Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(16):4993-5004. doi:10.7150/jca.55526 This issue Cite
Research Paper
Oxidized-Desialylated Low-Density Lipoprotein Inhibits the Antitumor Functions of Lymphokine Activated Killer Cells
Jesús S Aguilar Díaz de león1, Honor L Glenn2, Mark Knappenberger3, Chad R Borges1 ![]()
1. School of Molecular Sciences and The Biodesign Institute - Center for Personalized Diagnostics, Arizona State University, P.O. Box 876401, Tempe, AZ 85287, USA.
2. School of Life Sciences and The Biodesign Institute - Center for Immunotherapy, Vaccines and Virotherapy, Tempe, AZ 85287, USA.
3. School of Life Sciences and The Biodesign Institute - Center for Personalized Diagnostics, Arizona State University, P.O. Box 876401, Tempe, AZ 85287, USA.
Received 2020-11-5; Accepted 2021-5-19; Published 2021-6-16
Abstract

Elevated concentrations of circulating low density lipoprotein (LDL) that is abnormally oxidized and desialylated is both a precursor to and a hallmark of atherosclerosis. Peripheral blood mononuclear cells (PBMCs) treated in vitro with interleukin-2 (IL-2) become lymphokine activated killer (LAK) cells, the primary effectors of which are NK cells and NKT cells. LAK cells display antitumor functions such as increased cytotoxicity and IFN-γ production, and they have been evaluated as a potential cancer therapeutic. Atherosclerotic processes may influence innate immunity against cancer. Because prior studies have shown that low density lipoprotein (LDL) reduces T-cell and NK cell antitumor functions, we asked whether oxidized-desialylated LDL affects the functionality of LAK cells in vitro. We show here that LAK cells take up oxidized-desialylated LDL to a significantly greater extent than native LDL over a period of 72 hours. This resulted in a significant downregulation of LAK cell cytotoxicity against K562 cells. In particular, the expression of IFN-γ, CD56, and NKG2D were reduced upon oxidized-desialylated LDL treatment of LAK cells and, conversely, their expression was enhanced with native LDL. It was also observed that as the number of CD56 and NKG2D positive cells decreased upon treatment with oxidized-desialylated LDL, the number of CD3 positive cells increased in proportion. Additionally, only a slight inhibition of LAK cell cytotoxicity was observed with desialylation alone of LDL, and no significant inhibition was observed with oxidation alone of LDL. Thus, this study describes a new role of oxidized-desialylated LDL as an inhibitor of the antitumor functions of LAK cells. These observations have implications for how atherosclerosis processes, namely oxidation and desialylation of LDL, may influence LAK cell antitumor activity.
Keywords: lymphokine activated killer (LAK) cells, atherosclerosis, oxidized-desialylated low-density lipoprotein (LDL), cancer
Introduction
Among the immunotherapy methods being considered for anticancer therapy, adoptive transfer of lymphokine-activated killer (LAK) cells is one of the biotherapy approaches that has been translated into clinical studies [1-5]. In vitro culture of peripheral blood mononuclear cells (PBMCs) with interleukin-2 (IL-2) for several days results in a population of LAK cells with upregulated cytotoxicity against cancer cells and IFN-γ production [6]. LAK cells are composed mostly of NK cells (CD3-CD56+) and NKT cells (CD3+CD56+), and unlike cytotoxic T cells, LAK cells do not require the major histocompatibility complex (MHC) pathway to perform their antitumor activities [7]. This feature makes LAK cells an important anticancer biotherapy for those cancer cells that lack MHC and can't be targeted by T cells. The pathway through which LAK cells target and kill transformed cells is through the Natural Killer Group 2D (NKG2D) receptor [8]. The NKG2D receptor is expressed on both NK cells and NKT cells, and it acts as an activation signal when it recognizes its ligands on cancer cells [9]. When NK cells and NKT cells are activated through the NKG2D receptor, they secrete apoptosis inducing effectors that kill the cancer cells. Ligands for NKG2D include MICA, ULBP4, and ULBP1, which are implicated in NKG2D mediated recognition of multiple cancers [8, 9].
Several clinical trials have successfully activated and expanded LAK cells using IL-2 [1, 2, 7, 10-12]. When LAK cells were first introduced by Rosenberg et al in 1985 [10, 11], eleven of 25 patients showed significant cancer reduction, with one showing complete tumor regression. Recent studies have shown that the effectiveness of IL-2/LAK immunotherapy can be improved with locoregional administration, rather than by systemic administration of LAKs [12]. However, the cytotoxicity activity of LAK cells is ephemeral after they are administered to patients. Studies have shown that LAK cell immunotherapy is more effective with the continuous administration of IL-2 [13]. After LAK cell administration to patients, LAK cells need to be continuously activated to work. IL-2 activates LAK cells, but it can also inhibit them by inducing the expansion of regulatory T cells [14]. Moreover, administration of IL-2 can cause adverse effects on patients, such as vascular leak syndrome [15]. Additionally, there are inhibitory factors that inhibit the antitumor functions of LAK cells such as MHC [16], and sialic acid overexpression on cancer cells [17, 18].
Although there are many factors that might be involved in LAK cell inhibition, studies suggest that low-density lipoproteins, free cholesterols, and lipids, can also affect lymphocyte antitumor functions [19-23]: Low-density lipoprotein uptake inhibits the antitumor functions of γδT cells, which express NK receptors that determine their antitumor cytotoxicity [19]. Inhibition of cholesterol esterification increases CD8+ T cell cytotoxic functions [20]. NK cells with increased intracellular lipid accumulation display decreased cytotoxicity effects [21, 22]. Oxidation of low-density lipoprotein by polymorphonuclear leukocytes inhibits NK activity [23]. Oxidation and desialylation of LDL are two important features in the development of atherosclerosis that appear to facilitate the increase of LDL uptake by macrophages [24, 25], smooth muscle cells [26], fibroblasts [27], and aorta cells [28]. In vitro studies have shown that oxidized-desialylated LDL samples induce an increase in intracellular accumulation of triglycerides, free cholesterol, and cholesterol esters in these cells, as compared to native LDL samples [28]. In vivo studies suggest that desialylation of LDL appears to be an early event that leads to smaller, denser, more electronegative, and oxidized LDL particles, all of which are referred to as multiple modified LDL, which appear to be a hallmark of atherosclerosis [29, 30]. Although the link between multiple modified LDL and cancer immunity remains unclear, several studies have suggested that increased lipid accumulation and increased LDL uptake lead to the impairment of lymphocyte cytotoxic functions against cancer cells [31]. However, to our knowledge, there is still no evidence on whether multiple modified LDL affects LAK cell antitumor activity.
In this study, we asked whether the oxidized and/or desialylated forms of LDL modulate LAK cell cytotoxicity towards leukemia cells (K562 cells). We found that LAK cells take up oxidized-desialylated LDL to a greater extent than native LDL, and it causes them to be less cytotoxic against cancer cells. Moreover, oxidized-desialylated LDL uptake drives the downregulation of cytotoxicity associated proteins CD56 and NKG2D, as well as impairment of IFNγ production.
Oxidized-desialylated LDL becomes abundant under the conditions that foster atherosclerosis. Atherosclerosis shares multiple pathways with cancer, and it has been suggested that atherosclerosis promotes tumor development [32]. Here we found that oxidation and desialylation are two important posttranslational modifications on LDL that make it an important inhibitor of LAK cell antitumor activity. Thus the results presented here provide a link between factors involved in atherosclerosis and the progression of cancer.
Results
Oxidized-desialylated LDL inhibits LAK cell cytotoxicity in vitro
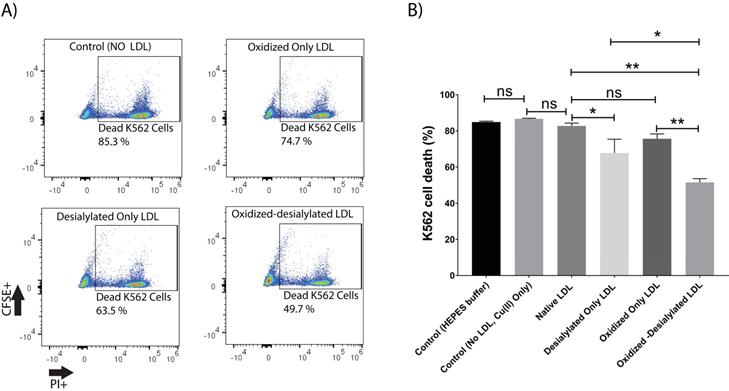
This study was initiated by investigating the capacity of desialylated LDL to inhibit LAK cell cytotoxicity. For this purpose, PBMCs were activated and expanded in vitro for 8 days in the presence of IL-2 to generate lymphokine activated killer (LAK) cells. LAK cells are composed of NK cells (CD3-CD56+) and NKT cells (CD3+CD56+). Analysis of IL-2 treated PBMCs by flow cytometry revealed an induced population of cells of which about 6% were NK cells and another 4% were NKT cells (Supplementary Fig. S1). This population of cells, referred to as LAK cells, were optimally cytotoxic against K562 cells in a 4-hour killing assay at a 10:1 effector to target ratio (Supplementary Fig. S2). Native LDL was desialylated with neuraminidase enzyme and added to the LAK cells at day 5 at 50 μg/ml and incubated for 72 hours. Our glycan node analysis procedure [33-38] was used to verify LDL desialylation prior to adding the LDL samples to the LAK cells. An increase in the relative abundance of terminal galactose, a decrease in 3-linked galactose, and a near-complete loss of 6-linked galactose revealed a major decrease in the number of terminal sialic acid residues present on LDL (Supplementary Fig. S3). Only a small inhibition of LAK cell cytotoxicity was observed every time we performed a K562 killing assay after having exposed the LAK cells to desialylated LDL (Figure 1). Because previous research has demonstrated that desialylation of LDL appears to be an early event that leads to its oxidation in atherosclerosis [39], we induced oxidation of desialylated LDL with Cu(II) ions. A thiobarbituric acid reactive substances (TBARS) assay revealed that Cu(II)-treated native and desialylated LDL were approximately 50% more oxidized than their untreated LDL counterparts (Supplementary Fig. S4). Desialylated and oxidized LDL was then added to the LAK cells at 50 μg/ml (5 mg/dL) and incubated for 72 hours to see how LDL with both modifications affected the cytotoxicity of LAK cells. Significant inhibition of LAK cell cytotoxicity was observed when LDL was both oxidized and desialylated (Figure 1). A lesser degree of inhibition was observed when LAK cells were treated with merely desialylated LDL. Oxidation alone did not induce a significant inhibition of LAK cell cytotoxicity (Figure 1). To ensure that inhibited K562 cell killing was not simply due to loss of LAK cells, LAK cell viability was checked by incubating the LAK cells with oxidized-desialylated LDL for 72 hours and labeling with Sytox Blue. Cell viability was determined using flow cytometry. Results revealed that treatment of LAK cells with oxidized-desialylated LDL induced a small toxic effect (about 4%) (Supplementary Fig. S5). By comparison, the magnitude of the inhibition effect observed in Figure 1 suggests that the inhibition of K562 cell killing by pre-treating LAK cells with oxidized-desialylated LDL is not simply due to the toxicity of oxidized-desialylated LDL toward LAK cells.
Oxidized-desialylated LDL inhibits LAK cell cytotoxicity in vitro. A) Activated and expanded LAK cells were cultured in serum free X-VIVO 10 media in a 24 well plate with 0.1 µg/ml IL-2 in the absence or presence of native LDL, oxidized only LDL, desialylated only LDL, or oxidized-desialylated LDL at 50 µg/ml for 72 hours. Then LAK cells were washed three times with X-VIVO 10 serum free media to remove residual external LDL and incubated in a 4-hr killing assay with K562 cells at a 10:1 effector to target ratio. Percent cytotoxicity was determined by flow cytometry. B) Quantification of K562 cell death. **indicates a statically significant difference between native LDL (control) and oxidized-desialylated LDL treated LAK cells, and oxidized only LDL vs oxidized-desialylated LDL treated LAK cells (p<0.0001).*Indicates significant difference between native LDL and desialylated only LDL, and desialylated only LDL vs oxidized-desialylated (p <0.001) treated LAK cells, n = 5 per group. ns indicates not statistically significant. Statistical significance determined using two-way ANOVA with Tukey posthoc test. Error bars represent standard deviation.
Modality of K562 Cell Death
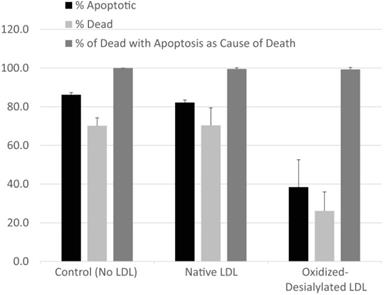
The type of cell death experienced by K562 cells in the presence of LAK cells exposed to various forms of LDL was evaluated by fluorescence microscopy using CellEvent Caspase-3/7 fluorogenic substrate (CE) which is specific for caspase-3/7 activity and indicative of apoptosis, in combination with CellTrace Violet, a cytoplasmic stain which was used to label K562 cells, and propidium iodide (PI) to identify dead cells. Cells and modified LDL were prepared as described in Materials and Methods. K562 cells, pre-labeled with CellTrace Violet, were incubated with a 10-fold excess of LAK cells that had been exposed to native LDL, oxidized-desialylated LDL, or no LDL at all (control) in assay medium containing CE indicator and PI. In a parallel set of control experiments, the same set of incubations were conducted in the absence of CellTrace Violet and CE dye in order to ensure that neither label altered total cell death. Neither CellTrace nor CE induced cell death of any kind (data not shown). After 4 hrs, samples were imaged by fluorescence microscopy. Images were analyzed to determine the percentage of K562 cells that 1) were experiencing apoptosis (determined as 100 × [# of CellTrace Violet & CE-labeled cells] / [# of CellTrace Violet-labeled K562 cells]), 2) had died (determined as 100 × [# of CellTrace Violet & PI-labeled cells] / [# of CellTrace Violet-labeled K562 cells]), or 3) had died via apoptosis (determined as 100 × [# of CellTrace Violet & CE & PI-labeled cells] / [# of CellTrace Violet & PI-labeled cells]) (Figure 2). A minimum of 130 K562 cells were analyzed in each image. Essentially all K562 cell death was due to apoptosis. Moreover, while oxidized-desialylated LDL inhibited K562 cell killing as expected, the type of cell death remained at 99-100% apoptosis regardless of whether the LAK cells were exposed to native LDL, oxidized-desialylated LDL or no LDL at all. Control incubations of K562 cells in the absence of LAK cells revealed < 1% total cell death.
LAK cells kill target cells via apoptosis. Target cells (K562 cells pre-labeled with CellTrace Violet) and effector (LAK) cells were incubated together for 4 hrs in the presence of CE dye (apoptosis indicator) and PI (cell death indicator). Percentage of apoptotic K562 cells was determined as 100 x [# of CellTrace & CE-labeled cells] / [# of CellTrace labeled cells]). The percentage of dead K56 was determined as 100 x [# of CellTrace & PI-labeled cells] / [# of CellTrace-labeled cells]). The percentages of K562 that died via apoptosis was determined as 100 x [# of CellTrace & CE & PI-labeled cells] / [# of CellTrace & PI-labeled cells]). A minimum of 130 K562 cells were analyzed in each field. Error bars represent standard deviation from two fields of a representative experiment. No significant differences in the percentage of cells that had died due to apoptosis were detected between LDL treatments (Kruskal-Wallis test).
Enhanced Uptake of Oxidized-Desialylated LDL by LAK Cells
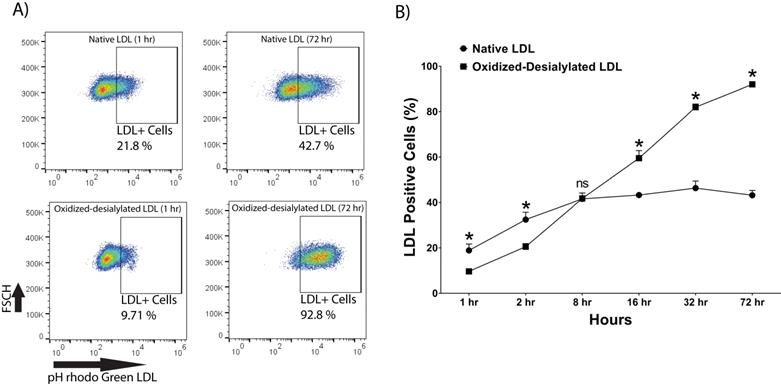
pHrodo Green-labeled LDL was desialylated with neuraminidase enzyme and oxidized with 10 μM Cu(II) ions. LAK cells were then incubated with native and oxidized-desialylated forms of pHrodo green LDL (Figure 3). During the first two hours of incubation, LAK cells took up native LDL faster than oxidized-desialylated LDL. After 8 hours of incubation, the uptake of native and oxidized-desialylated LDL was the same, with no significant difference. However, after 16, 32, and 72 hours, oxidized-desialylated LDL was taken up to a greater extent than native LDL in a time-dependent manner. After 72 hours, a clear increase in oxidized-desialylated LDL uptake was observed (Figure 3B). Non-significant differences in uptake were observed for oxidized LDL and, separately, desialylated LDL compared to native LDL (data not shown).
Enhanced uptake of oxidized-desialylated LDL by LAK cells. LAK cells were cultured in serum free X-VIVO 10 media in a V-bottom 96 well plate with IL-2 in the absence or presence of native pHrodo Green Conjugate LDL or oxidized-desialylated pHrodo Green Conjugate LDL at 10 µg/ml for 1, 2, 8, 16, 32, and 72 hours. The percentage of LDL positive cells was measured by flow cytometry. A) Qualitative flow data showing uptake of oxidized-desialylated LDL and native LDL, which shows the differences in uptake between the two forms of LDL. FSCH stands for forward scatter cell signal height which facilitates selection of single cells. B) Time course of native LDL and oxidized-desialylated LDL uptake by LAK cells. * Indicates statically significant differences between native LDL (control) and oxidized-desialylated LDL treated LAK cells (p<0.001). Statistical significance determined using multiple t-tests (one per group) and corrected for multiple comparisons using the Holm-Sidak method. n = 3 per time point. Error bars represent standard deviation. For some points, errors bars are shorter than the symbol, and error bars are not shown.
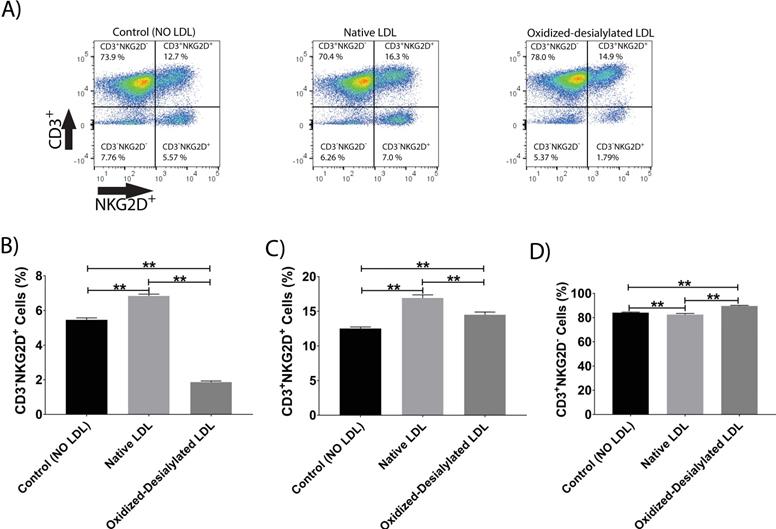
Oxidized-Desialylated LDL downregulates the cytotoxicity receptors CD56 and NKG2D
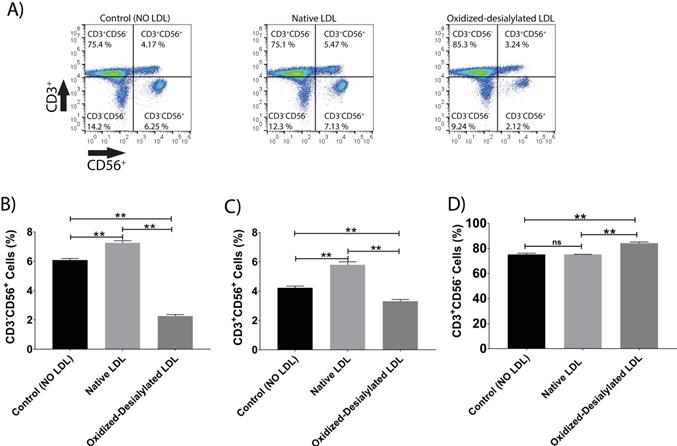
LAK cells express the surface markers CD56, CD3, and NKG2D. CD56 and NKG2D are cytotoxicity associated receptors expressed on both NK cells and NKT cells, and CD3 is an activating receptor expressed on NKT cells and T cells [40]. Because LAK cell anti-cancer (anti-K562) activity was reduced in the 4-hour killing assay upon LAK cell uptake of oxidized-desialylated LDL over a 72-hour period, we tested whether the expression levels of these receptors were also affected. After gating all the live cells and plotting them against CD3 and NKG2D, a decrease in the proportion of CD56+ cells and CD3+CD56+ cells was observed (Figure 4A-C). Interestingly, it was observed that as the number of CD3-CD56+ cells and CD3+CD56+ cells decreased, the number of CD3+-only cells increased (Figure 4D). This inverse relationship implies that oxidized-desialylated LDL causes LAK cells to differentiate into CD3+-only rather than CD56+ cells, making them less cytotoxic against K562 cancer cells. It was also observed that native LDL induced a small increase in the number of CD56+ cells, although in the 4-hour killing assay no change in cytotoxicity was observed.
Oxidized-desialylated LDL downregulates cytotoxicity receptor CD56 and upregulates the CD3 receptor. Activated and expanded LAK cells were cultured in serum free X-VIVO 10 media in a V-bottom 96 well plate with IL-2 in the absence or presence of native LDL or oxidized-desialylated LDL at 50 µg/ml for 72 hours. Then LAK cells were washed three times with X-VIVO 10 serum free media, reconstituted in PBS, and labeled with anti-CD56 antibodies, as well as with the dead staining dye SytoxBlue. A) Live cells were gated and plotted against CD3 and CD56. B) Oxidized-desialylated LDL decreased the number of CD3-CD56+ cells. C) The number of NKT cells (CD3+ CD56+) also decreased significantly. D) The number of CD3 positive cells increased significantly upon oxidized-desialylated LDL treatment of LAK cells. n = 5 per group. Error bars represent standard deviation. ** indicates statically significant differences (p<0.0001), determined using one-way ANOVA with Tukey posthoc test.
Additionally, it was observed that native LDL induced a small increase in the expression of NKG2D receptor (Figure 5A-C). However, oxidized-desialylated LDL downregulated the percentage of CD3-NKG2D+ cells while slightly increasing the percentage of CD3+NKG2D+ cells (Figure 5A-C). A small increase in the number of CD3+ only cells was also observed in the oxidized-desialylated treated samples (Figure 5D).
Oxidized-desialylated LDL downregulates activating receptor NKG2D. LAK cells were cultured and washed as described in Fig. 3. Then LAK cells were labeled with anti-NKG2D antibodies, as well as with the dead staining dye SytoxBlue. A) Live cells were gated and plotted against CD3 and NKG2D. B) Oxidized-desialylated LDL decreased the number of CD3-NKG2D+ cells. C) Oxidized-desialylated LDL increased the number of CD3+ cells relative to the LDL-free control, but decreased them relative to treatment with native LDL. D) Oxidized-desialylated LDL increased the percentage of CD3+NKG2D- cells relative to both other groups. n = 5 per group. **Indicates statistically significant differences (p<0.0001), determined using one-way ANOVA with Tukey posthoc test. Error bars represent standard deviation.
Oxidized-desialylated LDL impairs Interferon Gamma (IFN-γ) production
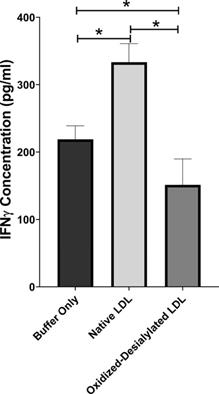
Interferon gamma (IFN-γ) is a pleiotropic cytokine with antitumor functions. IFN-γ is secreted by NK cells and NKT cells, and it is often considered the major effector of immunity [41]. Because oxidized-desialylated LDL caused an inhibition of LAK cell cytotoxicity and downregulation of the cytotoxicity receptors, we investigated whether production of IFN-γ was altered during the process. An ELISA was performed to quantitively measure soluble IFN-γ in LAK cell supernatants that had been treated with native LDL or oxidized-desialylated LDL. Results showed that oxidized-desialylated LDL decreased the production of IFN-γ. Previous research has shown that native LDL downregulates the production of IFN-γ in γδT cells [19] - prompting expectations of a similar result. Unexpectedly, however, native LDL significantly enhanced the production of IFN-γ in LAK cells (Figure 6).
Oxidized-desialylated LDL impairs IFNγ production. LAK cells were cultured in serum free X-VIVO 10 media with 0.1 µg/ml IL-2 in the presence or absence of native LDL or oxidized-desialylated LDL for 72 hours. After 72 hours, cell supernatants were removed, centrifuged at 330 g for 8 minutes. Soluble IFNγ was measured in undiluted cell supernatants by ELISA. *Indicates statically significant differences between native LDL (control) and oxidized-desialylated LDL treated LAK cells (p<0.001). Statistical significance determined using one-way ANOVA with Turkey posthoc test. n = 4 per group. Error bars represent standard deviation.
Discussion
Cancer and atherosclerosis are distinct diseases that share several molecular pathways, with some studies suggesting that atherosclerosis can lead to cancer development [32]. Characteristic features of both diseases include inflammation, uncontrolled cell proliferation, and oxidative stress [42]. Atherosclerosis is characterized by chronic inflammation, diabetes, hypertension, dyslipidemia, and obesity, all of which are predisposing risk factors for cancer [42]. At the molecular level, inflammation leads to the production of inflammatory cytokines, such as IL-6, IL-1β, and TNF, which have been associated with cardiovascular disease and several different types of cancer [43]. High concentrations of biomarkers for reactive oxygen species such as oxidized LDL and myeloperoxidase (MPO), have been found in the blood plasma of cancer patients and atherosclerosis patients, and the origin of both diseases is associated with oxidative stress [44, 45]. Additionally, it has been found that the metabolism of unstable atherosclerotic plaques is similar to that of cancer cells [46].
Oxidation and desialylation of low-density lipoprotein are hallmark modifications of LDL in atherosclerosis that contribute to the initiation and progression of the disease [29]. These modifications are believed to be needed to facilitate increased LDL uptake by atherosclerotic cells and cause them to have increased intracellular triglycerides, free cholesterol, and cholesterol esters [29, 30]. Although little is known about oxidized-desialylated LDL in cancer, literature suggests that oxidized LDL is involved in tumor development [47]. Long term exposure of human vascular smooth muscle cells (hVSMC) to chemically oxidized LDL promotes the overexpression of osteopontin, a glycoprotein involved in cancer metastasis [48]. Inflammation is a characteristic feature of both atherosclerosis and cancer, and studies have shown that oxidized LDL triggers inflammation. Macrophages take up oxidized LDL via scavenger receptors which, once inside the cell, induce cholesterol crystals that ultimately activate the inflammasome to release IL-1β and TNF cytokines, which are mediators of inflammation in both cancer and atherosclerosis [49-52].
Although it has been suggested that oxidized-LDL plays a role in cancer development [53], little is known about the role that oxidized and desialylated LDL plays in antitumor immunity. It has been previously shown that accumulation of native LDL in γδ T cells leads to reduced antitumor function [19]. A different study showed that when LDL becomes oxidized by polymorphonuclear lymphocytes, it inhibits NK cell cytotoxicity [23]. However, to our knowledge the role of LDL that is both oxidized and desialylated on the cytotoxicity of LAK cells has been unknown until now. Oxidation and desialylation are characteristic features of LDL in atherosclerosis [29]; the meaning of this in cancer immunity is still not well understood. We found that oxidized-desialylated LDL is taken up by LAK cells more extensively than native LDL after 16 hours of incubation, which leads to reduction of LAK cell cytotoxicity. This reduction in cytotoxicity was not, however, accompanied by a shift in the mechanism of K562 cell death; it was fully apoptotic under all experimental conditions tested (Figure 2). This observation was in line with previous studies on the mechanism by which LAK cells kill cancer cells [54-56].
The data presented here showed that the expression of the cytotoxicity-associated receptors CD56 and NKG2D, which determine the ability of LAK cells to mediate cancer cell lysis [57], was downregulated upon exposure of LAK cells to oxidized-desialylated LDL. This occurred in pre-expanded LAK cells that were kept on IL-2 stimulation during the incubation with oxidized-desialylated LDL. Thus IL-2 stimulation was not able to compensate for the inhibitory effect of oxidized-desialylated LDL. We also found an impairment in the production of IFNγ, a pleiotropic cytokine secreted by LAK cells. These results, however, differ according to the use of different cytotoxic lymphocytes. For instance, a different study found that native LDL can lower the expression of CD56 and NKG2D cytotoxicity receptors and IFN-γ in γδT cells [19]. However, we observed the opposite in that native LDL induced a small increase in the expression of CD56, NKG2D, and IFN-γ in LAK cells. Nevertheless, previous studies and our work show that enhanced uptake of LDL by cytotoxic immune cells can impair their antitumor capacity.
Multiple modified LDL has been extensively studied in the context of atherosclerosis, and how it can contribute to the development of this disease [58]. Although it is unknown how LDL becomes multiply modified in atherosclerosis, our findings suggest that the desialylated and oxidized LDL that is produced as a byproduct of atherosclerosis may at least partially inhibit the ability of the immune system to effectively surveil against cancer cells. Thus, the data presented here suggests at least one particular mechanistic link between atherosclerosis and cancer. Future research investigating the link between multiple modified LDL and cancer immunity will help establish additional details on the complex connection between these two devastating chronic diseases.
Materials and Methods
Cell culture and in vitro cytotoxicity assay
For LAK cell culture and expansion, peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation (Ficoll-Paque-GHC-17-1440-02; GE Healthcare) from Trima Residual Apheresis collection kits (RE202-Blood Centers of the Pacific). Trima residuals were centrifuged at 1200 g for 20 minutes at 15 °C. Isolated PBMCs were cultured in serum free X-VIVO 10 medium with Gentamicin L-Gln and Phenol Red (04-380Q; Lonza) for 5 days in the presence of interleukin-2 (IL-2, 589102; BioLegend) at 0.1 μg/ml. At day 5, fresh media was added along with fresh IL-2, and LAK cell populations were cultured for 72 hours in the presence or absence of native low-density lipoprotein (LDL, 12-16-120412; 50 μg/ml, Athens Research & Technology) or oxidized-desialylated low-density lipoprotein, and LAK cell cytotoxicity was evaluated in an in vitro killing assay. Cells were counted via hemocytometer and Trypan blue solution (0.4%). K562 leukemia cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS, SER-500, Zen-Bio). For the in vitro killing assays, about 800,000 LAK cells were washed three times in 2 ml serum free X-VIVO 10 media and seeded in a V-bottom 96 well plate. K562 cells were washed two times with 5 ml 1X PBS and stained with CFSE green dye (0.25 μmol/L in 1X PBS; Thermo Scientific) for 10 minutes at room temperature. Eighty microliters of target K562 cells at 106 cells/ml were added to a V-bottom 96 well plate containing 100 μl of the LAK cells at a 10:1 effector to target ratio, and incubated for 4 hours at 37 °C and 5% CO2. Immediately afterward cells were centrifuged at 800 × g for 10 minutes and reconstituted in 700 μl of 1X PBS, and to this was added 2 μl of a 1 mg/ml propidium iodide solution (P3566; Thermo Scientific) which was incubated for 20 minutes and subsequently analyzed by flow cytometry.
Cell Death Analysis
About 2 × 106 K562 cells were incubated with CellTrace Violet (C34571, Thermo Fisher Scientific) at a final working concentration of 20 μM and incubated for 30 minutes. Then, labeled K562 cells were washed once with 5 ml sterile 1X PBS and resuspended in RPMI medium containing 10% FBS, 5 μM CellEvent Caspase-3/7 Green Detection Reagent (C10723, Thermo Fisher Scientific) and 1 μg/ml propidium iodide. LAK cells were incubated with different forms of LDL and prepared as described above, and then they were reconstituted in X-VIVO 10 media containing 5 μM CellEvent and 1 μg/ml propidium iodide. These LAK cells were co-incubated with CellTrace-loaded K562 cells for 4 hours at a 10:1 ratio, as described above. After incubation, samples were imaged on a Nikon Ti-2, inverted fluorescence microscope using a 20 lens with N.A. of 0.75. Each fluorescent channel and differential interference contrast (DIC) images were captured sequentially. Excitation/emission wavelengths for the blue (CellTrace), green (CellEvent) and red (propidium iodide) channels were 405/450, 488/525, and 561/600 nm, respectively. Two random fields were imaged for each sample.
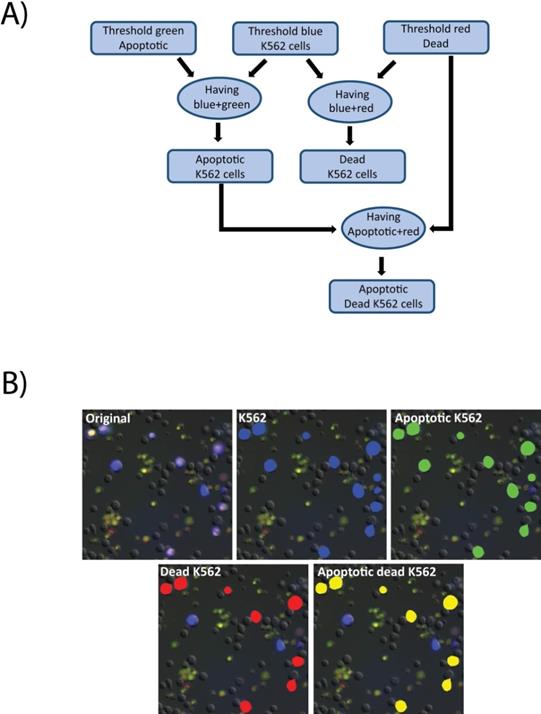
Images were analyzed using the Nikon Elements software. First a threshold was applied to the raw images in the blue channel to detect all K562 cells. Then the green and red channels were thresholded to define positive signal indicating apoptotic and dead cells, respectively. The “having” command was then used to select all detected K562 cells that contained any above-threshold green signal. The results were identified as apoptotic K562 cells. Similarly, having was employed to detect K562 cells with above-threshold red signal, to select dead K562 cells. Finally, having was applied to identified apoptotic K562, and red signal to identify K562 cells that were dead via apoptosis (Figure 7A). A cropped field in shown in Figure 7B indicating the output of the analysis steps. The initial image is an overlay of the fluorescence and DIC channels (7B, original), identified K562 cells are identified by a blue mask (7B, K562), the results of K562 cells that also contain the apoptosis indicator are masked in green (7B, Apoptotic K562), K562 positive for propidium iodide, indicating loss of membrane integrity are masked in red (7B, Dead K562), and the K562 positive for both indicators are indicated in yellow (7B, Apoptotic K562).
Cell death image analysis pipeline. A) A macro was created in the Nikon Elements software for image analysis. First the images were thresholded in the green, blue, and red channels representing, respectively, the apoptotic cells (labeled with CellEvent), the K562 cells (labeled with CellTrace Violet), and the dead cells as defined as loss of membrane integrity (labeled with propidium iodide). The having command was used to identify apoptotic K562 cells and dead K562 cells. Having was then used again to identify K562 cells that were dually positive for apoptosis and death indicators. B) Raw images (original) were processed through the pipeline to identify K562 cells (blue mask), apoptotic cells (green mask), dead K562 cells (red mask) and Apoptotic, dead K5562 (yellow mask).
Desialylation of low-density lipoprotein and glycan node analysis
In vitro desialylation of native low-density lipoprotein (LDL) was performed by incubating 50 μl of a 7.11 mg/ml LDL stock with 100 μl of 0.1M sodium acetate buffer pH 5, plus 50 μl of neuraminidase enzyme from Clostridium perfringens (1 U/ml, 11585886001; SIGMA) or 50 μl of water for the control. Total volume was brought up to 250 μl with water, and samples were incubated at 37 0C for 24 hours. To remove neuraminidase enzyme, samples were spin filtered using Amicon Ultra-0.5 Centrifugal 100 kD spin filter devices (UFC510024; Fisher Scientific). Samples were reconstituted in 450 μl of 10 mM HEPES in 0.15 M NaCl pH 7 buffer each time for a total of 4 spin throughs. To verify complete desialylation of LDL, the glycan node analysis method [33-38] was used. Glycan node analysis is a procedure based on glycan methylation analysis by which pooled glycans within whole biological samples are deconstructed in a way that conserves their monosaccharide and linkage information [33]. For this procedure, 60 μg of LDL were aliquoted, and to this was added 270 μl of DMSO and 105 μl of iodomethane. Then samples were added to sodium hydroxide beads for 11 minutes to drive permethylation. The rest of the steps for glycan linkage analysis of intact, complex biospecimens are described elsewhere [33-38]. An increase in the relative abundance of terminal galactose and a near-complete loss of 6-linked galactose signals (which arise solely due to terminal sialylation of glycans) were used to verify desialylation of LDL [33].
Oxidation of desialylated low-density lipoprotein and TBARS assay
A 200 μM copper II chloride solution was prepared using acetic acid pH 4. Desialylated LDL and/or native LDL at 1.7 mg/ml was incubated with 10 μM copper II chloride overnight at 4 °C to generate oxidized-desialylated and/or oxidized only LDL. For the control, native LDL was incubated with a blank acetic acid solution pH 4 at 4 °C overnight. Oxidation was verified with the thiobarbituric acid reactive substances (TBARS) assay, as described by Aguilar Diaz de leon & Borges [59]. Briefly, to 100 μl of an LDL sample (60 μg) or calibration standards were added 200 μl of 8.1% sodium dodecyl sulfate, 1.5 ml of 20% acetic acid solution adjusted to pH 4 with NaOH, and 1.5 ml of 0.8 % aqueous solution of thiobarbituric acid. Final volume was brought up to 4 ml with water. The mixture was heated at 95 °C for 1 hour and centrifuged at 1600 g at 4 °C. One hundred fifty microliters of supernatant was transferred to a 96 well plate, and the absorbance was measured immediately at 532 nm.
Flow-cytometry analysis
For antibody staining, LAK cells and PBMCs were labeled with monoclonal antibodies: anti-CD3, anti-CD56, and APC anti-human CD314 (NKG2D) (1D11, BioLegend). The percentage of CD3, CD56, and NKG2D positive cells was evaluated by flow cytometry in a ThermoFisher Attune NxT flow cytometer and FlowJo software. For LDL uptake measurements, 900,000 LAK cells were seeded in a 96 well plate with 225 μl X-VIVO 10 media and incubated with either native or oxidized-desialylated pHrodo Green LDL (L34355, Thermo Scientific) at 10 μg/ml for 1, 2, 8, 16, 32, and 72 hours. LAK cells were washed three times with 2 ml serum free X-VIVO 10 media, reconstituted in 700 μl 1X PBS, and immediately analyzed by flow cytometry.
Enzyme Linked Immunosorbent Assay (ELISA) of Interferon Gamma (IFNϒ)
IFNγ was quantitatively measured in undiluted samples of LAK culture supernatant. The wells of a high binding flat bottom 96-well plate (Costar, Washington, D.C.) were first coated overnight at 4 °C with 100 µl of 1 µg/mL solution of α-Human IFNγ monoclonal antibody (Clone 1-D1K, Mabtech, Sweden) diluted in bicarbonate buffer (1 to 100 dilution), pH 9.8. The wells were then washed 5 times with 200 μl PBS-T buffer. Next, 200 µl of 5% milk in PBS-T blocking buffer was incubated in each well for 1 hour at room temperature. Standards were diluted in PBS-T containing 5% powered milk and ranged from 2 ng/mL to 34 pg/mL of recombinant human IFNγ (R&D Systems, Minneapolis, MN). One hundred microliters of cell culture supernatants were plated in triplicate at dilutions of 1:10, 1:100, and 1:500 in 200 μl PBS-T 5% milk and incubated for 2 hours at room temperature. Samples were washed 5 times with 200 μl PBS-T. Biotinylated, secondary α-Human IFNγ monoclonal antibody (Clone 7-B6-1, Mabtech, Sweden) was diluted to 1 µg/mL in PBS-T containing 5% powered milk and 100 µl was incubated in each well overnight at 4 °C. Samples were once again washed 5 times with PBS-T, after which Precision Protein StrepTactin-HRP Conjugate (Bio-Rad, Hercules, CA) was diluted 1:250 in PBS-T containing 5% powered milk and 100 µl incubated in each well for 30 minutes at room temperature. After 5 more washes with PBS-T, 100 µl of TMB substrate (Thermo Scientific, Waltham, MA) was added to each well and allowed to incubate until a visible gradient developed in the standards, which was about 5 minutes. Subsequently, 100 µl of 2N sulfuric acid stop solution was added and the plate was read at 450 nm immediately.
Statistical analysis
Statistical analysis was performed using two-way ANOVA with multiple comparison test for comparison of experimental groups with cytotoxicity assays, TBARS assays, and flow cytometry analysis. For glycan linkage analysis, each uniquely linked monosaccharide was quantified by integrating extracted ion chromatogram peak using QuanLynx software. Integrated peaks were exported to an Excel spreadsheet. Statistical significance between glycan nodes was determined using two-tailed t-tests. Statistical analysis was performed using GraphPad.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Dr. Douglas Lake for helpful comments on the manuscript. JAD was supported in part by the Initiative for Maximizing Student Development (IMSD) program. Microscopy was done in the Advanced Light Microscopy Facility at Arizona State University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. García-Muñoz R, López-Díaz-de-Cerio A, Feliu J, Panizo A, Giraldo P, Rodríguez-Calvillo M, Grande C, Pena E, Olave M, Panizo C, Inogés S. Follicular lymphoma: in vitro effects of combining lymphokine-activated killer (LAK) cell-induced cytotoxicity and rituximab- and obinutuzumab-dependent cellular cytotoxicity (ADCC) activity. Immunol Res. 2016;64:548-557
2. López-Díaz de Cerio A, García-Muñoz R, Pena E, Panizo Á, Feliu J, Giraldo P, Rodríguez-Calvillo M, Martínez-Calle N, Grande C, Olave MT, Andrade-Campos M, Bandrés E, Núñez-Córdoba JM, Inogés S, Panizo C. Maintenance therapy with ex vivo expanded lymphokine-activated killer cells and rituximab in patients with follicular lymphoma is safe and may delay disease progression. Br J Haematol. 2020;189:1064-1073
3. Wrangle JM, Patterson A, Johnson CB, Neitzke DJ, Mehrotra S, Denlinger CE, Paulos CM, Li Z, Cole DJ, Rubinstein MP. IL-2 and beyond in Cancer Immunotherapy. J Interf Cytokine Res. 2018;38:45-68
4. Kjærgaard J, Hokland ME, Agger R, Skovbo A, Nannmark U, Basse PH. Biodistribution and tumor localization of lymphokine-activated killer T cells following different routes of administration into tumor-bearing animals. Cancer Immunol Immunother. 2000;48:550-560
5. Wu J, Lanier LL. Natural Killer Cells and Cancer. Adv Cancer Res. 2003;90:127-156
6. Kim GG, Donnenberg VS, Donnenberg AD, Gooding W, Whiteside TL. A novel multiparametric flow cytometry-based cytotoxicity assay simultaneously immunophenotypes effector cells: Comparisons to a 4 h 51Cr-release assay. J Immunol Methods. 2007;325:51-66
7. Saito H, Ando S, Morishita N, Lee KM, Dator D, Dy D, Shigemura K, Adhim Z, Nibu KI, Fujisawa M, Shirakawa T. A Combined Lymphokine-activated Killer (LAK) cell immunotherapy and adenovirus-p53 gene therapy for head and neck squamous cell carcinoma. Anticancer Res. 2014;34:3365-3370
8. Spear P, Wu MR, Sentman ML, Sentman CL. Nkg2d ligands as therapeutic targets. Cancer Immun. 2013;13:1-14
9. Eleme K, Taner SB, Önfelt B, Collinson LM, McCann FE, Chalupny NJ, Cosman D, Hopkins C, Magee AI, Davis DM. Cell Surface Organization of Stress-inducible Proteins ULBP and MICA That Stimulate Human NK Cells and T Cells via NKG2D. J Exp Med. 2004;199:1005-1010
10. Surgeons N, Disease C, Insti- N. Lymphokine-Activated Killer (LAK) Cells. Cell Immunol. 1985;134:296-313
11. Fagan EA, Feddleston ALW. Immunotherapy for cancer: The use of lymphokine activated killer (LAK) cells. Gut. 1987;28:113-116
12. Toh U, Yamana H, Sueyoshi S, Tanaka T, Niiya F, Katagiri K, Fujita H, Shirozou K, Itoh K. Locoregional cellular immunotherapy for patients with advanced esophageal cancer. Clin Cancer Res. 2000;6:4663-4673
13. Qian X, Wang X, Jin H. Cell transfer therapy for cancer: past, present, and future. J Immunol Res. 2014;2014:525913
14. Polhill T, Zhang GY, Hu M, Sawyer A, Zhou JJ, Saito M, Webster KE, Wang Y, Wang Y, Grey ST, Sprent J, Harris DCH, Alexander SI, Wang YM. IL-2/IL-2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J Am Soc Nephrol. 2012;23:1303-1308
15. Lotze MT, Matory YL, Rayner AA, Ettinghausen SE, Vetto JT, Seipp CA, Rosenberg SA. Clinical effects and toxicity of interleukin-2 in patients with cancer. Cancer. 1986;58:2764-2772
16. Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res. 2019;25:2392-2402
17. Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, Von Gunten S, Tzankov A, Tietze L, Lardinois D, Heinzelmann-Schwarz V, Von Bergwelt-Baildon M, Zhang W, Lenz HJ, Han Y, Amos CI, Syedbasha M, Egli A, Stenner F, Speiser DE, Varki A, Zippelius A, Läubli H. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128:4912-4923
18. Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. 2014;10:69-75
19. Rodrigues N V, Correia D V, Mensurado S, Nobrega-Pereira S, deBarros A, Kyle-Cezar F, Tutt A, Hayday AC, Norell H, Silva-Santos B, Dias S. Low-density lipoprotein uptake inhibits the activation and antitumor functions of human Vg9Vd2 T cells. Cancer Immunol Res. 2018;6:448-457
20. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CCY, Chang TY, Zhang T, Zhou P, Song BL, Liu W, Sun SC, Liu X, Li BL, Xu C. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531:651-655
21. Lagadari M, Truta-Feles K, Lehmann K, Berod L, Ziemer M, Idzko M, Barz D, Kamradt T, Maghazachi AA, Norgauer J. Lysophosphatidic acid inhibits the cytotoxic activity of NK cells: Involvement of Gs protein-mediated signaling. Int Immunol. 2009;21:667-677
22. Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, Tai LH. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer. 2019;19:11-14
23. Tanabe F, Sato A, Ito M, Ishida E, Ogata M, Sigeta S. Low-density lipoprotein oxidized by polymorphonuclear leukocytes inhibits natural killer cell activity. J Leukoc Biol. 1988;43:204-210
24. Grewal T, Bartlett A, Burgess JW, Packer NH, Stanley KK. Desialylated LDL uptake in human and mouse macrophages can be mediated by a lectin receptor. Atherosclerosis. 1996;121:151-163
25. Harada LM, Carvalho MD t, Passarelli M, Quintão EC r. Lipoprotein desialylation simultaneously enhances the cell cholesterol uptake and impairs the reverse cholesterol transport system: In vitro evidences utilizing neuraminidase-treated lipoproteins and mouse peritoneal macrophages. Atherosclerosis. 1998;139:65-75
26. Tertov V V, Orekhov AN. Metabolism of native and naturally occurring multiple modified low density lipoprotein in smooth muscle cells of human aortic intima. Exp Mol Pathol. 1997;64:127-145
27. Filipovic I, Buddecke E. Desialized Low-Density Lipoprotein Regulates Cholesterol Metabolism in Receptor-Deficient Fibroblasts. Eur J Biochem. 1979;101:119-122
28. Filipovic I, Schwarzmann G, Az W, Wiegandt H, Buddecke E. Sialic-Acid Content of Low-Density Lipoproteins Controls Their Binding and Uptake by Cultured Cells. Eur J Biochem. 1979;93:51-55
29. Orekhov AN, Ivanova EA, Melnichenko AA, Sobenin IA. Circulating desialylated low density lipoprotein. Cor Vasa. 2017;59:149-156
30. Alipov VI, Sukhorukov VN, Karagodin VP, Grechko AV, Orekhov AN. Chemical composition of circulating native and desialylated low density lipoprotein: what is the difference? Vessel Plus. 2017:107-115
31. Levitan I, Volkov S, Subbaiah P V. Comprehensive invited review. Oxidized LDL: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid Redox Signal. 2010;13:39-75
32. Tapia-Vieyra JV, Delgado-Coello B, Mas-Oliva J. Atherosclerosis and Cancer; A Resemblance with Far-reaching Implications. Arch Med Res. 2017;48:12-26
33. Borges CR, Rehder DS, Boffetta P. Multiplexed surrogate analysis of glycotransferase activity in whole biospecimens. Anal Chem. 2013;85:2927-2936
34. Zaare S, Aguilar JS, Hu Y, Ferdosi S, Borges CR. Glycan node analysis: A bottom-up approach to glycomics. J Vis Exp. 2016;2016:1-11
35. Ferdosi S, Rehder DS, Maranian P, Castle EP, Ho TH, Pass HI, Cramer DW, Anderson KS, Fu L, Cole DEC, Le T, Wu X, Borges CR. Stage Dependence, Cell-Origin Independence, and Prognostic Capacity of Serum Glycan Fucosylation, β1-4 Branching, β1-6 Branching, and α2-6 Sialylation in Cancer. J Proteome Res. 2018;17:543-558
36. Hu Y, Ferdosi S, Kapuruge EP, Diaz de Leon JA, Stücker I, Radoï L, Guénel P, Borges CR. Diagnostic and Prognostic Performance of Blood Plasma Glycan Features in the Women Epidemiology Lung Cancer (WELCA) Study. J Proteome Res. 2019;18(11):3985-3998
37. Aguilar Díaz de león JS, Borges C. Glycosylation Profiling of Glycoproteins Secreted from Cultured Cells Using Glycan Node Analysis and GC-MS. In: Methods in Molecular Biology. 2021;2271:317-330
38. Walker SA, Aguilar Díaz De león JS, Busatto S, Wurtz GA, Zubair AC, Borges CR, Wolfram J. Glycan Node Analysis of Plasma-Derived Extracellular Vesicles. Cells. 2020;9:1946
39. Summerhill VI, Grechko A V, Yet SF, Sobenin IA, Orekhov AN. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int J Mol Sci. 2019;20:3561
40. Del Zotto G, Marcenaro E, Vacca P, Sivori S, Pende D, Della Chiesa M, Moretta F, Ingegnere T, Mingari MC, Moretta A, Moretta L. Markers and function of human NK cells in normal and pathological conditions. Cytom Part B - Clin Cytom. 2017;92:100-114
41. Ni L, Lu J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018;7:4509-4516
42. Ross J, Stagliano N, Donovan M, Breitbart R, Ginsburg G. Atherosclerosis and Cancer: Common Molecular Pathways of Disease Development and Progression. Ann N Y Acad Sci. 2001;947:271-293
43. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-444
44. Panth N, Paudel KR, Parajuli K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv Med. 2016;2016:1-12
45. Khan A, Alsahli M, Rahmani A. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med Sci. 2018;6:33
46. Tomas L, Edsfeldt A, Mollet IG, Matic LP, Prehn C, Adamski J, Paulsson-Berne G, Hedin U, Nilsson J, Bengtsson E, Gonçalves I, Björkbacka H. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur Heart J. 2018;39:2301-2310
47. Delimaris I, Faviou E, Antonakos G, Stathopoulou E, Zachari A, Dionyssiou-Asteriou A. Oxidized LDL, serum oxidizability and serum lipid levels in patients with breast or ovarian cancer. Clin Biochem. 2007;40:1129-1134
48. Jiménez-Corona AE, Damián-Zamacona S, Pérez-Torres A, Moreno A, Mas-Oliva J. Osteopontin Upregulation in Atherogenesis Is Associated with Cellular Oxidative Stress Triggered by the Activation of Scavenger Receptors. Arch Med Res. 2012;43:102-111
49. DamiaÂn-Zamacona S, Toledo-Ibelles P, Ibarra-Abundis M, Uribe-Figueroa L, HernaÂndez-Lemus E, Macedo-Alcibia KP, Delgado-Coello B, Mas-Oliva J, Reyes-Grajeda JP. Early transcriptomic response to ldl and oxldl in human vascular smooth muscle cells. PLoS One. 2016;11:1-22
50. Guo H, Callaway JB, Ting JPY. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677-687
51. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nũez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-1361
52. Pittet MJ, Swirski FK. Monocytes link atherosclerosis and cancer. Eur J Immunol. 2011;41:2519-2522
53. Cedó L, Reddy ST, Mato E, Blanco-Vaca F, Escolà-Gil JC. HDL and LDL: Potential New Players in Breast Cancer Development. J Clin Med. 2019;8:853
54. Shemtov MM, Cheng DL, Kong L, Shu WP, Sassaroli M, Droller MJ LB. LAK cell mediated apoptosis of human bladder cancer cells involves a pH-dependent endonuclease system in the cancer cell: possible mechanism of BCG therapy. J Urol. 1995;154:269-274
55. Kwak JY, Han MK, Choi KS, Park IH, Park SY, Sohn MH, Kim UH, McGregor JR, Samlowski WE YC. Cytokines secreted by lymphokine-activated killer cells induce endogenous nitric oxide synthesis and apoptosis in DLD-1 colon cancer cells. Cell Immunol. 2000;203:84-94
56. Yamamoto T, Ueta E OT. Apoptosis induction by interleukin-2-activated cytotoxic lymphocytes in a squamous cell carcinoma cell line and Daudi cells - involvement of reactive oxygen species-dependent cytochrome c and reactive oxygen species-independent apoptosis-inducing factors. Immunology. 2003;110:217-224
57. Liu H, Wang S, Xin J, Wang J, Yao C, Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am J Cancer Res. 2019;9:2064-2078
58. Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, Daemen MJ, Demer LL, Hegele RA, Nicholls SJ, Nordestgaard BG, Watts GF, Bruckert E, Fazio S, Ference BA, Graham I, Horton JD, Landmesser U, Laufs U, Masana L, Pasterkamp G, Raal FJ, Ray KK, Schunkert H, Taskinen MR, van de Sluis B, Wiklund O, Tokgozoglu L, Catapano AL, Ginsberg HN. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313-2330
59. Aguilar Diaz De Leon J, Borges CR. Evaluation of oxidative stress in biological samples using the thiobarbituric acid reactive substances assay. J Vis Exp. 2020;2020:1-10
Author contact
![]() Corresponding author: Chad R. Borges, E-mail: chad.borgesedu.
Corresponding author: Chad R. Borges, E-mail: chad.borgesedu.