Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(3):840-848. doi:10.7150/jca.47918 This issue Cite
Research Paper
New novel non-MHC genes were identified for cervical cancer with an integrative analysis approach of transcriptome-wide association study
Haimiao Chen1#, Ting Wang1#, Shuiping Huang1,2 ![]() , Ping Zeng1,2
, Ping Zeng1,2 ![]()
1. Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China.
2. Center for Medical Statistics and Data Analysis, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China.
#These authors contributed equally to this work.
Received 2020-5-8; Accepted 2020-10-18; Published 2021-1-1
Abstract
Although genome-wide association studies (GWAS) have successfully identified multiple genetic variants associated with cervical cancer, the functional role of those variants is not well understood. To bridge such gap, we integrated the largest cervical cancer GWAS (N = 9,347) with gene expression measured in six human tissues to perform a multi-tissue transcriptome-wide association study (TWAS). We identified a total of 20 associated genes in the European population, especially four novel non-MHC genes (i.e. WDR19, RP11-384K6.2, RP11-384K6.6 and ITSN1). Further, we attempted to validate our results in another independent cervical cancer GWAS from the East Asian population (N = 3,314) and re-discovered four genes including WDR19, HLA-DOB, MICB and OR2B8P. In our subsequent co-expression analysis, we discovered SLAMF7 and LTA were co-expressed in TCGA tumor samples and showed both WDR19 and ITSN1 were enriched in “plasma membrane”. Using the protein-protein interaction analysis we observed strong interactions between the proteins produced by genes that are associated with cervical cancer. Overall, our study identified multiple candidate genes, especially four non-MHC genes, which may be causally associated with the risk of cervical cancer. However, further investigations with larger sample size are warranted to validate our findings in diverse populations.
Keywords: MetaXcan, cervical cancer, transcriptome-wide association study, Gene expression level, associated genes
Introduction
Cervical cancer is a common female carcinoma and is one of the leading causes of cancer death worldwide [1], especially in developing countries where it brings the major health burden due to limited screening programs available [2-4]. Cervical cancer has multiple complex etiologies and is caused by the combination of genetic risk factors and various external environmental exposures. For example, epidemiological evidence suggests that human papillomavirus (HPV) is one of the major risk factors of cervical cancer and contributes to almost all cases [5]. However, only ~1% of women finally develop cervical neoplasia with HPV infection [6] because of the strong heritable component and host genetic factors [7].
In the past few years several large-scale genome-wide association studies (GWAS) have discovered a lot of single nucleotide polymorphisms (SNPs) associated with the risk of cervical cancer (Table S1) and provide new insight into the genetic architecture of this type of cancer. However, understanding the underlying mechanism of those identified SNPs on cervical cancer remains considerably challenging as some of them are located within intergenic and the complex major histocompatibility complex (MHC) region [8-10]. The difficulty is that the causal genes mediating SNP effects on cervical cancer can be rarely ascertainable with GWAS data alone because of complicated linkage disequilibrium (LD) [11]. Therefore, the causal genes and pathways are still not well known and additional genomic functional analyses are required to elucidate the biological mechanism between genetic variants and the risk of cervical cancer [12, 13].
One reasonable explanation for this challenge is that GWAS-associated SNPs may be also associated with molecular-level traits (e.g. gene expression; those variants are also thus referred to as expression quantitative trait loci [eQTL]), which at least partially mediate the causal effects of SNPs on cervical cancer and hold the key to disentangle the genetic foundation of genetic susceptibility to cervical cancer and phenotypic variation. However, in large-scale cervical cancer GWASs genome-wide transcriptome profile is frequently unavailable or unmeasured for all individuals due to high cost and/or unavailability of specimen. Consequently, the association between gene expression and cervical cancer cannot be detected directly [14].
To investigate the relationship between unmeasured gene expression and cervical cancer in GWAS, we here bridge such a gap by efficiently imputing unmeasured expression based on external transcriptome reference datasets [15], where both gene expression levels and genotypes are available for a relatively small set of individuals. This is the basic idea behind prediXcan [15] — a kind of gene-based association analysis recently developed in the framework of transcriptome-wide association studies (TWAS) [14, 15]. Methodologically, prediXcan [15] can be viewed as a relatively independent two-stage inference procedure to prioritize causal genes by integrating transcriptome profile into the association between SNPs and phenotypes. The existence of rich transcriptome datasets from diverse human tissues [16-20] makes it feasible for such an integration. Recently, prediXcan was further extended so that it can be applicable with only summary statistics of GWAS and QTL; the resulting method is referred to as MetaXcan [21]. A similar summary-statistics based TWAS method termed FUSION was also proposed meanwhile [14]. The schematic framework of TWAS with prediXcan/MetaXcan is shown in Figure 1.
Schematic framework of TWAS using prediXcan with complete data sets or MetaXcan with only summary statistics results. TWAS can be viewed to be a relatively independent two-stage inference procedure: the first stage aims to estimate weights for cis-SNPs in an independent transcriptome reference panel using various genetic prediction models with complete data sets of genotypes and gene expressions (i.e. GTEx) (the top panel); the second stage aims to indirectly estimate the causal association between genetically predicted gene expression and a phenotype with weights of cis-SNPs obtained from the first stage (the bottom panel).
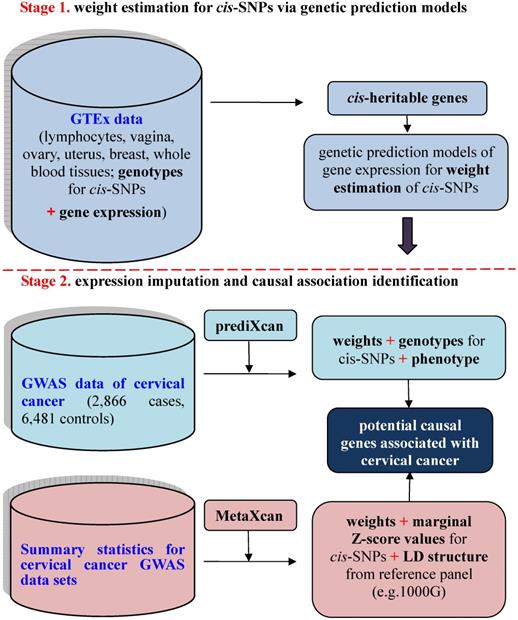
To identify the causal association between genes and cervical cancer, in this study we integrated the transcriptome from multiple GTEx tissues [17, 18] into the cervical cancer GWAS [22] using MetaXcan in the European (EUR) population. A total of 20 genes were identified, among which four novel non-MHC genes might be potentially causal genes associated with the susceptibility of cervical cancer. We also attempted to validate our results in another cervical cancer GWAS from the East Asian (EAS) population [23]. Functional analyses were further implemented for those TWAS-identified genes.
Materials and Methods
Cervical cancer GWAS datasets and MetaXcan analysis
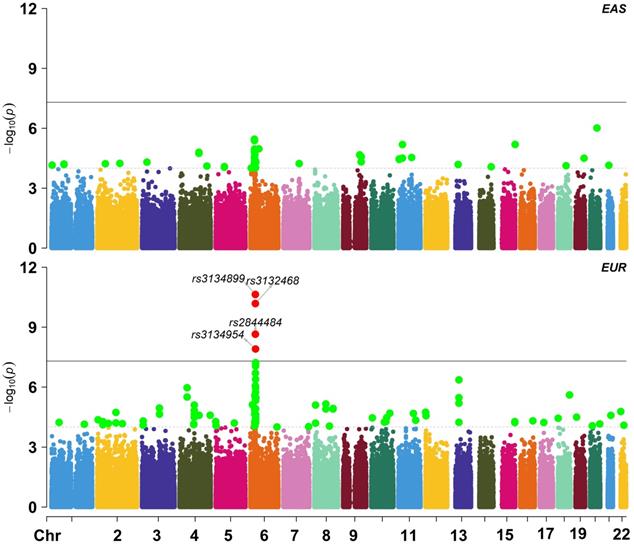
We obtained summary statistics of cervical cancer from [22], where a total of 9,347 (2,866 cases and 6,481 controls) individuals of EUR ancestry were included (Figure 2A). The TWAS analysis was implemented with MetaXcan [21] which was in prior trained with genotypes and gene expressions in 48 tissues from the GTEx Project (Table S2) [17, 18]. Another reference transcriptome reference panel from Depression Genes and Networks (DGN; 922 whole-blood samples) was also integrated [20]. The pre-calculated weights for cis-SNPs of each gene can be directly available at http://predictdb.org/ (October, 2019).
Manhattan plot for East Asian and European ancestry cohorts in the cervical cancer GWAS; EAS: East Asian ancestry; EUR: European ancestry.
Because it has been shown that integrating gene expressions from tissues that are mechanistically unrelated to diseases of interest can lead to spurious associations in TWAS [24], we followed previous work via PubMed retrieval [25] to determinate cervical-cancer related tissues (Table S3). We finally selected gynecological tissues (i.e. vagina, ovary, uterus and breast), whole blood and lymphocytes to be the most relevant tissues for cervical cancer (Supplementary File). In our analysis genes with false discover rate (FDR) < 0.05 were identified to be significant.
As will be shown below, a total of 20 novel genes were detected with MetaXcan in the EUR population. We further validated those genes in a latest cervical cancer GWAS from the EAS population (N = 3,314 with 1,320 case and 1,994 control) (Figure 2B) [23]. The corresponding transcriptome expression profile came from the tissue of peripheral blood on 98 Japanese individuals [26]. The LD matrix was computed with genotypes of 504 EAS individuals in the 1000 Genomes Project [27]. The analysis was implemented with a gene-based FUSION-type TWAS approach [14], which is mathematically equivalent to MetaXcan [21].
Functional analyses for identified genes
Next, combining with previous GWAS-identified genes (Table S1), we implemented three functional analyses for those TWAS-identified genes (Table 1 and Table S4). First, co-expression analysis was performed via weighted gene co-expression network analysis (WGCNA) [28]. The visualization of interconnection network was conducted in Cytoscape (http://www.cytoscape.org/) [29] with the topological overlap measurement (TOM) quantifying interconnection among genes [30]. Note that, TOM ranges between 0 and 1 indicating interconnection between two genes; greater TOM represents higher interconnection with the same set of genes [30]. However, co-expression analysis results from WGCNA may be hard to interpret since copy number variant (CNV) itself is a common feature in cancer [31]. In order to adjust for the variation in gene expression contributed by CNV, we also implemented an additional co-expression analysis with GRACE [32] — a recently proposed method which removes the influence of CNV before analysis. To implement the two analyses, we selected cervical cancer patients from TCGA who had both gene expression and copy number alteration, and finally kept 34 genes of 284 patients for GRACE and 48 genes of 296 patients for WGCNA. To apply GRACE, we quantile-normalized each gene expression and standardized each CNV.
TWAS-identified genes associated with cervical cancer across the relevant tissues
| Chr | position | Gene | Tissue | Gene type | R2 | z value | p value | FDR | |
|---|---|---|---|---|---|---|---|---|---|
| Low | Up | ||||||||
| 4 | 39,184,024 | 39,287,430 | WDR19 | GTEx WB | protein coding | 0.049 | 4.20 | 2.20E-05 | 2.05E-02 |
| 4 | 119,553,429 | 119,555,914 | RP11-384K6.2 | EBV TL | pseudogene | 0.100 | 4.21 | 2.69E-05 | 2.45E-02 |
| 4 | 119,553,429 | 119,555,914 | RP11-384K6.2 | ovary | pseudogene | 0.108 | 4.20 | 2.60E-05 | 1.79E-02 |
| 4 | 119,553,429 | 119,555,914 | RP11-384K6.2 | GTEx WB | pseudogene | 0.269 | 4.21 | 2.67E-05 | 2.05E-02 |
| 4 | 119,512,928 | 119,556,134 | RP11-384K6.6 | ovary | processed transcript | 0.076 | 4.18 | 2.60E-05 | 1.79E-02 |
| 4 | 119,512,928 | 119,556,134 | RP11-384K6.6 | GTEx WB | processed transcript | 0.251 | 4.24 | 2.96E-05 | 2.05E-02 |
| 21 | 35,014,706 | 35,272,165 | ITSN1 | GTEx WB | protein coding | 0.077 | -5.05 | 2.97E-05 | 2.05E-02 |
| 6 | 31,949,801 | 31,970,458 | C4A | vagina | protein coding | 0.094 | -4.57 | 4.46E-07 | 4.75E-04 |
| 6 | 31,973,413 | 31,976,228 | CYP21A1P | ovary | pseudogene | 0.110 | -4.94 | 4.93E-06 | 6.78E-03 |
| 6 | 31,694,815 | 31,698,394 | DDAH2 | DGN WB | protein coding | 0.190 | -4.92 | 7.71E-07 | 1.62E-03 |
| 6 | 30,620,896 | 30,640,814 | DHX16 | DGN WB | protein coding | 0.001 | -4.67 | 8.60E-07 | 1.62E-03 |
| 6 | 32,780,540 | 32,784,825 | HLA-DOB | ovary | protein coding | 0.600 | -4.62 | 2.97E-06 | 6.78E-03 |
| 6 | 32,780,540 | 32,784,825 | HLA-DOB | uterus | protein coding | 0.500 | -4.82 | 3.77E-06 | 8.17E-03 |
| 6 | 32,780,540 | 32,784,825 | HLA-DOB | GTEx WB | protein coding | 0.480 | 5.07 | 1.45E-06 | 2.67E-03 |
| 6 | 32,520,490 | 32,527,799 | HLA-DRB6 | vagina | pseudogene | 0.451 | -5.30 | 4.08E-07 | 4.75E-04 |
| 6 | 31,539,831 | 31,542,101 | LTA | DGN WB | protein coding | 0.107 | -5.67 | 1.15E-07 | 4.05E-04 |
| 6 | 31,686,425 | 31,689,622 | LY6G6C | DGN WB | protein coding | 0.003 | 5.13 | 1.47E-08 | 7.74E-05 |
| 6 | 31,367,561 | 31,384,016 | MICA | GTEx WB | protein coding | 0.189 | 4.49 | 2.96E-07 | 8.18E-04 |
| 6 | 31,462,658 | 31,478,901 | MICB | EBV TL | protein coding | 0.148 | 6.53 | 7.22E-06 | 9.86E-03 |
| 6 | 31,462,658 | 31,478,901 | MICB | GTEx WB | protein coding | 0.339 | 3.88 | 6.56E-11 | 3.63E-07 |
| 6 | 28,021,006 | 28,021,943 | OR2B8P | ovary | pseudogene | 0.247 | -3.87 | 1.03E-04 | 4.26E-02 |
| 6 | 28,083,406 | 28,084,329 | RP1-265C24.5 | ovary | pseudogene | 0.075 | 4.13 | 1.08E-04 | 4.26E-02 |
| 6 | 31,926,857 | 31,937,532 | SKIV2L | vagina | protein coding | 0.180 | -5.98 | 3.62E-05 | 2.57E-02 |
| 6 | 31,543,344 | 31,546,113 | TNF | DGN WB | protein coding | 0.082 | 4.91 | 2.27E-09 | 2.39E-05 |
| 6 | 30,876,019 | 30,894,236 | VARS2 | DGN WB | protein coding | 0.371 | -3.88 | 9.20E-07 | 1.62E-03 |
| 6 | 28,317,691 | 28,335,336 | ZKSCAN3 | ovary | protein coding | 0.144 | -4.18 | 1.03E-04 | 4.26E-02 |
Note: EBV TL: EBV transformed lymphocytes; WB: whole blood; DGN: Depression Genes and Networks. R2 shows the prediction accuracy of the cis-SNPs on gene expression in a tissue of GTEx.
To explore functional feature for those genes, we further performed functional enrichment analysis, including gene ontology (GO) and KEGG pathway, with DAVID 6.8 (https://david.ncifcrf.gov/) [33]. Enrichment analysis allows us to validate our findings by determining functional annotations for those identified genes. In order to detect interaction and association, we also conducted protein-protein interaction analysis in terms of the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING 11.0) database (https://string-db.org/) [34].
Results
New novel non-MHC genes associated with cervical cancer identified by TWAS
We here focus on the TWAS associations yielded from the six tissues that were considered to be most possibly relevant with cervical cancer (i.e. DGN and GTEx whole blood, EBV-transformed lymphocytes, ovary, uterus, breast and vagina in GTEx). Results based on all the available tissues in GTEx are relegated to Supplementary File; see also Figure S1 and Table S4. Specifically, a total of 20 distinct genes from 26 association pairs (FDR < 0.05) are discovered in terms of the six tissues that are relevant to cervical cancer (Table 1). Among those, both HLA-DOB and RP11-384K6.2 display associations with cervical cancer in three tissues; 80.0% (16/20) are located in the MHC region (Chr 6: 25,000,000-34,000,000) and 70.0% (14/20) are protein coding genes.
In particular, four non-MHC genes (i.e. WDR19, RP11-384K6.2 and RP11-384K6.6 in Chr 4 and ITSN in Chr 21) are identified. Importantly, each of these four genes is not nearby (within 1Mb upstream and downstream) any previous GWAS index SNPs or associated genes (Figure S2-S4 and Table S1). Moreover, their expression levels are only weakly correlated with each other; for example, the Pearson's correlation coefficient of expression levels for all pairs of the four genes ranges from -0.123 (between RP11.384K6.6 and ITSN1) to 0.304 (between RP11.384K6.6 and RP11.384K6.2) in terms of 376 EUR individuals in the Geuvadis Project [19], implying that the four non-MHC genes are likely new novel genes involved in the risk of developing cervical cancer. Furthermore, the four genes are still associated with cervical cancer using conditional TWAS analysis after controlling for the strongest SNP within respective locus (Table S5), supporting their independent roles on the susceptibility of cervical cancer.
Validation results of those associated genes in the EAS population
We further validate the associations discovered above using datasets from the EAS population and show the results in Table S6. In this validation analysis 18 out of 20 associated genes are found in the EAS population; among those, 4 genes (p = 1.47E-02 for WDR19, p = 2.65E-02 for HLA-DOB, p = 4.92E-02 for MICB and p = 1.78E-02 for OR2B8P) are identified to be also associated with cervical cancer at the nominal significance level of 0.05. Particularly, with regards to the four non-MHC genes, we observe that, besides WDR19, ITSN1 is also detected to be marginally related to cervical cancer (p = 0.081) in the EAS population. However, we cannot replicate other associated genes in the EAS population. We also found many population-specific associated genes (Figure 3) from the GTEx and DGN whole blood with TWAS, such as FLOT1 (p = 1.637E-10), HISTIH2BH (p = 1.037E-09).
(a) Manhattan plot of TWAS results for the EAS and EUR ancestry cohorts in the blood tissue from GTEx; (b) Manhattan plot of TWAS results for the EAS and EUR ancestry cohorts in the blood tissue from DGN.
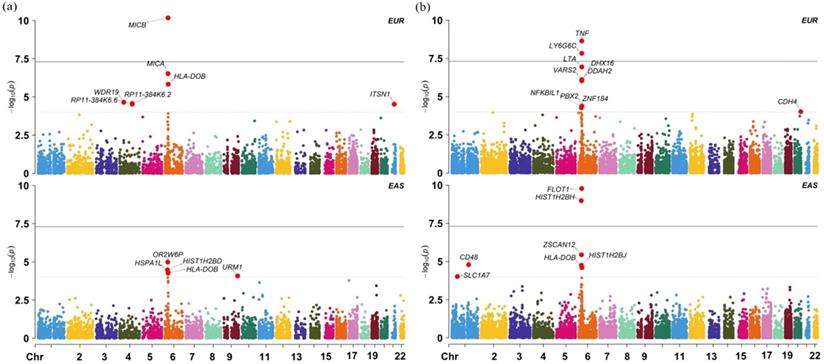
Results of gene co-expression network analysis
Taking both scale-free fit index and mean connectivity as reference, the soft-thresholding was determined to be 3 for WGCNA and GRACE. With WGCNA and using average linkage hierarchical clustering and module merging, all the genes were divided into five modules with different colors (Table S7). It means that most of the genes have high correlation with genes in the same module but weak correlation with genes in other modules. However, some genes (in grey) cannot be classified into any co-expression modules. With GRACE, the genes are divided into three modules (Table S7). The difference of co-expression genes between WGCNA and GRACE (TOM > 0.1) is shown in Figure S5-S6; and four potentially hub genes (e.g. HLA.DQA1, HLA.DQB1, HLA.DRB1 and SLAMF7) are discovered by WGCNA. Of interest, all of them have co-expressed genes, such as SLAMF7 and LTA.
Gene enrichment analysis and Protein-protein interaction network
According to DAVID 6.8, those GWAS- or TWAS identified genes are enriched in nine GO terms (Table S8), some of which are significantly associated with cervical cancer. The significant KEGG pathways for cervical cancer are presented in Table S9. For example, it is shown that “type I diabetes mellitus” is most significantly enriched (FDR = 2.21E-9). The persistent high-risk HPV infection was associated with increased risk for cervical cancer and numbers of infiltrating T cells, particularly CD8 T cells, and the presentation of HPV E6/E7 epitopes were associated with improved prognosis [35]. Other GO terms which obviously play important role in cervical cancer are also found, such as “MHC class II receptor activity”, “MHC class II protein complex” [36] and “peptide antigen binding” [37].
We further constructed the protein-protein interaction network to explore the biological function with STRING summary score above 0.4 (Figure S7). Each node represents a gene; the undirected link between two nodes is an edge, denoting the interaction between two genes. For example, strong interactions, such as HLA-DRB1, TNF, LTA, HLA-DOB, LILRB1, MICA, HLA-C, HLA-B and MICB, were previously detected to be associated with cervical cancer [38].
Discussion
In the present study we have systematically evaluated the association of genetically predicted gene expression across various human tissues with the risk of cervical cancer. With extensively computational genetic analyses our results can provide substantial new insights into the understanding of genetics and etiology for cervical cancer. Importantly, we identified a set of associated genes in the EUR population including four promising new non-MHC genes (i.e. WDR19, RP11-384K6.2, RP11-384K6.6 and ITSN1).
RP11-384K6.6 is a type of IncRNA and discovered to be regulated by multiple differential miRNAs, such as hsa-miR-16, hsa-miR-708 and hsa-miR-486-5p [39]. Interestingly, Serum miR-486-5p can inhibit the PTEN expression and activate the oncogenic PI3K/Akt pathway in cervical cancer [40], and further stimulate cell proliferation, migration and invasion. PTEN, located at 10q23.3, is a tumor-suppressor gene and was identified to be associated with the risk of cervical cancer [41, 42], posing an important role in controlling cell growth, including promoting apoptosis, down-regulating adhesion and suppressing cell migration [43, 44]. RP11-384K6.2 is regulated by hsa-miR-657, which was found to have tumor-associated putative target genes that were associated with cervical cancer, including CDC14B, TNFSF11 and PTPN2 [45].
WDR19 and ITSN1 are also likely susceptibility genes of cervical cancer. Specifically, WDR19 is the component of the IFT complex A (IFT-A). IFT-A modulates canonical Wnt/Wg-signalling, which was discovered to be involved in the risk of many cancers [46, 47]. ITSN1 belongs to the ITSN family and shares highly similar structures and functions with ITSN2 [48-50]. Importantly, ITSN2 interacts with Eps8, the down-regulation of P53 and P21Waf1/Cip1 in cervical cancer [51].
We further validated our main finding in an independent EAS GWAS dataset [23]. However, we only replicated the 4 genes including WDR19, HLA-DOB, MICB and OR2B8P. This failure may be due to the following reasons. First, we observe that substantial genetic heterogeneity exists in cervical cancer between the EUR and EAS populations. For example, the trans-ethnic genetic correlation of cervical cancer is estimated to be only rg = 0.038 (se = 0.015) with Linkage Disequilibrium Score regression [52] and 0.127 with Pearson's product-moment correlation, implying high ethnic difference in genetic foundation of cervical cancer. Indeed, we find many population-specific associated SNPs (Figure 2), such as rs3132468 (p = 0.095 in the EAS population but p = 6.56E-11 in the EUR population), rs2844484 (p = 1.12E-4 in the EAS population but p = 2.67E-9 in the EUR population) and rs3134954 (p = 0.014 in the EAS population but p = 1.24E-8 in the EUR). Second, except peripheral blood, we cannot access transcriptome profiles of other types of tissue (e.g. ovary) in our validation analysis. Given the distinct genetic architecture among various tissues [18] and diverse populations [53], it is unexpected that we cannot replicate genes that were discovered in non-peripheral-blood tissues. Note that, the two replicated newly associated genes (i.e. WDR19 and ITSN1) in the EAS population were exactly also identified in blood in the EUR population. Third, the EAS transcriptome dataset has smaller sample size compared with the GTEx transcriptome datasets; for example, the average sample size of GTEx is about 200, two-fold higher than the EAS transcriptome dataset. In addition, the EAS cervical cancer GWAS also has smaller sample size (3,314 vs. 9,341) compared with the EUR cervical cancer GWAS. Therefore, we have reduced power when identifying associated genes in the EAS population.
In the subsequent functional analyses with GRACE, we showed that both SLAMF7 and LTA were co-expressed in tumor samples consistent with the results by WGCNA. Moreover, we conducted GO enrichment and KEGG pathway analysis for both GWAS-identified and TWAS-identified genes, and found 9 GO terms and 16 pathways, respectively. We found that both WDR19 and ITSN1 were enriched in “plasma membrane”, which may be likely associated with cancer through its proteomics [54] or calcium channels [55]. In addition, KEGG analysis found that type I diabetes mellitus was the most significantly enriched pathway, in line with the finding that diabetes is a well-known risk factor of cervical cancer [56-59].
Our multi-tissue transcriptome imputation approach has a number of advantages. Transcriptome imputation methods allow the study of genetically regulated gene expression without directly measuring expression data from an appropriate cell type in diseases. What's more, transcriptome imputation aggregates SNP-level associations to individual genes, reducing the multiple testing burden and increasing statistical power. Moreover, TWAS methods utilize eQTL information from eQTL databases with uniform sample collection and enable replication across the ancestry. However, our study has several limitations despite of these interesting results. The weights of cis-SNPs in TWAS were estimated with relatively small sample size for the external transcriptome reference data [16-20], which may underline the power of the TWAS analysis. As the sample size increases for relevant tissues, we expect that more potentially associated genes would be discovered. Besides, as it has shown that the leverage of gene expression reference panels from tissues that are less mechanistically related to diseases of interest can lead to bias and spuriously associated genes [24], the true mechanism and relevant tissues are still completely known although we carefully selected six tissues considered to be related to cervical cancer.
Conclusion
This study discovers multiple candidate genes, especially four non-MHC genes, which may be causally associated with the risk of cervical cancer. However, further investigations with larger sample size are warranted to validate our findings in diverse populations.
Abbreviations
TWAS: transcriptome-wide association study; GWAS: genome-wide association studies; HPV: human papillomavirus; SNP: single nucleotide polymorphisms; MHC: major histocompatibility complex; LD: linkage disequilibrium; eQTL: expression quantitative trait loci; FDR: false discover rate; TOM: topological overlap measurement; CNV: copy number variants.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We are very grateful to Dr. Fumihiko Takeuchi for the help in providing us the summary statistics of the East Asia cervical cancer GWAS. We thank Harvard Dataverse for making the summary statistics of the European cervical cancer GWAS publicly available. We thank all the investigators and participants contributed to those studies. We appreciate the reviewer for the constructive comments and suggestions which substantially improved our manuscript. We also thank the GitHub repository https://github.com/hakyimlab which contains the implementations of MetaXcan described in this manuscript and provides a step-by-step guideline on running the complete MetaXcan analysis.
Funding
This study was supported by grants from the Natural Science Foundation of Jiangsu Province (BK20181472), Youth Foundation of Humanity and Social Science funded by Ministry of Education of China (18YJC910002), the China Postdoctoral Science Foundation (2018M630607 and 2019T120465), the QingLan Research Project of Jiangsu for Outstanding Young Teachers, the Six-Talent Peaks Project in Jiangsu Province of China (WSN-087), the Social Development Project of Xuzhou, the Postdoctoral Science Foundation of Xuzhou Medical University, the National Natural Science Foundation of China (81402765), the Statistical Science Research Project from National Bureau of Statistics of China (2014LY112), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX20_2501), Training Project for Youth Science and Technology Innovation Team at Xuzhou Medical University and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for Xuzhou Medical University. The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the manuscript. PZ had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Authors' contributions
PZ conceived the idea for the study. PZ and SH obtained the genetic data. PZ developed the study methods. PZ, HC and TW performed the data analyses. PZ and HC interpreted the results of the data analyses. PZ and HC wrote the manuscript with suggestions from other authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68:7-30
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
3. Arbyn M, Castellsagué X, de Sanjosé S, Bruni L, Saraiya M, Bray F. et al. Worldwide burden of cervical cancer in 2008. Ann Oncol. 2011;22:2675-86
4. Arbyn M, Weiderpass E, Bruni L, de Sanjosé S, Saraiya M, Ferlay J. et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. The Lancet Global Health. 2020;8:e191-e203
5. Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet (London, England). 2013;382:889-99
6. Schiffman M, Glass AG, Wentzensen N, Rush BB, Castle PE, Scott DR. et al. A long-term prospective study of type-specific human papillomavirus infection and risk of cervical neoplasia among 20,000 women in the Portland Kaiser Cohort Study. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2011;20:1398-409
7. Magnusson PK, Lichtenstein P, Gyllensten UB. Heritability of cervical tumours. International journal of cancer. 2000;88:698-701
8. Chen D, Juko-Pecirep I, Hammer J, Ivansson E, Enroth S, Gustavsson I. et al. Genome-wide association study of susceptibility loci for cervical cancer. Journal of the National Cancer Institute. 2013;105:624-33
9. Madeleine MM, Johnson LG, Smith AG, Hansen JA, Nisperos BB, Li S. et al. Comprehensive analysis of HLA-A, HLA-B, HLA-C, HLA-DRB1, and HLA-DQB1 loci and squamous cell cervical cancer risk. Cancer research. 2008;68:3532-9
10. Maciag PC, Schlecht NF, Souza PS, Franco EL, Villa LL, Petzl-Erler ML. Major histocompatibility complex class II polymorphisms and risk of cervical cancer and human papillomavirus infection in Brazilian women. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2000;9:1183-91
11. Gallagher MD, Chen-Plotkin AS. The Post-GWAS Era: From Association to Function. American journal of human genetics. 2018;102:717-30
12. Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C. et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;2015:895-907
13. Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371-5
14. Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245-52
15. Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091-8
16. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S. et al. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45:580-5
17. GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648-60
18. GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550:204-13
19. Lappalainen T, Sammeth M, Friedländer MR, t Hoen PAC, Monlong J, Rivas MA. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506-11
20. Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C. et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14-24
21. Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9:1825
22. Leo PJ, Madeleine MM, Wang S, Schwartz SM, Newell F, Pettersson-Kymmer U. et al. Defining the genetic susceptibility to cervical neoplasia—A genome-wide association study. PLoS Genet. 2017;13:e1006866
23. Takeuchi F, Kukimoto I, Li Z, Li S, Li N, Hu Z. et al. Genome-wide association study of cervical cancer suggests a role for ARRDC3 gene in human papillomavirus infection. Human molecular genetics. 2019;28:341-8
24. Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D. et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592-9
25. Hao X, Zeng P, Zhang S, Zhou X. Identifying and exploiting trait-relevant tissues with multiple functional annotations in genome-wide association studies. PLoS genetics. 2018;14:e1007186
26. Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M. et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50:390-400
27. The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68-74
28. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559
29. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498-504
30. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology. 2005;4:Article17
31. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B. et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics. 2013;45:1134-40
32. Cai L, Li Q, Du Y, Yun J, Xie Y, DeBerardinis RJ. et al. Genomic regression analysis of coordinated expression. Nature communications. 2017;8:2187
33. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
34. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research. 2019;47:D607-d13
35. Reeves E, Wood O, Ottensmeier C, King E, Thomas G, Elliott T. et al. HPV Epitope Processing Differences Correlate with ERAP1 Allotype and Extent of CD8 T-cell Tumor Infiltration in OPSCC. Cancer immunology research. 2019;7:1202-13
36. Tang A, Dadaglio G, Oberkampf M, Di Carlo S, Peduto L, Laubreton D. et al. B cells promote tumor progression in a mouse model of HPV-mediated cervical cancer. International journal of cancer. 2016;139:1358-71
37. Jabbar B, Rafique S, Salo-Ahen O, Ali A, Munir M, Idrees M. et al. Antigenic Peptide Prediction From E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Frontiers in immunology. 2018;9:3000
38. Chambuso R, Ramesar R, Kaambo E, Denny L, Passmore JA, Williamson AL. et al. Human Leukocyte Antigen (HLA) Class II -DRB1 and -DQB1 Alleles and the Association with Cervical Cancer in HIV/HPV Co-Infected Women in South Africa. Journal of Cancer. 2019;10:2145-52
39. Wei G. Bioinformatics analysis of microRNA comprehensive regulatory network in congenital microtia. International journal of pediatric otorhinolaryngology. 2015;79:1727-31
40. Li C, Zheng X, Li W, Bai F, Lyu J, Meng QH. Serum miR-486-5p as a diagnostic marker in cervical cancer: with investigation of potential mechanisms. BMC cancer. 2018;18:61
41. Su TH, Chang JG, Perng LI, Chang CP, Wei HJ, Wang NM. et al. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in cervical cancer. Gynecologic oncology. 2000;76:193-9
42. Harima Y, Sawada S, Nagata K, Sougawa M, Ostapenko V, Ohnishi T. Mutation of the PTEN gene in advanced cervical cancer correlated with tumor progression and poor outcome after radiotherapy. International journal of oncology. 2001;18:493-7
43. Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. Journal of the National Cancer Institute. 1999;91:1922-32
44. Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX. et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034-45
45. Ding H, Wu YL, Wang YX, Zhu FF. Characterization of the microRNA expression profile of cervical squamous cell carcinoma metastases. Asian Pacific journal of cancer prevention: APJCP. 2014;15:1675-9
46. Coombs GS, Covey TM, Virshup DM. Wnt signaling in development, disease and translational medicine. Current drug targets. 2008;9:513-31
47. Liu F, Millar SE. Wnt/beta-catenin signaling in oral tissue development and disease. Journal of dental research. 2010;89:318-30
48. Yamabhai M, Hoffman NG, Hardison NL, McPherson PS, Castagnoli L, Cesareni G. et al. Intersectin, a novel adaptor protein with two Eps15 homology and five Src homology 3 domains. The Journal of biological chemistry. 1998;273:31401-7
49. Mohney RP, Das M, Bivona TG, Hanes R, Adams AG, Philips MR. et al. Intersectin activates Ras but stimulates transcription through an independent pathway involving JNK. The Journal of biological chemistry. 2003;278:47038-45
50. Hussain NK, Jenna S, Glogauer M, Quinn CC, Wasiak S, Guipponi M. et al. Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nature cell biology. 2001;3:927-32
51. Chen YJ, Shen MR, Chen YJ, Maa MC, Leu TH. Eps8 decreases chemosensitivity and affects survival of cervical cancer patients. Molecular cancer therapeutics. 2008;7:1376-85
52. Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR. et al. An atlas of genetic correlations across human diseases and traits. Nature genetics. 2015;47:1236-41
53. Shang L, Smith JA, Zhao W, Kho M, Turner ST, Mosley TH. et al. Genetic Architecture of Gene Expression in European and African Americans: An eQTL Mapping Study in GENOA. Am J Hum Genet. 2020;106:496-512
54. Leth-Larsen R, Lund RR, Ditzel HJ. Plasma membrane proteomics and its application in clinical cancer biomarker discovery. Molecular & cellular proteomics: MCP. 2010;9:1369-82
55. Deliot N, Constantin B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochimica et biophysica acta. 2015;1848:2512-22
56. Kuo HY, Lin ZZ, Kuo R, Shau WY, Lai CL, Yang YY. et al. The Prognostic Impact of Type 2 Diabetes Mellitus on Early Cervical Cancer in Asia. The oncologist. 2015;20:1051-7
57. Kapp DS, Fischer D, Gutierrez E, Kohorn EI, Schwartz PE. Pretreatment prognostic factors in carcinoma of the uterine cervix: a multivariable analysis of the effect of age, stage, histology and blood counts on survival. International journal of radiation oncology, biology, physics. 1983;9:445-55
58. Jiamset I, Hanprasertpong J. Impact of diabetes mellitus on oncological outcomes after radical hysterectomy for early stage cervical cancer. Journal of gynecologic oncology. 2016;27:e28
59. Chen S, Tao M, Zhao L, Zhang X. The association between diabetes/hyperglycemia and the prognosis of cervical cancer patients: A systematic review and meta-analysis. Medicine. 2017;96:e7981
Author contact
![]() Corresponding authors: Shuiping Huang, Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China. E-mail: hspedu.cn; Ping Zeng, Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China; Phone: 86-516-8326 2659; Fax: 86-516-8326 2659; E-mail: zpstatedu.cn.
Corresponding authors: Shuiping Huang, Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China. E-mail: hspedu.cn; Ping Zeng, Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, Jiangsu, 221004, China; Phone: 86-516-8326 2659; Fax: 86-516-8326 2659; E-mail: zpstatedu.cn.