Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(14):4247-4256. doi:10.7150/jca.51657 This issue Cite
Research Paper
Downregulation of THRSP Promotes Hepatocellular Carcinoma Progression by Triggering ZEB1 Transcription in an ERK-dependent Manner
Qiong Hu1 ![]() , Xiaolu Ma2, Chuner Li1, Chenhao Zhou1, Jiayao Chen1, Xuechun Gu1
, Xiaolu Ma2, Chuner Li1, Chenhao Zhou1, Jiayao Chen1, Xuechun Gu1
1. Department of Laboratory Medicine, Zhoushan Hospital of Zhejiang Province, Zhoushan, Zhejiang, 316021, China.
2. Department of Clinical Laboratory, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical School, Fudan University, Shanghai, 200032, China.
Received 2020-8-7; Accepted 2021-5-1; Published 2021-5-19
Abstract

Background: Hepatocellular carcinoma (HCC) is a major leading cause of cancer mortality worldwide. Thyroid hormone responsive (THRSP) gene is primarily known for regulating responses to thyroid hormones, but its expression has been correlated with differential outcomes in some cancers. To date, however, its role in the progression of HCC remains unknown.
Methods: The mRNA and protein expression of THRSP was measured in HCC tissues and cell lines via qPCR and western blot assays. Lentiviral transfection was used to establish stable cell lines overexpressing THRSP and shRNA was used to silence THRSP. The effects of THRSP on cell growth were then determined in vivo and in vitro. Cell migration and invasion of HCC cells were investigated using transwell and wound healing assays.
Results: In tissue samples from patients, HCC tissues had decreased THRSP expression relative to adjacent healthy tissues. Further, patients with decreased THRSP protein and mRNA expression had worse outcomes. Knockdown of THRSP led to increased cell growth, migration, and invasion of HCC cells, and THRSP overexpression exerted an anti-tumor effect in vivo and in vitro. We found that increased expression of THRSP inhibited hepatocellular carcinogenesis by inhibiting the process of epithelial-to-mesenchymal transition through acting on the ERK/ZEB1 signaling pathway.
Conclusion: THRSP may act as a functional tumor suppressor and was frequently reduced in HCC tissue samples. We identified a novel pathway for the THRSP/ERK/ZEB1-regulated suppression of HCC tumorigenesis and invasion. Restoring THRSP expression may represent a promising approach for HCC therapies.
Keywords: Hepatocellular carcinoma, Thyroid hormone responsive gene, epithelial to mesenchymal phenotypes, HCC therapies
Introduction
Hepatocellular carcinoma (HCC), the most common type of primary liver cancer, ranks as the fifth leading cause of cancer mortality worldwide [1]. Surgical resection remains the preferred curative therapy for HCC patients. Recently, the potential combination of immunotherapies and targeted therapies has provided additional treatment alternatives for HCC. However, the high frequency of tumor metastasis or recurrence still exceeds 50%, largely due to the significant heterogeneity observed in HCC [2-5]. Therefore, it is essential to understand the biological mechanisms and related signaling pathways that are associated with HCC progression in order to improve the prognosis and survival of HCC patients.
The thyroid hormone responsive (THRSP), also known as spot 14, was originally identified as a regulator that responds to thyroid hormones [6]. This gene was important for epithelial cell proliferation and intracellular lipogenesis in the mammary glands [7]. Wellberg et al. previously reported that the THRSP exerted extensive amplification in breast cancers and was highly expressed in breast tumor tissues [8]. Indeed, the high expression of THRSP in primary invasive breast cancer was proved to predict the time to tumor recurrence, with greater expression associated with less recurrence-free survival. Moreover, THRSP can also mediate progesterone-induced adipogenesis and promote the growth of breast cancer cells [9-11]. Recently, studies have shown that THRSP is highly expressed in human liver tissues. It has been reported that THRSP, given its role in regulating lipid metabolism, plays an important role in the hepatocytes and is involved in the pathogenesis of nonalcoholic fatty liver disease [12-13]. However, the role or mechanism of THRSP in the progression of HCC remains unclear.
In this study, data from public databases and biological experiments were combined to investigate the mechanism by which THRSP influences liver cancer. In contrast with prior findings in breast cancer, we observed that THRSP expression was frequently reduced in HCC and that down-regulation of THRSP was positively correlated with recurrence, larger tumor size, and more aggressive phenotype. Mechanistically, THRSP inhibited hepatocellular carcinoma progression by inhibiting the ERK/ZEB1 pathway induced EMT process.
Materials and Methods
HCC tissues
We recruited 10 pairs of HCC tissue samples, and corresponding adjacent non-cancerous tissue samples, along with related clinical information from patients undergoing surgery at the department of oncology, Fudan University Shanghai Cancer Center between December 2019 and February 2020. Signed informed consents were obtained from all subjects.
Cell culture
L02, Huh7, HepG2 and Hep3B cells were purchased from the Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, Shanghai, China), and HCCLM3 cells were obtained from the Shanghai Zhongshan Hospital, Fudan university. All cell lines were confirmed to be free of mycoplasma contamination. The cells were maintained in Dulbecco's modifed Eagle's medium (DMEM, Gibco, Thermo Fisher Scientifc, Cambridge, MA, USA) supplemented with 10% bovine growth serum, 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco) and cultured in an incubator at 37 °C, with 5% CO2.
Lentivirus
Lentiviruses for THRSP overexpression were constructed by Merdobio Corporation (Shanghai, China). Those carrying the THRSP shRNA, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For stable cell line construction, Huh7 cells were transfected with THRSP-overexpressing or Hep3B cells with THRSP-silencing lentivirus at a multiplicity of infection (MOI) of 20, followed by selection using 2.5 µg/ml puromycin for 2 months. Non-stable clones were removed due to poor tolerance to puromycin.
Western blot
Western blotting was performed following previous studies [14]. The utilized antibodies included anti-THRSP (1:500, sc-516102; Santa Cruz), anti-phospho-ERK (Tyr202/Tyr204) (1:1000, 4377S; CST), anti-ERK (1:1000, 4695S; CST), anti-Slug (1:1000, 9585S; CST), anti-Snail (1:1000, 3879S; CST), anti-Twist (1:1000, 46702S; CST), anti-ZEB1 (1:1000, 70512S; CST), anti-ZEB2 (1:1000, 3396S; CST). anti-TGF-beta (1:1000, 5544S; CST), anti-AKT (1:1000, 4685S; CST), anti-phospho-AKT (1:1000, 4060S; CST), anti-E-cadherin (1:1000, 14472S; CST), and anti-N-cadherin (1:1000, 13116S; CST), anti-Vimentin (1:1000, 5741S; CST), Anti-GAPDH (1:3000, 5174S; CST) was used as a loading control.
Real-time PCR
Total mRNA was extracted using the RNA Extraction Kit (Takara Bio Inc., Shiga, Japan) following the manufacturer's protocol. Reverse-transcription assays were performed using the PrimeScript™ RT Reagent Kit (Takara Bio Inc., Shiga, Japan). Then qRT-PCR was carried out using SYBR Premix Ex Taq™ (Takara Bio Inc., Shiga, Japan). All the primers were listed in Supplementary Table 1.
Cell counting Kit-8 (CCK-8) assays
Stably infected cells were cultured in 96-well plates and maintained in incubator at 37 °C for overnight. Next day, the cell supernatant was removed and replaced with a fresh medium, containing the CCK-8 reagent (10 μL/well) and the complete DMEM medium (90 μL/well). After 2 h, the absorbance value was measured at 450 nm.
Wound-healing assays
Stably infected cells were cultured in 6-well plates and maintained until cell fusion occurred. The bottom of the plate was scratched with a 100 μL pipette tip. The floating cells were washed out and pictures of wound closure were taken at 0 and 24 h, respectively.
Colony formation assays
Stably infected cells were seeded in 6-well plates. After 14 days, the cells were fixed with 4% paraformaldehyde and stained with purple crystal. The size of the surviving colonies was observed and calculated. Photographs of colonies were collected.
Transwell migration and invasion assays
The invasion and migration ability of cells were studied using Matrigel-coated (BD Biosciences, Franklin Lakes, NJ, USA) or non-Matrigel-coated Transwells. The serum was used as chemoattractant. Thus, Stable cell lines were cultured in the upper chamber of the transwells and supplemented with serum-free medium. DMEM medium supplemented with 10% FBS was added into the lower plate wells. After 24 hours of incubation, the upper filter with the cancer cells was removed and the cells that had migrated to the lower chamber were fixed, stained, and counted.
Mouse model of HCC
Male BALB/c nude mice (4-6 weeks old) were purchased from the Experimental Animal Center of Shanghai Institute. Subcutaneous xenograft tumors were established as follows: 5 × 106 cells were diluted in 0.15 mL of phosphate buffered saline and then injected subcutaneously into the flanks of nude mice. The size of the subcutaneous tumor was measured once per week as: length×width×width×0.5. After 6 weeks, the animals were sacrificed. The volume of the tumor was measured. Animal care and experimental protocols were conducted under guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at Shanghai Cancer Center, Fudan University.
Statistical analysis
All experiments were repeated at least three times in order to confirm the results. The data are reported as mean ± standard error of the mean (SEM). The Student's t-test or Wilcoxon matched-pairs test used SPSS 22.0 (SPSS, Chicago, IL, USA) software and Prism 6.0 (GraphPad Software, La Jolla, CA, USA). The difference was considered significant at P < 0.05.
Results
Low expression of THRSP predicts poor prognosis in HCC
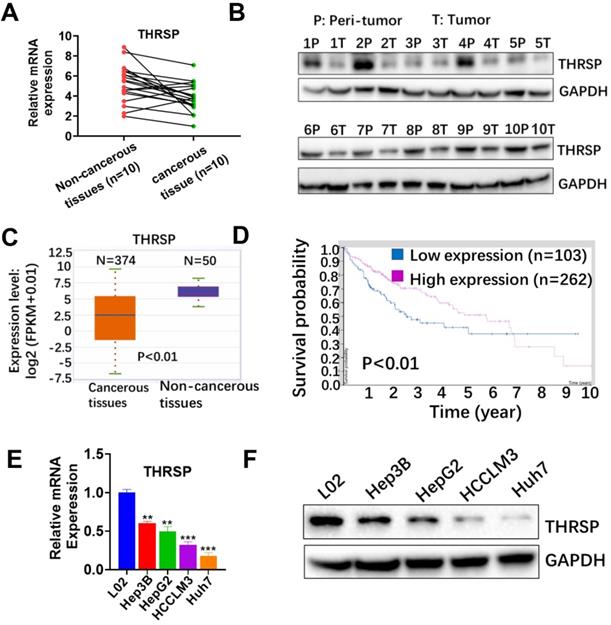
To verify the clinical significance of THRSP in HCC, we first determined the expression of THRSP in both cancer tissues and paired adjacent non-cancerous tissues using qRT-PCR and western blot (WB). We found that mRNA and protein expression of THRSP were significantly decreased in the majority of cancerous tissues, compared with paired non-cancerous tissues (Fig. 1A and 1B). These results were consistent with data from the TCGA database (http://starbase.sysu.edu.cn/panCancer). According to TCGA database, mRNA expression of THRSP was down-regulated in 374 patients with liver cancer, compared with 50 normal samples (P<0.01, Fig 1C). Additionally, HCC patients with low THRSP expression had worse overall survival than patients with high THRSP expression according to TCGA database (Protein Atlas database (https://www.proteinatlas.org; Fig. 1D). Moreover, downregulation of THRSP was validated by qPCR and WB analyses in liver cancer cell lines relative to normal liver cells (Fig. 1E and 1F). These data indicated that THRSP was frequently decreased in HCC and low THRSP levels were related with poor prognoses.
THRSP is downregulated in human HCC and predicted poor prognosis. A. mRNA levels of THRSP were analyzed in 10 HCC tissue samples, compared with adjacent non-cancerous samples. B. Protein levels of THRSP were analyzed in HCC tissue samples and adjacent non-cancerous tissues. C. Relative THRSP expression levels in 374 HCC patients and 50 normal samples from The Cancer Genome Atlas database. D. A Kaplan-Meier analysis of overall survival (OS) in patients with different expression level of THRSP. E. Relative THRSP levels in L02 and four HCC cells were analyzed using qRT-PCR assay. F. Relative THRSP levels in L02 and four HCC cells were analyzed using western blotting. Data are means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
THRSP suppresses cell proliferation in HCC
To further determine the biological effect of THRSP in HCC cells, we transfected HCCLM3 and Huh7 cell lines (THRSP-low) with lentivirus to overexpress THRSP. In addition, stable models of THRSP knockdown in Hep3B and HepG2 cell lines (THRSP-high) were established with short hairpin RNAs (shRNA). The efficiency of transfection was determined by RT-PCR and WB assays. We found that THRSP was successfully overexpressed (Fig. 2A), and shTHRSP-3 led to significant silencing, as expected (Fig. 2B). Therefore, shTHRSP-3 was used for following experiments. Meanwhile, the normal cells were used as control groups (Ctrl). In vitro assays showed that the overexpression of THRSP restrained the activity of HCCLM3 and Huh7 cells and inhibited proliferation, as shown in a CCK8 experiment (Fig. 2C). In contrast, downregulation of THRSP promoted proliferation of Hep3B and HepG2 cells (Fig. 2D). Similar result was also observed in Huh7 cells. Colony formation analyses demonstrated that overexpression of THRSP can decrease the number of clones of HCCLM3 and Huh7 cell lines, however silencing THRSP expression increased the number of clones formed by Hep3B and HepG2 cell lines (Fig. 2E).
THRSP inhibits HCC cell proliferation and tumorigenesis in vitro and in vivo. A, B. Western blot analysis of the transfection efficiency of THRSP in HCC cell lines. C. Proliferation rate was analyzed using CCK-8 assay of HCC cells with THRSP knockdown. D. Proliferation rate was analyzed using CCK-8 assay of HCC cells with THRSP overexpression. E. Proliferation rate was analyzed by clone formation assay of HCC cells with THRSP overexpression or silenced. F. Downregulation of THRSP decreased Hep3B cell subcutaneous growth, whereas THRSP overexpression had the opposite effect. G. The EMT-related markers and proliferation-related markers were analyzed in the indicated tissues using WB assays. Data are means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
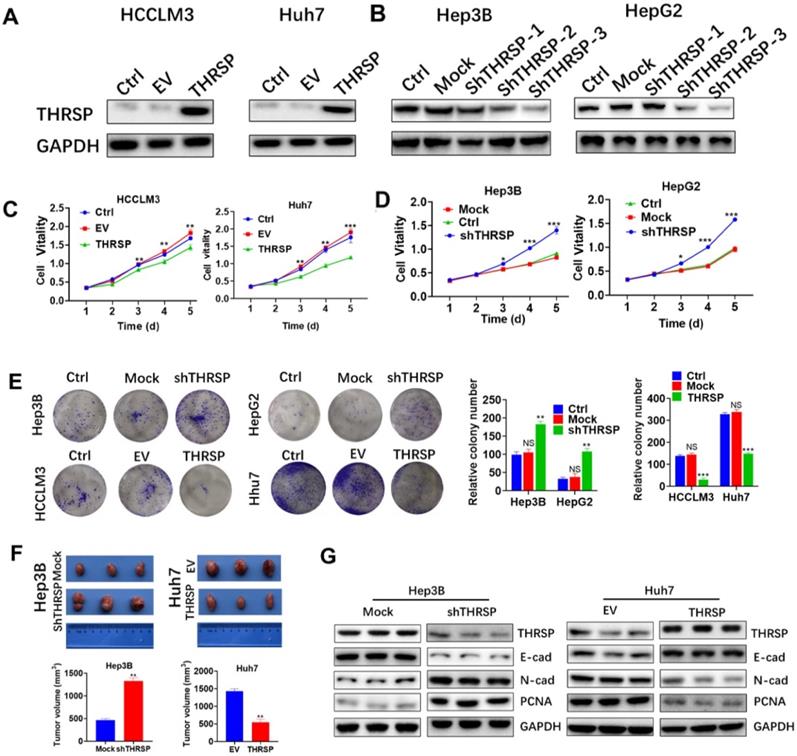
To further confirm the above findings in vivo, subcutaneous xenograft tumors were constructed in nude mice. Significantly larger volumes of HCC tumors were found in the Hep3B-shTHRSP group, whereas smaller tumors were observed in the Huh7-THRSP group, compared with their corresponding control group respectively (Fig. 2F). To further demonstrate that THRSP suppressed tumor cell growth, we measured molecular markers of cell proliferation and invasion in the subcutaneous xenograft tumors. We found THRSP knockdown decreased expression of E-cadherin, but increased expression of N-cadherin and PCNA. In contrast, THRSP overexpression increased expression of E-cadherin, and inhibited expression of N-cadherin and PCNA. (Fig. 2G). In summary, our results suggested that THRSP inhibits HCC growth both in vitro and in vivo.
THRSP suppresses cell migration and invasion of HCC
The impact of THRSP on the invasiveness and the mobility of HCC cells was explored. Wound-healing experiments showed that downregulation of THRSP increased mobility (as indicated by regrowth into the scratched area) in Hep3B and Huh7 cells, but overexpression of THRSP inhibited scratch regrowth (Fig. 3A). Further, THRSP-knockdown cells showed increased invasiveness and the mobility in Transwell assays, and THRSP overexpression decreased the invasive and migratory behavior of HCC cells (Fig. 3B and 3C).
THRSP inhibits HCC cell migration and invasion. A. The migration of cancer cells was determined by the wound-healing assay. B, C. The migration and invasion ability were determined using transwell assays in the indicated cell lines. Scale bars: 100μm. D. mRNA levels of EMT-related markers were measured by qPCR. E. Protein levels of EMT-related markers and proliferation-related markers were measured by western blot. Data are means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
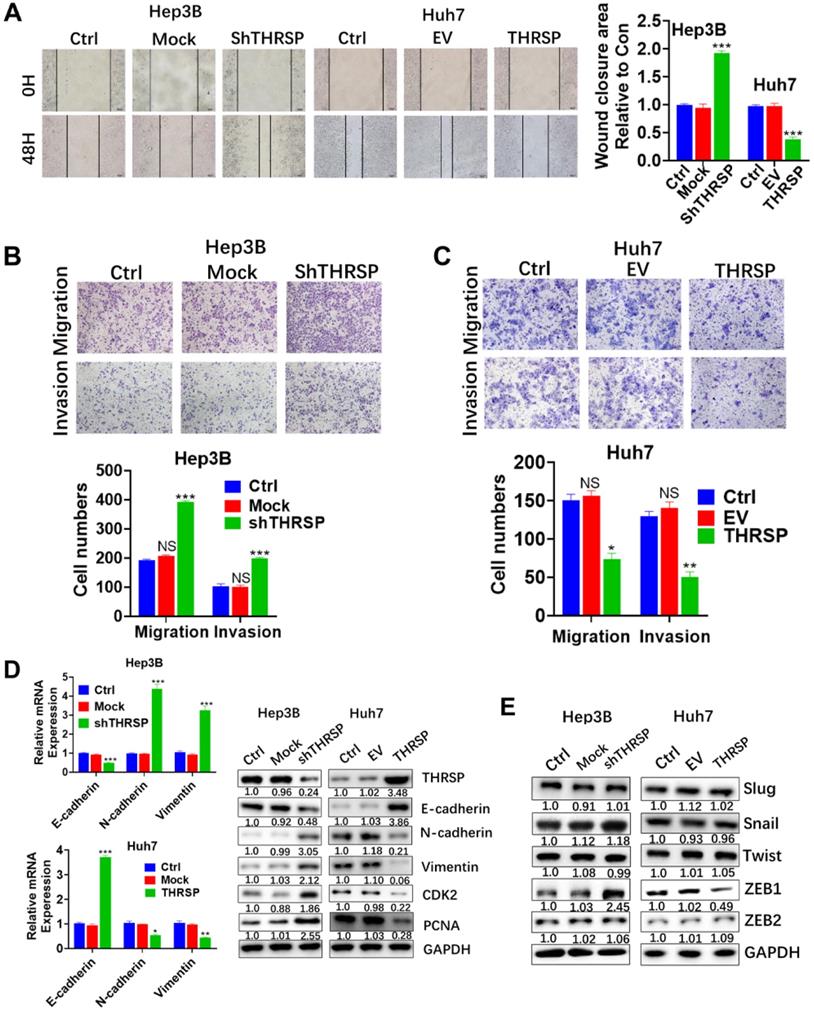
Epithelial to mesenchymal transformation (EMT) is responsible for the migration and invasion of individual cancer cells [15]. To clarify the underlying role of THRSP in the process of EMT, qPCR and WB were carried out to determine the expression of EMT-related markers. When THRSP was downregulated in Hep3B cells, the expression of E-cadherin (an epithelial marker) was decreased and the expression of N-cadherin and vimentin (mesenchymal markers) were increased, as were cell proliferation-related markers, CDK2, and PCNA. Conversely, THRSP overexpression increased the expression of E-cadherin and reduced that of N-cadherin and vimentin in Huh7 cells, as well as CDK2 and PCNA, compared to control cells (Fig. 3D). Moreover, we analyzed expression of the upstream transcription regulators of EMT, such as snail, slug, ZEB1/2, and twist, which are important for cell migration and invasion [16-18]. Of these, only ZEB1 expression was decreased or increased with overexpression or downregulation of THRSP, respectively (Fig. 3E).
THRSP-induced HCC progression was dependent on ERK phosphorylation
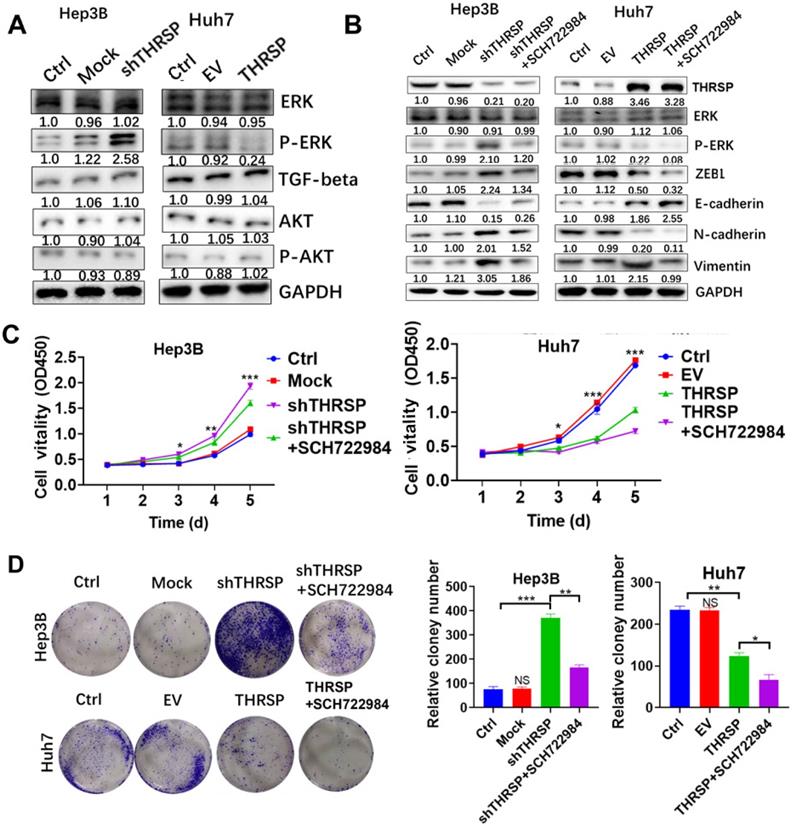
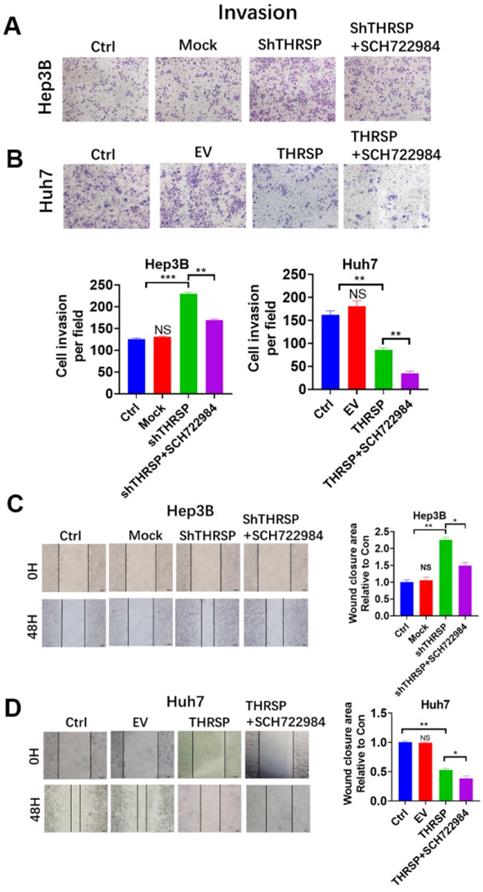
Next, we sought to clarify the molecular mechanism by which THRSP regulates EMT. We found that the expression of phosphorylated ERK was increased after THRSP silencing and decreased after THRSP overexpression. However, there were no changes in p-AKT or TGF-β1 (Fig. 4A). We used SCH722984, a novel specific inhibitor of ERK1/2 [19, 20], to explore the mechanisms of THRSP-induced HCC progression. Western blots demonstrated that the activation of p-ERK was inhibited after treating Hep3B-shTHRSP cells with SCH722984 (Fig. 4B). Consistently, the same effect was observed in Huh7-THRSP cells (Fig. 4B). CCK8 assays showed that the survival rate of cells was dramatically reduced after treatment with SCH722984 (Fig. 4C). Similarly, this was also confirmed in colony formation assays (Fig. 4D). When treated with SCH722984, the invasion and migration of Hep3B-shTHRSP and Huh7-THRSP cells were partially inhibited (Fig. 5A and 5B). Wound healing assays also confirmed that THRSP suppressed cell migration (Fig. 5C and 5D). These data indicate that the ERK signaling pathway is vital for THRSP-induced HCC progression and that ZEB1 is a key molecule in THRSP-induced EMT.
THRSP reverses EMT process in an ERK/ZEB1 dependent manner. A. Protein levels of signaling pathways were analyzed by western blot after modulation of THRSP expression. B. Western blot analysis of indicated markers in liver cancer cells. C. Proliferation rate was analyzed using CCK-8 assay in indicated HCC cells with SCH722984 treatment. D. Proliferation rate was analyzed using colony formation assay in indicated HCC cells with SCH722984 treatment. Data are means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
THRSP abolished cell migration of HCC in an ERK/ZEB1 dependent manner. A. Transwell analysis of Hep3B cell lines in THRSP knockdown cells with SCH722984 treatment. B. Transwell analysis of Huh7 cell lines in THRSP overexpression cells with SCH722984 treatment. C. Wound healing analysis of Hep3B cell lines in THRSP knockdown cells with SCH722984 treatment. D. Wound healing analysis of Huh7 cell lines in THRSP overexpression cells with SCH722984 treatment. Scale bars: 100 µm; Data are means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
This study is the first to demonstrate a role of THRSP in the progression of HCC. Our evidence suggests that the expression of THRSP was significantly decreased in HCC tissues and HCC cell lines compared with adjacent non-cancerous tissues and normal liver cell lines. Patients with low levels of THRSP had shorter survival time than those with high levels of THRSP. Consistent with these clinical findings, we found that THRSP inhibited the proliferation, invasion, and migration of liver cancer cells in vitro. Further, in vivo assays confirmed that the growth of liver cancer cells was significantly suppressed.
EMT is a critical process in tumorigenesis, especially tumor migration and invasion [15]. A variety of biological and pathological processes are attributed to EMT, such as tissue regeneration, embryo formation, and tumorigenesis [21, 22]. Increasing amounts of evidence have demonstrated that EMT is responsible for the migration and invasion of different cancer types, including HCC [23, 24]. However, there is a need for better understandings of the specific mechanisms of EMT progression. Additionally, targeted strategies for the clinical management of HCC in response to patient-specific EMT-related biomarkers remain an area for improvement [25]. Previous studies have shown that P53 has an inhibitory effect on the EMT process [26]. We found that downregulation of THRSP decreased epithelial markers and increased mesenchymal markers, thereby promoting EMT in HCC cell lines regardless of P53 mutation states. This indicated THRSP might inhibit the aggressive phenotype of HCC in an independent P53 manner. Importantly, among several transcription factors studied that is involved in the EMT process, only ZEB1 appeared to be affected by THRSP. This is the first report of the potential mechanism of THRSP in HCC.
Numerous previous studies have indicated that cancer signaling pathways are aberrantly activated to increase EMT and enhance carcinogenesis [27-29]. We investigated several key pathways, including ERK1/2, TGF-β, and AKT signaling, and found that, when THRSP was silenced or overexpressed, only ERK signaling was activated, with altered protein levels of phosphorylated ERK but no change in total ERK protein expression. The ERK pathway is associated with the process of EMT [29, 30]; the over-activation or inactivation of ERK signaling is frequently found in various cancers, including HCC [31-33]. In our study, an underlying link between THRSP and the activation of ERK signaling in HCC cells was established. When THRSP-overexpressing cells were treated with an inhibitor of ERK1/2, the EMT process and tumorigenesis were partially inhibited. This suggests that the ERK pathway is important for THRSP-regulated HCC processes.
We found that THRSP reversed the EMT process in a manner that was dependent on the ERK/ZEB1 signaling pathway. Consistently, Sheng et al. have previously reported that the ERK/ZEB1 signaling pathway is essential for EMT [34]. MAP kinase is known as an extracellular signal-regulated kinase (ERK) that acts as an integration point for a variety of biochemical signals and participates in multiple cellular processes such as proliferation, invasion, and transcriptional regulation. After activation, it moves into the nucleus to phosphorylate nuclear targets [32]. Additionally, it can act as a transcriptional repressor, independent of its kinase activity [35]. ZEB1 is a member of the zinc-finger E-box-binding homeobox family and is a transcription factor that regulates EMT. According to prior reports, ZEB1 promotes tumor invasion and metastasis by inducing EMT in many cancers. ZEB1 recruits a variety of chromatin enzymes from the E-cad promoter, which is a key mechanism for regulating EMT [36]. Our in vitro assays showed that THRSP decreased ZEB1 expression but did not impact other transcription factors that also control EMT such as slug, snail, twist, or zeb2. ERK signaling was the only EMT-related pathway that appeared to be regulated by THRSP. Our experiments confirm that ERK modulates the expression of ZEB1. However, the specific regulatory relationship between ERK and ZEB1 remains a topic for further study.
In conclusion, we found that THRSP may act as tumor suppressor gene in HCC. Our results indicated that THRSP was decreased in HCC tissues and cell lines, and THRSP expression was negatively correlated with HCC progression and prognosis. THRSP inhibited cell proliferation, migration, and invasion in an ERK/ZEB1-dependent manner. This is the first report of the role of THRSP in HCC, and these findings suggest that restoration of THRSP expression might represent a promising therapeutic strategy towards HCC treatment.
Abbreviations
THRSP: Thyroid hormone responsive; ZEB1: zinc finger E-box binding homeobox 1; HCC: hepatocellular carcinoma; mRNA: messenger RNA; qPCR: Quantitative real-time polymerase chain reaction; MOI: multiplicity of infection; ERK: extracellular signal-regulated kinase; p-ERK: phosphorylated extracellular signal-regulated kinase; EMT: epithelial to mesenchymal transformation; ZEB2: Zinc finger E-box binding homeobox 2; TGF-beta: transforming growth factor-beta; MAP kinase: Mitogen-activated protein kinase; DMEM: Dulbecco's Modified Eagle's medium; CCK-8: Cell Counting Kit-8; FBS: fetal bovine serum; WB: western blot; shRNA: short hairpin RNA; PCNA: Proliferating cell nuclear antigen; CDK2: Cyclin-dependent kinase 2.
Supplementary Material
Supplementary table S1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82002618).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Yang JD, Hainaut P, Gores GJ. et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589-604
3. Reig M, Da Fonseca LG, Faivre S. New trials and results in systemic treatment of HCC. J Hepatol. 2018;69:525-533
4. Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106-117
5. Dhanasekaran R, Nault JC, Roberts LR. et al. Genomic Medicine and Implications for Hepatocellular Carcinoma Prevention and Therapy. Gastroenterology. 2019;156:492-509
6. Ren J, Xu N, Zheng H. et al. Expression of Thyroid Hormone Responsive SPOT 14 Gene Is Regulated by Estrogen in Chicken (Gallus gallus). Sci Rep. 2017;7:10243
7. Na H, Lee H, Lee MH. et al. Deletion of exons 3 and 4 in the mouse Nr1d1 gene worsens high-fat diet-induced hepatic steatosis. Life Sci. 2016;166:13-19
8. Wellberg EA, Rudolph MC, Lewis AS. et al. Modulation of tumor fatty acids, through overexpression or loss of thyroid hormone responsive protein spot 14 is associated with altered growth and metastasis. Breast Cancer Res. 2014;16:481
9. Chou WY, Ho CL, Tseng ML. et al. Human Spot 14 protein is a p53-dependent transcriptional coactivator via the recruitment of thyroid receptor and Zac1. Int J Biochem Cell Biol. 2008;40:1826-1834
10. Kinlaw WB, Quinn JL, Wells WA. et al. Spot 14: A marker of aggressive breast cancer and a potential therapeutic target. Endocrinology. 2006;147:4048-4055
11. Martel PM, Bingham CM, McGraw CJ. et al. S14 protein in breast cancer cells: direct evidence of regulation by SREBP-1c, superinduction with progestin, and effects on cell growth. Exp Cell Res. 2006;312:278-288
12. Zeng N, Huang R, Li N. et al. MiR-451a attenuates free fatty acids-mediated hepatocyte steatosis by targeting the thyroid hormone responsive spot 14 gene. Mol Cell Endocrinol. 2018;474:260-271
13. Chella Krishnan K, Kurt Z, Barrere-Cain R. et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018;6:103-115.e7
14. Yang WJ, Sun YF, Jin AL. et al. BCL11B suppresses tumor progression and stem cell traits in hepatocellular carcinoma by restoring p53 signaling activity. Cell Death Dis. 2020;11:895
15. Barriga EH, Mayor R. Adjustable viscoelasticity allows for efficient collective cell migration. Semin Cell Dev Biol. 2019;93:55-68
16. Ma Y, Zhang H, Xiong C. et al. CD146 mediates an E-cadherin-to-N-cadherin switch during TGF-β signaling-induced epithelial mesenchymal transition. Cancer Lett. 2018;430:201-214
17. Kim TW, Lee SY, Kim M. et al. DSGOST regulates resistance via activation of autophagy in gastric cancer. Cell Death Dis. 2018;9:649
18. Yao L, Conforti F, Hill C. et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019;26:943-957
19. Wong DJ, Robert L, Atefi MS. et al. Antitumor activity of the ERK inhibitor SCH772984 [corrected] against BRAF mutant, NRAS mutant and wild-type melanoma. Mol Cancer. 2014;13:194
20. Morris EJ, Jha S, Restaino CR. et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742-750
21. Shu DY, Butcher E, Saint-Geniez M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int J Mol Sci. 2020;21:4271
22. Sun NY, Yang MH. Metabolic Reprogramming and Epithelial-Mesenchymal Plasticity: Opportunities and Challenges for Cancer Therapy. Front Oncol. 2020;10:792
23. Bai Y, Sha J, Kanno T. The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial-Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma. Cancers (Basel). 2020;12:555
24. Abudoureyimu M, Zhou H, Zhi Y. et al. Recent progress in the emerging role of exosome in hepatocellular carcinoma. Cell Prolif. 2019;52:e12541
25. Giannelli G, Koudelkova P, Dituri F. et al. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798-808
26. Yang-Hartwich Y, Tedja R, Roberts CM. et al. p53-Pirh2 Complex Promotes Twist1 Degradation and Inhibits EMT. Mol Cancer Res. 2019;17:153-164
27. Wang R, Lu X, Yu R. Lycopene Inhibits Epithelial-Mesenchymal Transition and Promotes Apoptosis in Oral Cancer via PI3K/AKT/m-TOR Signal Pathway. Drug Des Devel Ther. 2020;14:2461-2471
28. Yang W, Zhang C, Li Y. et al. Phosphorylase Kinase β Represents a Novel Prognostic Biomarker and Inhibits Malignant Phenotypes of Liver Cancer Cell. Int J Biol Sci. 2019;15:2596-2606
29. Ma XL, Shen MN, Hu B. et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110β and predicts poor prognosis. J Hematol Oncol. 2019;12:37
30. Olea-Flores M, Zuñiga-Eulogio MD, Mendoza-Catalán MA. et al. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial-Mesenchymal Transition in Cancer. Int J Mol Sci. 2019;20:2885
31. Neuzillet C, Tijeras-Raballand A, de Mestier L. et al. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141:160-171
32. Min L, He B, Hui L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin Cancer Biol. 2011;21:10-20
33. Pascale RM, Simile MM, Peitta G. et al. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers (Basel). 2019;11:1450
34. Sheng W, Shi X, Lin Y. et al. Correction to: Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res. 2020;39:167
35. Chandra Mangalhara K, Manvati S, Saini SK. et al. ERK2-ZEB1-miR-101-1 axis contributes to epithelial-mesenchymal transition and cell migration in cancer. Cancer Lett. 2017;391:59-73
36. Goossens S, Vandamme N, Van Vlierberghe P. et al. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim Biophys Acta Rev Cancer. 2017;1868:584-591
Author contact
![]() Corresponding author: Qiong Hu, Department of Laboratory Medicine, Zhoushan Hospital of Zhejiang Province, Zhoushan, Zhejiang, 316021, China. Tel & Fax: +86-0580-2292905, E-mail: hq4340com. Qiong Hu is also the first author of this work.
Corresponding author: Qiong Hu, Department of Laboratory Medicine, Zhoushan Hospital of Zhejiang Province, Zhoushan, Zhejiang, 316021, China. Tel & Fax: +86-0580-2292905, E-mail: hq4340com. Qiong Hu is also the first author of this work.