherapy resistance in malignancies.
Keywords: PTEN, HOIP, Apoptosis, Ubiquitin
Introduction
Chemotherapy has been the major cancer treatment in advanced tumor stages [1]. Most of the genotoxic drugs, such as cisplatin, are reactive, interact with DNA and induce DNA damage [2]. Besides, they also induce tumor cell anti-proliferation and cancer cell apoptosis [3]. Despite the progression of chemotherapy during the past 40 years, the improvement of long-term survival is still limited [4, 5]. The side effects, including myelosuppression and nephrotoxicity, are still the restriction factors for the efficiency of chemotherapy [6]. Based on the facts, it is a common aim for the oncological doctors to treat patients with improved efficiency, reduced side effects and effective control of acquired resistance.
There is a growing concern about the chemotherapy resistance, which compromises the therapy efficiency and severely hampers the long-term survival for cancer patients. Several intrinsic factors, including gene mutations, translocations, epigenetic changes, are proved to contribute drug resistance [7]. Decreased drug uptake and increased efflux by the cell membrane transporters are also important mechanisms for compromised chemotherapy efficacy [8]. Besides, the activation of pro-survival signaling and suppression of apoptotic pathways, including NFKB signaling and PI3K-AKT pathways, are proved to be important mediators for the development of chemotherapy resistance [9, 10]. Although extensive efforts are put in investigating the potential mechanisms, still little is known about the determinants and pathways in mediating resistance. Hence, the chemotherapy resistance mechanisms and the development of corresponding overcome strategies are still subject to intensive studies.
Several studies have identified the PI3K-AKT pathway is hyper-activated in human cancers and facilitates chemotherapy resistance, which effect is counteracted by PTEN [11]. The PTEN (Phosphatase and tensin homologue) is characterized as an important tumor suppressor, which inhibits growth factor signaling via PI3 kinase. The PTEN protein dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3) and counter-balances the phosphorylation effect caused by AKT [12]. The inactivation or mutation of PTEN has been connected to several human cancers, including inherited cancer (Hamartoma Tumor Syndromes) [13]. Besides the genetic and epigenetic inactivation of PTEN, several studies also implicate the protein level of PTEN is always decreased through post-translational modifications, such as phosphorylation and ubiquitination [14, 15]. Several ubiquitin ligases are shown to potentiate cancer progression via promoting PTEN ubiquitination and degradation, such as XIAP and RNF126 [16, 17].
HOIP (HOIL-1-interacting Protein) belongs to the RING family protein family, which was firstly recognized as one component of the linear ubiquitin assembly complex (LUBAC) [18]. Our previous studies showed that HOIP could facilitate estrogen signaling and suppress P53 pathway for breast cancer progression [19, 20]. Interestingly, our further work reveals that HOIP modulates cisplatin-induced cell death regardless of P53 status, which means that HOIP could regulate chemotherapy sensitivity independent of P53 pathway. Here, our data show that HOIP correlates with poor chemotherapy outcome in several cancers. Molecular studies indicate that HOIP could associate with PTEN and facilitate its poly-ubiquitination and degradation, which ultimately prohibits chemotherapy-related inhibition in human cancers.
Results
HOIP correlates with chemotherapy sensitivity in several types of human cancers
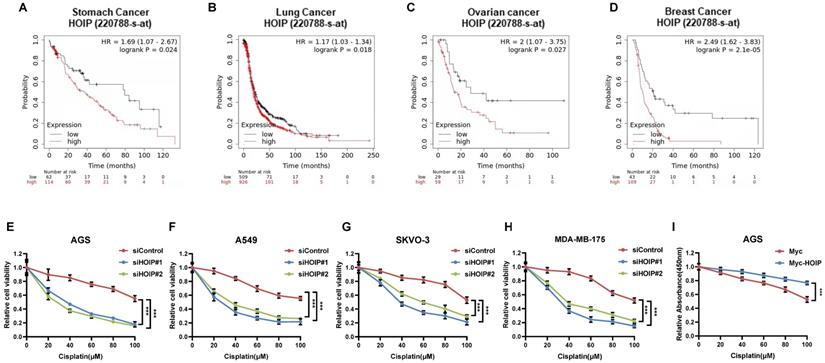
Since previous paper showed the correlation between HOIP and cisplatin resistance, we further analyze the HOIP effect in relation to the prognosis of chemotherapy patients [21]. From the public available database, we observe that HOIP expression level correlates with poor survival in gastric cancer patients, who endured chemotherapy (Fig. 1A). Such conclusion is also confirmed in ovarian cancer, non-small cell lung cancer and breast cancer (Fig. 1B-D). Besides, we tested the correlation between HOIP expression and cisplatin sensitivity in a series of cancer cell lines, while HOIP was depletion by two independent siRNAs. The data showed that HOIP depletion could significantly sensitize cisplatin-induced cell death in AGS, A549, SKVO-3 and MDAMB175 cells (Fig. 1F-H). Besides, HOIP over-expression could further confer cisplatin-induced cell inhibition in AGS cells (Fig. 1I).
HOIP correlates with chemotherapy sensitivity in several types of human cancers. A. Kaplan-Meier graph of progression-free survival showed that HOIP relates to poor prognosis in gastric cancer patients treated with chemotherapy. B. Kaplan-Meier graph of progression-free survival showed that HOIP relates to poor prognosis in lung cancer patients treated with chemotherapy. C. Kaplan-Meier graph of progression-free survival showed that HOIP relates to poor prognosis in ovarian cancer patients treated with chemotherapy. D. Kaplan-Meier graph of progression-free survival showed that HOIP relates to poor prognosis in breast cancer patients treated with chemotherapy. E. HOIP depletion sensitized cisplatin-mediated cell inhibition in AGS cells. AGS cells were transfected with 50 nM siHOIP or siControl. After 24 hours, cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay. F. HOIP depletion sensitized cisplatin-mediated cell inhibition in A549 cells. A549 cells were transfected with 50 nM siHOIP or siControl. After 24 hours, cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay. G. HOIP depletion sensitized cisplatin-mediated cell inhibition in SKVO-3 cells. SKVO-3 cells were transfected with 50 nM siHOIP or siControl. After 24 hours, cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay. H. HOIP depletion sensitized cisplatin-mediated cell inhibition in MDAMB175 cells. MDAMB175 cells were transfected with 50 nM siHOIP or siControl. After 24 hours, cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay. I. HOIP over-expression conferred resistance in cisplatin-induced cell inhibition in AGS cells. AGS cells were transfected with 2 ug HOIP plasmid or empty vector. After 24 hours, cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay.
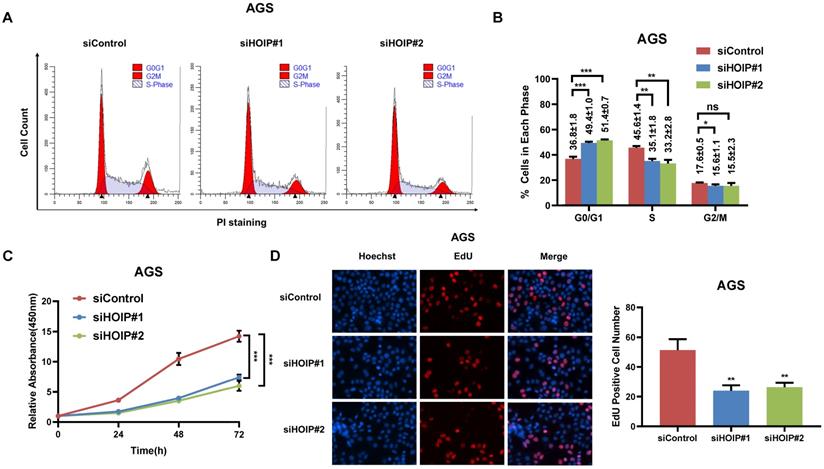
HOIP deletion causes cell cycle arrest and inhibits cell growth in cancer
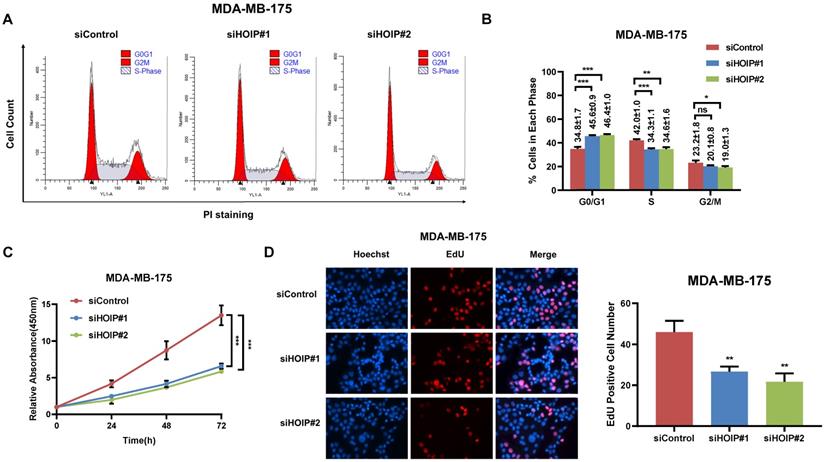
We utilize AGS cell as the model for the cell proliferation assays. The cell cycle analysis via flowcytometry shows that HOIP depletion significantly decreases the proportion of cells in S phase and increases the proportion of cells in G1 phase (Fig. 2A & B). The CCK8 assay implicates that HOIP depletion significantly inhibits cell growth in AGS cells (Fig. 2C). Besides, the EdU staining assay also indicates HOIP depletion significantly decreased the cell numbers of EdU incorporation in AGS cells (Fig. 2D). Besides, we further examined the HOIP depletion effect in MDAMB175 cells. The cell cycle analysis via flowcytometry shows that HOIP depletion significantly decreases the proportion of cells in S phase and increases the proportion of cells in G1 phase in MDAMB175 cells (Fig. 3A & B). The CCK8 assay implicates that in MDAMB175 cells, HOIP depletion significantly inhibits cell growth (Fig. 3C). Besides, the EdU staining assay also indicates HOIP depletion significantly decreased the cell numbers of EdU incorporation in MDAMB175 cells (Fig. 3D).
HOIP deletion causes cell cycle arrest and inhibits cell growth in gastric cancer. A and B. The cell cycle analysis of HOIP knockdown effect in AGS cells. AGS cells were transfected with 50nM HOIP siRNA or 50nM control siRNA. There were two independent siRNA be used. After 24 hours, cells were harvested, fixed via 70% ethanol and stained with propidium iodide. The cells were subject to FACS analysis. Experiments were done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for cell proportion comparison. The representative histograms and cell cycle phases were shown in Figure 2A and 2B respectively. C. HOIP depletion inhibited the cell proliferation in AGS cells. AGS cells were transfected with 50nM HOIP siRNA or 50nM control siRNA. There were two independent siRNA be used. After 24 hours, the CCK-8 assay was used to determine the cellar metabolic activity at indicated time points after transfection. Experiments were done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for cell growth comparison. D. HOIP depletion inhibited the number of EdU positive AGS cells. AGS cells were transfected with siControl or siHOIP. After 24 hours, EdU was added into the medium for 2 hours incubation. The absolute cell number was counted to indicate cell proliferation activity.
HOIP deletion causes cell cycle arrest and inhibits cell growth in MDAMB175 cells. A and B. The cell cycle analysis of HOIP knockdown effect in MDAMB175 cells. MDAMB175 cells were transfected with 50 nM HOIP siRNA or 50nM control siRNA. There were two independent siRNA be used. After 24 hours, cells were harvested, fixed via 70% ethanol and stained with propidium iodide. The cells were subject to FACS analysis. Experiments were done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for cell proportion comparison. The representative histograms and cell cycle phases were shown in Figure 3A and 3B respectively. C. HOIP depletion inhibited the cell proliferation in MDAMB175 cells. MDAMB175 cells were transfected with 50 nM HOIP siRNA or 50 nM control siRNA. There were two independent siRNA be used. After 24 hours, the CCK-8 assay was used to determine the cellar metabolic activity at indicated time points after transfection. Experiments were done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for cell growth comparison. D. HOIP depletion inhibited the number of EdU positive MDAMB175 cells. MDAMB175 cells were transfected with siControl or siHOIP. After 24 hours, EdU was added into the medium for 2 hours incubation. The absolute cell number was counted to indicate cell proliferation activity.
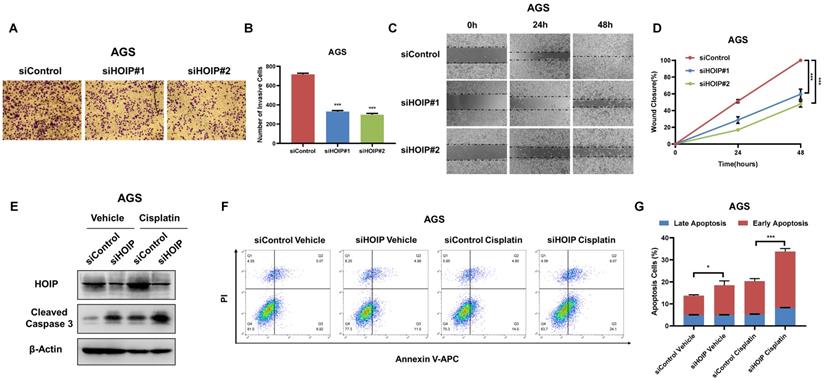
HOIP deletion inhibits migration and causes cell apoptosis in gastric cancer
Besides, HOIP silencing also inhibit cell invasion and migration capacity (Fig. 4A-D). We further utilize AGS cell as the model for the cell death assays. The immuno-blotting showed that HOIP depletion could increase cleaved caspase-3 level in AGS cells (Fig. 4E). We further confirm the conclusion in the Annexin V/PI double staining coupled with FACS analysis. The data shows that HOIP depletion increases the proportion of apoptotic cells in AGS cell line (Fig. 4F & G).
HOIP deletion inhibits cell invasion and causes cell apoptosis in gastric cancer. A and B. HOIP promotes cell invasion in AGS cells. AGS cells were transfected with indicated 50 nM HOIP siRNA (mix of #1 and #2) or 50 nM control siRNA. Trans-well was used to check the migration capacity. The cell number was counted and Data are presented as ±SD. **P<0.01, ***P<0.001 (student's t-test). C and D. Wound-healing assay of AGS cells were transfected with indicated 50 nM HOIP siRNA (mix of #1 and #2) or 50 nM control siRNA. Quantification of wound closure at the indicated time points. Data are presented as ±SD. **P<0.01, ***P<0.001 (student's t-test). E. HOIP depletion increased cleaved caspase-3 protein levels in AGS cells. AGS cells were transfected with siControl or siHOIP for 24 hours. Then cells were harvested for western blot analysis. HOIP and cleaved caspase-3 protein levels were determined by Western blot. Actin was used as internal control. F and G. HOIP depletion promoted apoptosis in AGS cells. AGS cells were transfected with siControl or siHOIP. After 24 hours, cells were stained with PI and Annexin V. Then cells were subject to FACS analysis for the proportion of apoptotic cells. Each group was done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for comparison.
HOIP modulates cell proliferation and apoptosis via PTEN in AGS cells
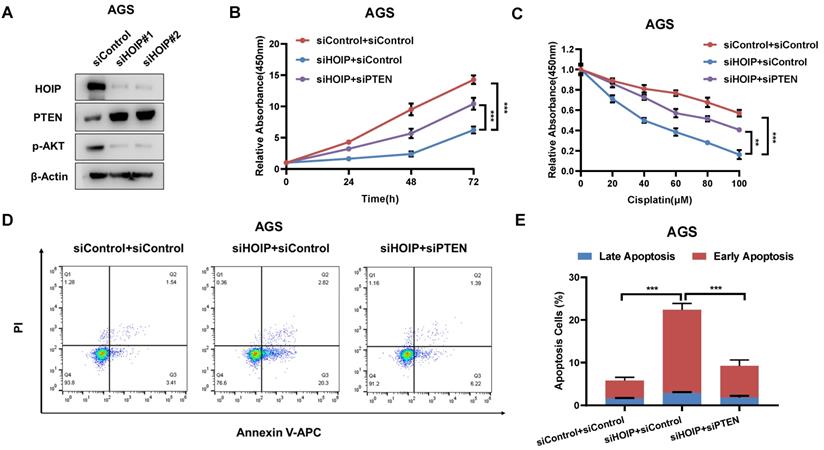
In order to investigate the logic link between the cancer phenotype and PTEN in HOIP function, we carry out several rescue experiments. The immuno-blotting assay shows that HOIP depletion increases PTEN protein level, but decreases the phosphorylation of AKT (S473) (Fig. 5A). Figure 4B shows that HOIP depletion could inhibit AGS cell proliferation, which effect could be at least partially rescued by PTEN knocking-down (Fig. 5B). Besides, the HOIP depletion could sensitize cisplatin-caused cell inhibition, which effect could be partially rescued by PTEN silencing (Fig. 5C). The annexin V/PI staining coupled with FACS analysis shows that PTEN silence could at least partially rescue cell apoptosis caused by HOIP depletion in AGS cells (Fig. 5D & 5E).
HOIP modulates cell proliferation and apoptosis via PTEN in AGS cells. A. HOIP depletion increase PTEN protein level, but decreases phosphor-AKT (Ser-473) in AGS cells. AGS cells were transfected with siControl or siHOIP. After 48 hours, cells were harvested for western blot analysis. HOIP, PTEN and phosphor-AKT (Ser-473) protein levels were determined by Western blot. Actin was used as internal control. B. Cell growth inhibition by HOIP silencing could be partially rescued by PTEN depletion in AGS cells. AGS cells were transfected with 50nM HOIP siRNA, 50nM control siRNA or 50nM siHOIP+siPTEN. After 24 hours, the CCK-8 assay was used to determine the cellar metabolic activity at indicated time points after transfection. Experiments were done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for cell growth comparison. C. HOIP depletion sensitized cisplatin-induced inhibition in AGS cells, which effect could be partially rescued by PTEN depletion. AGS cells were transfected with 50nM HOIP siRNA, 50nM control siRNA or 50nM siHOIP+siPTEN. The cells were treated with cisplatin for indicated concentration for 24 hours. The cell viability was determined via CCK8 assay. D and E. HOIP depletion promoted apoptosis, which effects could be partially rescued by PTEN depletion in AGS cells. AGS cells were transfected with 50nM HOIP siRNA, 50 nM control siRNA or 50nM siHOIP+siPTEN. After 24 hours, cells were stained with PI and Annexin V. Then cells were subject to FACS analysis for the proportion of apoptotic cells. Each group was done in triplicates. *P<0.05; ** P<0.01; ***P<0.001 for comparison.
HOIP associates with PTEN and modulates PTEN stability
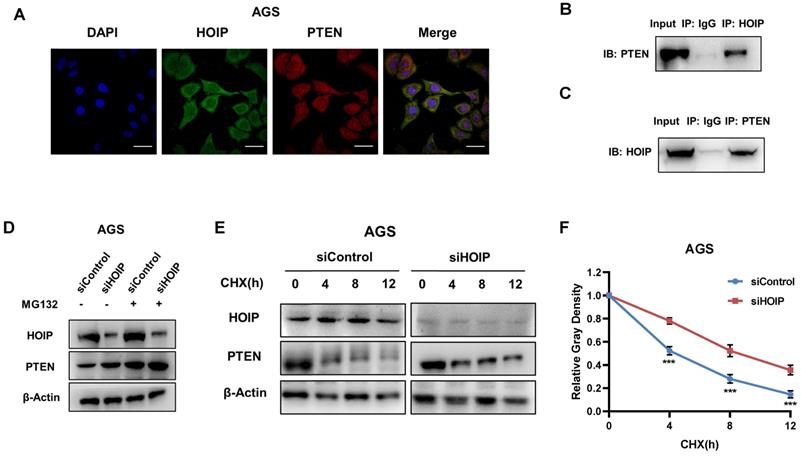
We further investigate the regulatory mechanism between HOIP and PTEN. The immuno-staining shows that HOIP is mainly located in the cytosol, while PTEN is located both in cytosol and nucleus (Fig. 6A). The endogenous immuno-precipitation shows that HOIP could interact with PTEN in AGS cells (Fig. 6B & C). HOIP depletion could increase PTEN protein level, which effect could be diminished via the proteasome inhibitor MG132 (Fig. 6D). This might indicate that HOIP modulates PTEN via protein stability. Then we utilized cycloheximide, the protein synthesis inhibitor, to measure the protein stability. Figure 6E & F shows that HOIP depletion significantly increased endogenous PTEN half-life (Fig. 6E & F).
HOIP associates with PTEN and modulates PTEN stability. A. Intracellular localization analysis of HOIP and PTEN by immunofluorescence assay. AGS cells were cultured in normal medium before fixation. Intracellular localization of PTEN (red) and HOIP (green) were shown. Nuclei (blue) were stained with 40,6-diamidino-2-phenylindole (DAPI). B and C. Co-IP assay reveals association between endogenous HOIP and PTEN in AGS cells. AGS cells were harvested with RIPA lysis buffer. CO-IP was performed using antibody as indicated. D. In the presence of the proteasome inhibitor MG132, HOIP could not further increase PTEN protein levels. AGS cells were transfected with 50 μM siControl or siHOIP. After 24 h, cells were treated with 10 µM MG132/vehicle for 6h. Cell lysates were prepared for Western blot analysis. The results are representative for three independent experiments. E and F. HOIP depletion increases PTEN half-life in AGS cells. AGS cells were transfected with 50nM siControl or siHOIP. After 24 h, cells were treated with 100 µM cycloheximide/vehicle for indicated times. Cell lysates were prepared for Western blot analysis. The results are representative for three independent experiments. The PTEN relative density was measured by Image J software.
HOIP promotes PTEN K48-linked poly-ubiquitination and degradation
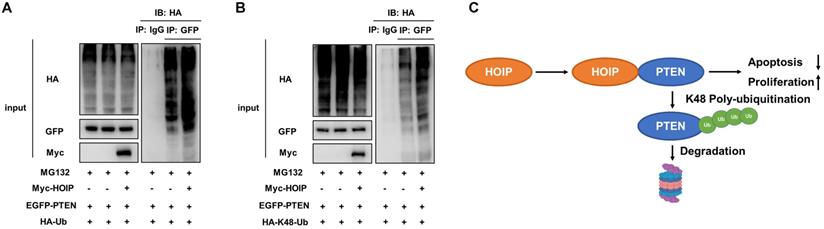
As on kind of E3 ubiquitin ligase, previous studies show that HOIP could facilitate several ubiquitination manners, such as K48-linked ubiquitination, linear ubiquitination and mono-ubiquitination. We further investigate the effect of HOIP on PTEN ubiquitination. The ubiquitin-based immuno-precipitation assay shows that HOIP could facilitate the overall and K48-linked poly-ubiquitination level of PTEN (Fig. 7A & B).
HOIP promotes PTEN K48-linked poly-ubiquitination and degradation. A. HOIP increases total poly-ubiquitination of PTEN. HEK293 cells were transfected with 2 µg EGFP-PTEN plasmid, 0.5 µg HA-Ub plasmid and 0.5 µg Myc-tag or Myc-HOIP plasmids. The cell extracts were immunoprecipitated with HA antibody. The total poly-ubiquitinated PTEN was detected via western blotting analysis. B. HOIP increases K48-linked poly-ubiquitination of PTEN. HEK293 cells were transfected with 2 µg EGFP-PTEN plasmid, 0.5 µg HA-K48 Ubi plasmid and 0.5 µg Myc-tag or Myc-HOIP plasmids. The cell extracts were immunoprecipitated with HA antibody. The total poly-ubiquitinated PTEN was detected via western blotting analysis. C. The hypothetical model for HOIP regulating PTEN in cancers: HOIP interacts with PTEN protein and directly promotes PTEN K48-linked poly-ubiquitination and degradation, which subsequently promotes cell cycle arrest and apoptosis.
Discussion
In our study, we identify the RING finger protein HOIP could interact with PTEN protein and promote PTEN poly-ubiquitination and degradation in human cancers (Fig. 7C). In addition, HOIP depletion could induce cell cycle arrest and cisplatin-induced apoptosis, which effect could be partially rescued by PTEN silencing. These data implicate that targeting HOIP could be a promising strategy to reverse or alleviate chemotherapy resistance.
Chemotherapy is the most important and commonly used regimen for human cancers, especially for recurrent or metastatic cases. The potential mechanisms for chemotherapy resistance are complicated. For example, cancer cells could increase the efflux or decrease the influx of chemotherapy drug, which protect the damage to the cells. Besides, the activation of DNA repair process and autophagy are also critical when the cancer cells endure chemotherapy stress [22]. Interestingly, several studies implicate that the activation of pro-survival and inactivation of apoptotic pathway also play critical roles in chemotherapy [23]. For example, the normal function of P53 is necessary for cisplatin-induced cell death, while the mutation or deletion of P53 could lead the tolerance of DNA damage and impair the cellular apoptotic response caused by chemotherapy [24]. Besides, the activation of AKT signaling and the loss of PTEN also prove to be important in mediating chemotherapy resistance [25].
PTEN is a ubiquitously expressed tumor suppressor, which counteracts the activation of AKT through its lipid phosphatase activity and inhibits MAPK signaling via its protein phosphatase activity [12, 26]. PTEN abnormalities are widely observed in human cancers, such as loss of heterozygosity (LOH) or gene mutations [27, 28]. However, recent studies also report that the post-translational modifications of PTEN also have functional impacts in cancer behaviors, such as cancer progression and drug resistance [29]. For example, the HECT family E3 ubiquitin ligase SMURF1 could promote PTEN poly-ubiquitination and degradation, which subsequently facilitates cancer progression in glioblastoma [30]. In our current study, we implicate the RING Family E3 ligase HOIP in mediating cancer growth and cisplatin resistance via facilitating PTEN degradation. Our study provides a novel regulation mechanism in modulating PTEN stability by HOIP and provides potential targets to re-sensitize chemotherapy resistance in human cancers.
HOIP gene was firstly cloned from MCF-7 cells in 2004 [31]. Further studies revealed that HOIP protein is highly expressed in muscles, testis and heart [32]. The whole-body knockout of HOIP will lead to embryonic lethality by TNFR-mediated cell death [33]. The most well studied function of HOIP is that it could form the linear ubiquitination assembly complex (LUBAC) with RBCK1 and SHARPIN, which facilitates the linear ubiquitination of IKKγ and the signaling transduction of NFKB [34]. Our previous study showed that HOIP could promote ER alpha signaling and suppress P53 signaling in breast cancer cells [35]. Although HOIP could inhibit wild type P53 function and facilitate cell survival, our current study shows that HOIP could still promote cell growth and inhibit apoptosis in P53 mutant background. Our molecular experiments show that HOIP could exert its pro-survival function via mediating PTEN degradation.
In conclusion, our study demonstrates the E3 ligase HOIP as a regulator of PTEN/AKT signaling in human cancer cells. HOIP suppresses PTEN protein level and promotes breast cancer cell growth and anti-apoptosis. As a newly discovered modulator of PTEN, HOIP could be a promising target to sensitize chemotherapy or reverse chemotherapy resistance in human cancers.
Materials and Methods
Cell culture
AGS, MDAMB175, HEK293, A549 and SKVO-3 cells are acquired form American Type Culture Collection (ATCC). A549 and AGS cells are maintained with RPMI-1640 (42401, Life Technologies) supplemented with 2 mM L-glutamine (25030, Life Technologies) and 10% FBS. MDAMB175, HEK293 and SKVO-3 cells are culture with Dulbecco's Modified Eagle's Medium that contains 4.5 g/L glucose and 4 mM L-glutamine (DMEM, 41965, Life Technologies) supplemented with 10% Fetal Bovine Serum (FBS, 10270, Life Technologies). All cell lines are characterized by cell line authentication. The cell line authentication via Short Tandem Repeat (STR) is performed via PowerPlex 21 system. The STR data of A549, AGS, MDAMB175, SKVO-3 and HEK293 cell lines are found consistent with STR data in ATCC.
Western blot
Standard western-blot assays are used to analyze protein expression in cells. The following antibodies are used for assays: anti-GFP (AB290, Abcam, 1:1000), anti-HA (901514, Biolegend, 1:1000) anti-Myc (AB32, Abcam, 1:1000), anti-Actin (3700, Cell Signaling Technology, 1:1000), anti-HOIP (Ab46322, Abcam, 1:1000), anti-PTEN (9552, Cell Signaling Technology, 1:1000), anti-Phospho-AKT (9271, Cell Signaling Technology, 1:1000) and anti-Cleaved Caspase-3 (9661, Cell Signaling Technology, 1:1000). Protein signals are detected with an ECL kit (Millipore Co., Billerica, Massachusetts, USA).
Plasmids and siRNA
The Myc-HOIP plasmid is acquired from our previous study [19]. The PTEN plasmid is acquired from the Addgene platform. The HA-K48 and HA-K63 Ubi plasmids were used in previous study [36]. The plasmids are transfected with Lipofectamin 2000 (1662298, Invitrogen). For siRNA transfection, the HOIP siRNA sequences are provided in the previous study [20]. The PTEN siRNA sequences are shown: 5-GUU AGC AGA AAC AAA AGG AGA UAU CAA-3, 5-UUG AUA UCU CCU UUU GUU UCU GCU AAC-3. The siControl sequence is 5-UUC UCC GAA CGU GUC ACG UTT-3, 5-ACG UGA CAC GUU CG GAGA ATT-3.
Quantification of cell viability
AGS, MDAMB175, A549 and SKVO-3 cells are transfected with siHOIP or siControl into 24-well plates. Twenty-Four hours after transfection, the cells number was countered and 4000 cells were seeded into 96-well plates. The relative cell viability was measured at indicated time points. Cell numbers are determined using the CCK8 cell proliferation reagent with the absorbance at 450 nm. For cisplatin-induced cell death assay, AGS, MDAMB175, A549 and SKVO-3 cells are transfected with siHOIP or siControl into 24-well plates. Twenty-Four hours after transfection, the cells number was countered and 20000 cells were seeded into 96-well plates. Different concentrations of cisplatin are added for 24 hours. Cell viability is determined using the CCK8 cell proliferation reagent with the absorbance at 450 nm.
EdU staining assay
For ethynly-deoxyuridine (EdU) labeled DNA, cells were incubated with EdU for 2 hours. Later on, the cells were fixed in cell culture plates with 4% formalin. The EdU positive cells were counted with statistical analysis.
Flow cytometry assay
For the cell cycle analysis, the AGS cells were transfected with 50 uM siHOIP or siControl. After 24 hours, cells were fixed via 70% ethanol and stained with propidium iodide. For the apoptosis assay, the AGS cells were transfected with 50 uM siHOIP or siControl. Twenty-four hours post-transfection, cells were stained with propidium iodide and annexin V. The BD LSR flow was used to measure the fluorescence intensity.
Co-immunoprecipitation assay
Immunoprecipitation is performed as described in previous study [37]. The AGS total cell lysis is pre-cleared with rabbit IgG for 2 h and subsequently immunoprecipitated with HOIP antibody (Ab46322, Abcam) over night, while rabbit IgG (Santa Cruz) is used as the negative control. The bounded protein is analyzed by Anti-PTEN (9552, Cell Signaling Technology). The bound proteins are analyzed by western blotting.
Protein stability assays
About 105 AGS cells are seeded into twenty-four well plates and transfected with 50 μM HOIP siRNA or siControl. After 48h, cells are treated with 100μM cycloheximide (C7698, Sigma) for indicated time points. Samples are subject to western blot for PTEN degradation.
Poly-ubiquitination detection assay
To directly detect the enriched K48-ubiquitinated and total ubiquitination PTEN from the cell extracts, HEK293 cells are transfected with K48 Ubi or Ub plasmids together with EGFP-PTEN plasmid and Myc-HOIP or Myc-vector. After 24 h, the cells are treated with 20 μM MG132 for 7 hours, then total protein is extracted and pre-cleared with 30μl protein A (santa cruz, SC-2001) for 4 h. The supernatant is collected and immunoprecipitated by PTEN antibody. Western blot with HA antibody is performed to detect total poly-ubiquitinated PTEN or K48-linked poly-ubiquitinated PTEN.
Statistical analysis
Statistical analysis is performed by GraphPad Prism 7 software or SPSS version 23.0 (SPSS, Inc., IL). Data are expressed as mean ± s.e.m. Differences between two independent groups were tested with Student's t-test. Kaplan-Meier analysis with log-rank test was applied for survival analysis. Differences are considered to be statistically significant when *P < 0.05; **P < 0.01; ***P < 0.001.
Abbreviations
PTEN: Phosphatase and tensin homologue; HOIP: HOIL-1-interacting Protein; PIP3: Phosphatidylinositol 3,4,5-trisphosphate; LUBAC: the linear ubiquitin assembly complex; NFKB: Nuclear factor kappa B; ATCC: American Type Culture Collection; STR: Short Tandem Repeat; IP: Immuno-precipitation.
Acknowledgements
We thank all the members of the Henan Key Laboratory of Immunology and Targeted Drugs, School of Laboratory Medicine for sharing valuable material and research support.
Funding
The project were supported from the Program for Science & Technology Innovation Talents in Universities of Henan Province (Grant No. 21HASTIT049, Ting Zhuang); the Joint Fund of the National Natural Science Foundation of China (No. U1604190, Jian Zhu); the Taishan Scholar Program of Shandong Province (NO.202103175, Jian Zhu); the National Natural Science Foundation of China (Grant No. U1804167, 81770721, 81570624, Zhiguo Niu and Qingsong Huang); Shandong Provincial National Natural Science Foundation (ZR2016HQ44, Yinlu Ding); Key Scientific and Technological Projects of Henan Province (Grant No.202102310024; Qingsong Huang).
Ethics
This study was reviewed and approved by the Ethical Board at Jiangsu University. This usage of clinical samples was reviewed and approved by the Ethical Board at the Jiangsu University with written informed consents from all the patients.
Author Contributions
Wenrong Xu, Yinlu Ding and Jian Zhu conceived the designed that study. Zhiguo Niu, Shuxiao Dong and Xin Li performed the molecular and cellular biology of the study. Zhiguo Niu, Shuxiao Dong and Xin Li performed the cellular phenotype assays. Jianhui Gao, Huijie Yang, Cheng Yan, Shu Zhuo and Hui Qian performed the bioinformatics data analysis. Ting Zhuang and Qingsong Huang wrote the manuscript and approved the manuscript. Jian Zhu, Yinlu Ding and Wenrong Xu offered the funding support and project supervision during the revision stage.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-378
2. Tchounwou PB, Dasari S, Noubissi FK, Ray P, Kumar S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J Exp Pharmacol. 2021;13:303-328
3. Ghosh S. Cisplatin. The first metal based anticancer drug. Bioorg Chem. 2019;88:102925
4. Wada N, Akamaru Y, Wada R, Munakata K, Takiuchi D, Azama T. et al. Adjuvant Chemotherapy and the Prognosis of ypStage Gastric Cancer. Gan To Kagaku Ryoho. 2019;46(1):85-87
5. Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd FA, Stephens RJ. et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26(21):3552-3559
6. Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem Res Toxicol. 2019;32(8):1469-1486
7. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27-36
8. Zhu H, Luo H, Zhang W, Shen Z, Hu X, Zhu X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des Devel Ther. 2016;10:1885-1895
9. Kuang CM, Fu X, Hua YJ, Shuai WD, Ye ZH, Li Y. et al. BST2 confers cisplatin resistance via NF-kappaB signaling in nasopharyngeal cancer. Cell Death Dis. 2017;8(6):e2874
10. Zhang Y, Bao C, Mu Q, Chen J, Wang J, Mi Y. et al. Reversal of cisplatin resistance by inhibiting PI3K/Akt signal pathway in human lung cancer cells. Neoplasma. 2016;63(3):362-370
11. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer. At the bench and bedside. Semin Cancer Biol. 2019;59:125-132
12. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8(3):187-198
13. Macken WL, Tischkowitz M, Lachlan KL. PTEN Hamartoma tumor syndrome in childhood: A review of the clinical literature. Am J Med Genet C Semin Med Genet. 2019;181(4):591-610
14. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20(14):5010-5018
15. Okahara F, Ikawa H, Kanaho Y, Maehama T. Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J Biol Chem. 2004;279(44):45300-45303
16. Van Themsche C, Leblanc V, Parent S, Asselin E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J Biol Chem. 2009;284(31):20462-20466
17. Xu H, Ju L, Xiong Y, Yu M, Zhou F, Qian K. et al. E3 ubiquitin ligase RNF126 affects bladder cancer progression through regulation of PTEN stability. Cell Death Dis. 2021;12(3):239
18. Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012;31(19):3833-3844
19. Zhu J, Zhao C, Kharman-Biz A, Zhuang T, Jonsson P, Liang N. et al. The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor alpha and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene. 2014;33(34):4340-4351
20. Zhu J, Zhao C, Zhuang T, Jonsson P, Sinha I, Williams C. et al. RING finger protein 31 promotes p53 degradation in breast cancer cells. Oncogene. 2016;35(15):1955-1964
21. MacKay C, Carroll E, Ibrahim AFM, Garg A, Inman GJ, Hay RT. et al. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014;74(8):2246-2257
22. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M. et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838
23. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY). 2016;8(4):603-619
24. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921-8946
25. Pu X, Hildebrandt MA, Lu C, Lin J, Stewart DJ, Ye Y. et al. PI3K/PTEN/AKT/mTOR pathway genetic variation predicts toxicity and distant progression in lung cancer patients receiving platinum-based chemotherapy. Lung Cancer. 2011;71(1):82-88
26. Ebbesen SH, Scaltriti M, Bialucha CU, Morse N, Kastenhuber ER, Wen HY. et al. Pten loss promotes MAPK pathway dependency in HER2/neu breast carcinomas. Proc Natl Acad Sci U S A. 2016;113(11):3030-3035
27. Alvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: It's all about diversity. Semin Cancer Biol. 2019;59:66-79
28. Yehia L, Keel E, Eng C. The Clinical Spectrum of PTEN Mutations. Annu Rev Med. 2020;71:103-116
29. Gao C, Yuan X, Jiang Z, Gan D, Ding L, Sun Y. et al. Regulation of AKT phosphorylation by GSK3beta and PTEN to control chemoresistance in breast cancer. Breast Cancer Res Treat. 2019;176(2):291-301
30. Xia Q, Zhang H, Zhang P, Li Y, Xu M, Li X. et al. Oncogenic Smurf1 promotes PTEN wild-type glioblastoma growth by mediating PTEN ubiquitylation. Oncogene. 2020;39(36):5902-5915
31. Thompson HG, Harris JW, Lin L, Brody JP. Identification of the protein Zibra, its genomic organization, regulation, and expression in breast cancer cells. Exp Cell Res. 2004;295(2):448-459
32. Ehrlund A, Anthonisen EH, Gustafsson N, Venteclef N, Robertson Remen K, Damdimopoulos AE. et al. E3 ubiquitin ligase RNF31 cooperates with DAX-1 in transcriptional repression of steroidogenesis. Mol Cell Biol. 2009;29(8):2230-2242
33. Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014;9(1):153-165
34. Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. 2015;212(6):939-951
35. Zhu J, Zhuang T, Yang H, Li X, Liu H, Wang H. Atypical ubiquitin ligase RNF31: the nuclear factor modulator in breast cancer progression. BMC Cancer. 2016;16:538
36. Wang Z, Kong Q, Su P, Duan M, Xue M, Li X. et al. Regulation of Hippo signaling and triple negative breast cancer progression by an ubiquitin ligase RNF187. Oncogenesis. 2020;9(3):36
37. Xue M, Zhang K, Mu K, Xu J, Yang H, Liu Y. et al. Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis. 2019;8(5):30
Author contact
![]() Corresponding authors: Wenrong Xu, Jiangsu Key Laboratory of Medical Science and Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, 212013, China. E-mail: iclsedu.cn; Jian Zhu, Department of general surgery, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250033, China. E-mail: zhujian1204com; Yinlu Ding, Department of general surgery, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250033, China. E-mail: dingyinlucom.
Corresponding authors: Wenrong Xu, Jiangsu Key Laboratory of Medical Science and Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, 212013, China. E-mail: iclsedu.cn; Jian Zhu, Department of general surgery, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250033, China. E-mail: zhujian1204com; Yinlu Ding, Department of general surgery, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250033, China. E-mail: dingyinlucom.