Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(24):7253-7263. doi:10.7150/jca.50835 This issue Cite
Research Paper
Surfactin from Bacillus subtilis induces apoptosis in human oral squamous cell carcinoma through ROS-regulated mitochondrial pathway
Thi Thuy Tien Vo1, Ju-Fang Liu2, Ching-Zong Wu1, Wei-Ning Lin3, Yuh-Lien Chen4, I-Ta Lee1 ![]()
1. School of Dentistry, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan.
2. School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan.
3. Graduate Institute of Biomedical and Pharmaceutical Science, Fu Jen Catholic University, New Taipei City, Taiwan.
4. Department of Anatomy and Cell Biology, College of Medicine, National Taiwan University, Taipei, Taiwan.
Received 2020-7-19; Accepted 2020-10-12; Published 2020-10-21
Abstract

Recently, ambient air particulate matter (PM) has been shown to increase the risk of oral cancer. The most common malignant tumor in the oral cavity is oral squamous cell carcinoma (OSCC). Recent studies have revealed that surfactin, a cyclic lipopeptide generated by Bacillus subtilis, has anti-inflammatory and anti-cancer properties. However, the exact anti-cancer effects of surfactin on human OSCC and underlying molecular mechanisms remain largely unknown. In the present study, we found that treatment of SCC4 and SCC25 cells (human OSCC cell lines) with surfactin reduced the viability of SCC4 and SCC25 cells by induction of apoptosis. Surfactin-induced apoptosis was associated with caspase activation and poly(ADP-ribose) polymerase (PARP) cleavage and was regulated by the mitochondrial pathway, exemplified by mitochondrial depolarization, mitochondrial-derived reactive oxidative species (ROS) production, cytochrome c release, up-regulation of Bad and Bax, and down-regulation of Bcl-2. Surfactin induced NADPH oxidase-dependent ROS generation, which appeared essential for the activation of the mitochondrial pathway. Surfactin-induced mitochondrial-derived ROS generation was associated with JNK1/2 activation. After treatment with surfactin, ROS caused JNK1/2-dependent cell death of SCC4 and SCC25 cells. Taken together, our findings suggest that surfactin induces mitochondria associated apoptosis of human OSCC cell lines, and surfactin may be a potential chemotherapeutic agent for future OSCC treatment.
Keywords: apoptosis, surfactin, particulate matter, oral squamous cell carcinoma, reactive oxidative species
Introduction
In many parts of the world, new cases of oral cancer and deaths are increasing. Known risk factors include smoking, drinking, human papillomavirus (HPV) and betel quid chewing [1]. It is additionally believed that exposure to heavy metals, as well as emissions from petroleum and chemical plants, is also related to the development of oral cancer. To date, it is well known that air pollution, especially ambient air particulate matter (PM), is harmful to the respiratory and cardiovascular system [2]. The combined effects of household and ambient air pollution cause approximately 7 million premature deaths every year, mainly due to heart disease, stroke, lung cancer, chronic obstructive pulmonary disease, and acute respiratory infections, leading to increased mortality [3,4]. The composition of PM is highly complicated, including nitrate, sulfate, ammonia, and so on. Compared to PM10, PM2.5 can cause greater damage to human health. PM2.5 generally penetrates the lung barrier and enters the blood system. Most studies in the past had explored the relationship between betel nuts, cigarettes, or alcohol consumption and oral cancer, but few investigations have examined the relationship between air pollution and oral cancer. The most common malignant tumor in the oral cavity is oral squamous cell carcinoma (OSCC). Despite considerable advances in cancer treatment, surgical resection followed by adjuvant radiation therapy and chemotherapy remains the conventional regimen that is widely accepted in this field. Besides, a variety of new treatment strategies, including immunotherapy, nanotechnology, and molecular-targeted therapy, has emerged as potential options, but still display limited success without substantial clinical effect [5-7].
Natural products have the characteristics of safety and uncommon side effects. Therefore, they are increasingly used as a positive source for development of novel cancer preventive and therapeutic drugs [8,9]. For instances, curcumin, resveratrol, apigenin, eugenol, and genistein are well-known cancer preventive agents, and some of their analogues have synergic effects targeting multiple pathways, including cyclooxygenase-2 (COX-2), signal transducer and activator of transcription-3 (STAT-3) signaling, and reactive oxidative species (ROS) [10-14]. Among them, surfactin is a bacterial cyclic lipopeptide generated by Bacillus subtilis [15]. Recently, surfactin has been shown to possess some properties including anti-cancer, anti-bacterial, and anti-viral activities [16]. Park et al. indicated that surfactin could reduce 12-O-tetradecanoylphorbol-13-acetate (TPA)-mediated breast cancer cell migration/invasion via the inhibition of matrix metallopeptidase-9 (MMP-9) levels [15]. In addition, Wang et al. also proved that surfactin could promote apoptosis of HepG2 cells via the ROS signaling [17]. Despite its various activities, the molecular basis behind the pharmacological activities of surfactin and its therapeutic effects on OSCC remain largely unknown. Here, we studied the anti-cancer effects of surfactin on PM-treated human OSCC and the novel mechanisms underlying these processes. In the present study, we report that the pro-apoptotic effect of surfactin in SCC4 and SCC25 human OSCC cell lines is regulated by increasing ROS production, activation of the JNK1/2 phosphorylation cascade, and the mitochondrial pathways. This study therefore provides evidence, for the first time, that surfactin can be considered as a potential chemotherapeutic agent for the treatment of human OSCC.
Methods
Materials
Anti-GAPDH, anti-phospho-JNK1/2, anti-β-actin, anti-caspase-3, anti-caspase-7, anti-caspase-9, anti-poly(ADP-ribose) polymerase (PARP), anti-Bax, anti-Bad, anti-Bcl-2, anti-cytochrome c, anti-Akt, and anti-COX IV antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). SP600125, Z-VAD-FMK, MitoTEMPO, surfactin, and urban PM (SRM 1648a) were purchased from Sigma (St. Louis, MO, USA). N-acetyl-L-cysteine (NAC) and diphenyleneiodonium chloride (DPI) were taken from Biomol (Plymouth Meeting, PA).
Cell culture
SCC4 and SCC25 human OSCC cell lines were kindly provided by Dr. J. F. Liu (School of Oral Hygiene, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan). SCC4 and SCC25 cells were grown in DMEM/F12 supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and 0.4 μg/ml hydrocortisone. Cells were maintained as monolayer cultures in a humidified atmosphere of 5% CO2 at 37°C. In addition, human gingival fibroblasts (HGFs) and human oral keratinocytes (HOKs) were purchased from ScienCell Research Laboratories (San Diego, CA). They were cultured in fibroblast medium (ScienCell Research Laboratories, San Diego, CA) and oral keratinocyte medium (ScienCell Research Laboratories, San Diego, CA), respectively, supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C in 5% CO2. Experiments were performed with cells from passages 3 to 8. Prior to treatment or stimulation with reagents, cells were serum starved for 24 h.
Cell viability
The cell viability of SCC4 and SCC25 cells in response to surfactin was assessed using PrestoBlue Cell Viability Reagent (Invitrogen, CA, USA) according to the manufacturer's protocol.
Western blot
SCC4 and SCC25 cells were grown to confluence in 6-well plates and then treated with surfactin for the indicated time intervals. The cells were washed, scraped, collected, and centrifuged at 45000 × g at 4°C for 1 h to yield the whole cell extract, as previously described [18]. Samples were denatured, subjected to SDS-PAGE using a 12% running gel, and transferred to nitrocellulose membrane. Membranes were incubated with anti-caspase-3, anti-caspase-7, anti-caspase-9, anti-PARP, anti-Bcl-2, anti-Bax, anti-Bad, or anti-phospho-JNK1/2 antibody for 24 h, followed by incubation with anti-mouse or anti-rabbit horseradish peroxidase antibody for 1 h. The immunoreactive bands were detected by ECL reagents developed by Hyperfilm-ECL.
Caspase activity determinations
Caspase activity in cell lysates was measured using the manufacturer's protocols (caspase-3, -7, and -9 colorimetric assay kits; R&D Systems Inc., Minneapolis, MN, USA). Cells were treated with surfactin for 48 h and then lysed in lysis buffer [50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 10 mM EGTA, 10 mM digitonin, and 2 mM DTT]. The cell lysates (50 μg proteins) were incubated with caspase-3, -7, and -9 specific substrates (Ac-DEVD-pNA and Ac-LEHD-pNA) at 37°C for 1 h. Caspase activity and absorbance were measured with an enzyme-linked immunosorbent assay reader at OD405. All results were obtained from three independent experiments.
Cytosolic and mitochondrial protein extraction
To obtain cytosolic and mitochondrial fractions, cells were treated with a digitonin buffer (20 mM Hepes-KOH, pH 7.3, 110 mM KAc, 5 mM NaAc, 2 mM MgAc2, 1 mM EGTA, and 200 μg/ml digitonin) on ice for 10 min to permeabilize the cell membrane. The cell lysate was then centrifuged at 10000 × g at 4°C for 15 min. The supernatant was collected as a cytosolic fraction, and the pellet (mitochondria-containing fraction) was resuspended in 1X-SDS-loading buffer. Protein content was estimated according to a commercial protein assay (Bio Rad, Milan, Italy), and the samples were either analyzed immediately or stored at -80°C. Total, cytosolic, and mitochondrial extracts were then analyzed by Western blot.
Mitochondrial membrane potential detection
Mitochondrial membrane potential (ΔΨm) was detected by a fluorescent dye JC-1 (Sigma, St. Louis, MO). The change from red fluorescence to green fluorescence in the JC-1 assay can be used to detect the decline in mitochondrial membrane potential. Furthermore, this transition can also be used as an early detection indicator of apoptosis. After being treated with various concentrations of surfactin for 48 h, the SCC4 cells in 6-well plate were washed with PBS twice, and then 1 ml of serum-free DMEM/F-12 medium was added followed by 1 ml of JC-1 staining working solution in each well. The plate was incubated for 20 min in the incubator at 37°C with 5% CO2. The plate was observed and photographed under a fluorescence microscope (Carl Zeiss, Gottingen, Germany). The wavelengths of excitation and emission were 514 nm and 529 nm for detection of JC-1 monomers, respectively. The values of 585 nm and 590 nm were used to detect JC-1 aggregates. The relative ratio of red and green fluorescence represented the change of mitochondrial membrane potential (ΔΨm). Five groups of data of each well were recorded.
Determination of NADPH oxidase activity by chemiluminescence assay
After incubation, cells were gently scraped and centrifuged at 400 × g for 10 min at 4°C. The cell pellet was resuspended with 35 μl of ice-cold RPMI-1640 medium per well, and the cell suspension was kept on ice. To a final 200 μl volume of pre-warmed (37°C) RPMI-1640 medium containing either NADPH (1 μM) or lucigenin (20 μM), 5 μl of cell suspension (0.2 × 105 cells) were added to initiate the reaction followed by immediate measurement of chemiluminescence in an Appliskan luminometer (Thermo®) in out-of-coincidence mode. Appropriate blanks and controls were established, and chemiluminescence was recorded. Neither NADPH nor NADH enhanced the background chemiluminescence of lucigenin alone (30-40 counts per min). Chemiluminescence was continuously measured for 12 min, and the activity of NADPH oxidase was expressed as counts per million cells.
Measurement of intracellular ROS and mitochondrial ROS generation
CellROX Green Reagent and MitoSOX Red mitochondrial superoxide indicator (Molecular Probes, Eugene, OR) were used in these experiments. For the purpose of these experiments, SCC4 cells were washed with warm Hank's Balanced Salt Solution (HBSS) and incubated in HBSS or cell medium containing 5 μM CellROX Green Reagent or MitoSOX Red mitochondrial superoxide indicator at 37°C for 30 min. Subsequently, HBSS or medium containing CellROX Green Reagent or MitoSOX Red mitochondrial superoxide indicator was removed and replaced with fresh medium. SCC4 cells were then incubated with surfactin for the indicated times. Cells were washed twice with PBS and detached with trypsin/EDTA, and the fluorescence intensity of the cells was analyzed using a FACScan flow cytometer (BD Biosciences, San Jose, CA) at 485 nm excitation and 520 nm emission (CellROX Green Reagent) and 510 nm excitation and 580 nm emission (MitoSOX Red mitochondrial superoxide indicator), respectively.
Transient transfection with human siRNAs
Human scrambled, JNK1, and JNK2 siRNAs were taken from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Transient transfection of siRNAs was performed using a Lipofectamine 2000 Transfection Reagent (Invitrogen, CA, USA) according to the manufacturer's instructions.
DAPI staining
SCC4 and SCC25 cells (5000 cells/ml) in 24-well plates were incubated with surfactin (15 or 30 μM) for the indicated times. Cells in each treatment were individually fixed with 3.7% (vol/vol) formaldehyde for 15 min and then stained by DAPI (0.5 μg/ml) for determining cell chromatin condensation. All samples were examined and photographed using Nikon Eclipse TE300 inverted fluorescence microscope (Nikon Corp. Tokyo, Japan).
Statistical analysis
We analyzed the data with the GraphPad Prism program (GraphPad, San Diego, CA, USA). Quantitative data were expressed as the mean±S.E.M. and analyzed with one-way ANOVA followed with Tukey's post-hoc test. We considered P<0.05 as a significant difference.
Results
Surfactin decreases cell viability and induces apoptosis in SCC4 and SCC25 cells
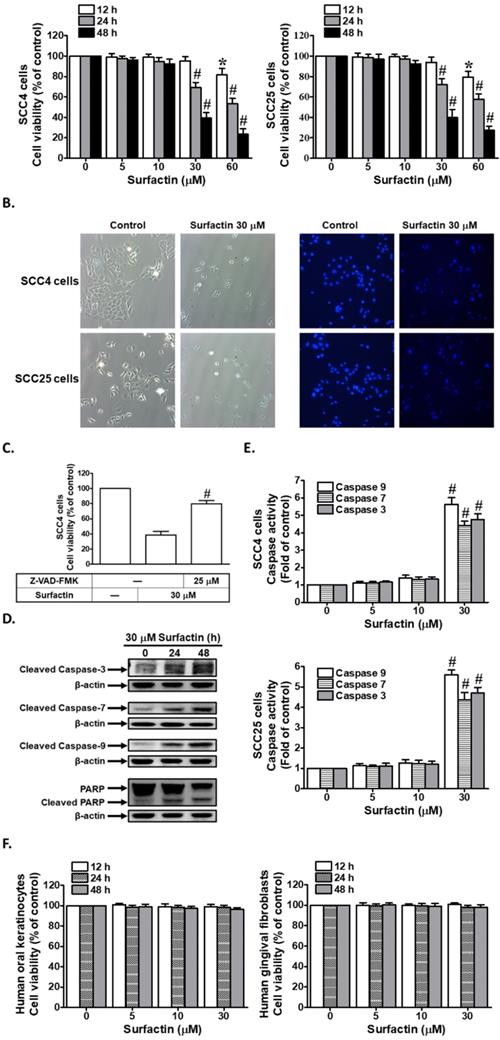
To examine the anti-cancer potential of surfactin on human OSCC, its effect was investigated on cell viability using the SCC4 and SCC25 human OSCC cell lines. Cells were treated with different concentrations of surfactin for the indicated times and then cell viability was assessed. As shown in Fig. 1A, surfactin markedly reduced cell viability in SCC4 and SCC25 cells in a concentration- and time-dependent manner. In addition, we also observed that surfactin could decrease cell viability in human osteosarcoma cell lines (HOS and U2OS) (Supplementary Fig. 1). These data suggest that surfactin may cause apoptosis in many cancer cells. On the other hand, the occurrence of apoptosis was evaluated by DAPI staining (Fig. 1B). Caspases play a central role in mediating most apoptotic responses [14]. To analyze the apoptotic pathway and the role of caspases involved in apoptosis of SCC4 cells regulated by surfactin, cell viability in SCC4 cells was examined in the presence of the pan-caspase inhibitor Z-VAD-FMK. As shown in Fig. 1C, 30 μM surfactin significantly reduced cell viability in SCC4 cells. Moreover, this observed effect by surfactin was abrogated in the presence of Z-VAD-FMK. This result demonstrated that apoptosis is associated with caspase activation. Caspase-9 is a highly specific protease that only cleaves a few proteins, whereas caspase-3 and caspase-7 contribute to the majority of cleavage that takes place during apoptosis [19]. Western blot results of the expression levels of caspase-3, -7, and -9 after exposure to surfactin for different times (Fig. 1D) showed the involvement of these effector caspases. Expression of PARP, a protein targeted and cleaved by caspases during apoptosis, was also examined. As shown in Fig. 1D, surfactin induced PARP cleavage in SCC4 cells. Moreover, in this study, we investigated the effects of surfactin on caspase-3, -7, and -9 activity in SCC4 and SCC25 cells. As shown in Fig. 1E, 30 μM surfactin markedly increased caspase-3, -7, and -9 activity. Finally, we investigated the effects of surfactin on cell viability of normal HOKs and HGFs. Interestingly, as shown in Fig. 1F, surfactin did not affect cell viability of these cells. Taken together, these data suggest that surfactin can decrease cell viability and induce apoptosis in SCC4 and SCC25 cells, which is regulated via caspase activation and PARP cleavage.
Surfactin reduces cell viability and induces apoptosis in SCC4 and SCC25 cells. (A) Cells were treated with various concentrations of surfactin for the indicated times and then the cell viability was measured. (B) Cells were treated with 30 μM surfactin for 48 h and then the occurrence of apoptosis was evaluated by DAPI staining. (C) SCC4 cells were pretreated with Z-VAD-FMK for 1 h and then incubated with surfactin for 48 h. Cell viability was assayed. (D) SCC4 cells were treated with surfactin for 24 or 48 h and then the cleaved caspase-3, -7, -9 and PARP protein levels were determined. (E) Cells were treated with various concentrations of surfactin for 48 h and then the caspase activity was analyzed. (F) Human oral keratinocytes and human gingival fibroblasts were treated with various concentrations of surfactin for the indicated times and then the cell viability was assayed. Data are expressed as mean±S.E.M. of four independent experiments. *P < 0.05; #P < 0.01, as compared with control (A, E). #P < 0.01, as compared with the cells exposed to surfactin alone (C).
Surfactin induces ROS-dependent apoptosis
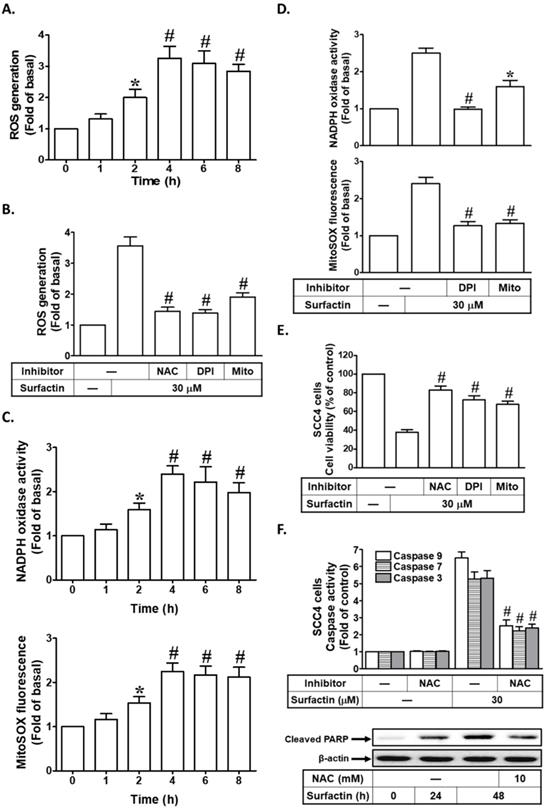
Generation of ROS by diverse anti-cancer drugs or phytochemicals has been closely related with the induction of apoptosis in cancers. Moreover, we investigated the effect of surfactin on the production of intracellular ROS in these cells. When ROS production was measured after exposure of SCC4 cells to surfactin at different times (1-8 h) by flow cytometric analysis of CellROX Green Reagent fluorescence intensity (Fig. 2A), it was observed that ROS intracellular levels were increased as early as 2 h after exposure. The sources for ROS include NADPH oxidases, mitochondrial electron transport enzymes, xanthine oxidase, cyclooxygenase, lipoxygenase, and uncoupled nitric oxide synthase [20]. Here, we showed that ROS production induced by surfactin was reduced effectively by pretreatment with NAC (a thiol-containing antioxidant), DPI (an NADPH oxidase inhibitor), or MitoTEMPO (a mitochondria-targeted antioxidant) in SCC4 cells (Fig. 2B). In addition, we also found that surfactin enhanced NADPH oxidase activity and mitochondrial superoxide production in a time-dependent manner, which were reduced by pretreatment with DPI or MitoTEMPO (Figs. 2C and D). Next, we investigated whether increased production of ROS was critical for surfactin-induced apoptosis, wherein cells were pretreated with NAC, DPI, or MitoTEMPO, and then incubated with surfactin for 48 h. The results in Fig. 2E showed that apoptosis induced by surfactin was reduced in the presence of NAC, DPI, or MitoTEMPO. Finally, we investigated the roles of ROS in surfactin-indcued caspase activation and PARP cleavage. As shown in Fig. 2F, surfactin-induced caspase-3, -7, -9 activation and PARP cleavage were reduced by pretreatment with NAC in SCC4 cells. Thus, these results indicate that surfactin induces apoptosis by a mechanism requiring the increase of intracellular ROS levels in SCC4 cells.
Surfactin induces ROS-dependent apoptosis. (A) SCC4 cells were treated with surfactin (30 μM) for the indicated times and then the ROS generation was measured. (B) SCC4 cells were pretreated with NAC (10 mM), DPI (1 μM), or MitoTEMPO (10 μM) and then incubated with surfactin for 4 h. The ROS generation was measured. (C) SCC4 cells were treated with 30 μM surfactin for the indicated times and then the NADPH oxidase activity and MitoSOX fluorescence were measured. (D) SCC4 cells were pretreated with DPI (1 μM) or MitoTEMPO (10 μM) for 1 h and then incubated with surfactin for 4 h. The NADPH oxidase activity and MitoSOX fluorescence were measured. (E) Cells were pretreated with NAC (10 mM), DPI (1 μM), or MitoTEMPO (10 μM) and then incubated with surfactin for 48 h. The cell viability was measured. (F) Cells were pretreated with NAC (10 mM) and then incubated with surfactin for 48 h. The caspase activity was analyzed. In addition, SCC4 cells were pretreated with NAC and then incubated with surfactin for 24 or 48 h. The protein expression of cleaved PARP was determined. Data are expressed as mean±S.E.M. of three independent experiments. *P < 0.05; #P < 0.01, as compared with control (A, C). *P < 0.05; #P < 0.01, as compared with the cells exposed to surfactin alone (B, D, E, F).
Surfactin promotes apoptosis via the mitochondrial pathway
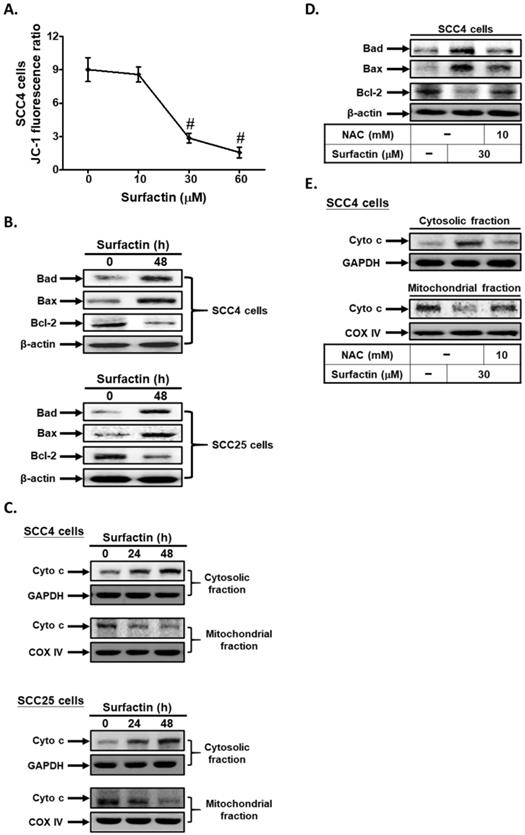
Mitochondrial changes, including permeability transition pore opening and the collapse of ΔΨm, result in the release of cytochrome c into the cytosol, which subsequently causes apoptosis by the activation of caspases [21]. Here, we used a fluorescent dye JC-1 to detect mitochondrial membrane potential (ΔΨm) in surfactin-treated SCC4 cells. The relative ratio of red and green fluorescence represented the change of mitochondrial membrane potential (ΔΨm). As shown in Fig. 3A, the JC-1 fluorescence ratio significantly reduced at 48 h in a dose-dependent manner. The mitochondrial-mediated pathway of apoptosis is regulated by the Bcl-2 family of antiapoptotic (Bcl-2, Bcl-XL, Bcl-1) and proapoptotic proteins (Bax, Bad, and Bak) [14]. Here, we proved that surfactin increased Bad and Bax expression and reduced Bcl-2 expression in SCC4 and SCC25 cells (Fig. 3B). In addition, surfactin was also found to cause a mitochondrial membrane permeability transition, as determined by Western blot of the release of cytochrome c from the mitochondria into the cytosol in cells exposed to the drug for 24 or 48 h (Fig. 3C). Finally, we observed that pretreatment with NAC markedly decreased surfactin-induced Bad and Bax expression and up-regulated surfactin-reduced Bcl-2 expression in SCC4 cells (Fig. 3D). On the other hand, pretreatment with NAC also reduced surfactin-induced the release of cytochrome c from the mitochondria into the cytosol (Fig. 3E). Thus, these data suggest that surfactin induces apoptosis via the mitochondrial pathway in SCC4 and SCC25 cells.
Surfactin promotes apoptosis through the mitochondrial pathway. (A) Cells were treated with various concentrations of surfactin for 48 h and then the JC-1 fluorescence ratio was measured. (B) Cells were treated with surfactin for 48 h and then the protein expression of Bad, Bax, or Bcl-2 was determined. (C) Cells were treated with surfactin for 24 or 48 h and then the cytosolic and mitochondrial fractions were prepared and subjected to Western blot using an anti-cytochrome c antibody. GAPDH was used as a marker protein for cytosolic fractions. COX IV was used as a marker protein for mitochondrial fractions. (D) Cells were pretreated with NAC and then treated with surfactin for 48 h. The protein expression of Bad, Bax, or Bcl-2 was determined. (E) Cells were pretreated with NAC and then treated with surfactin for 48 h. The cytosolic and mitochondrial fractions were prepared and subjected to Western blot using an anti-cytochrome c antibody. Data are expressed as mean±S.E.M. of three independent experiments. #P < 0.01, as compared with control.
ROS induces JNK1/2 activation leading to apoptosis
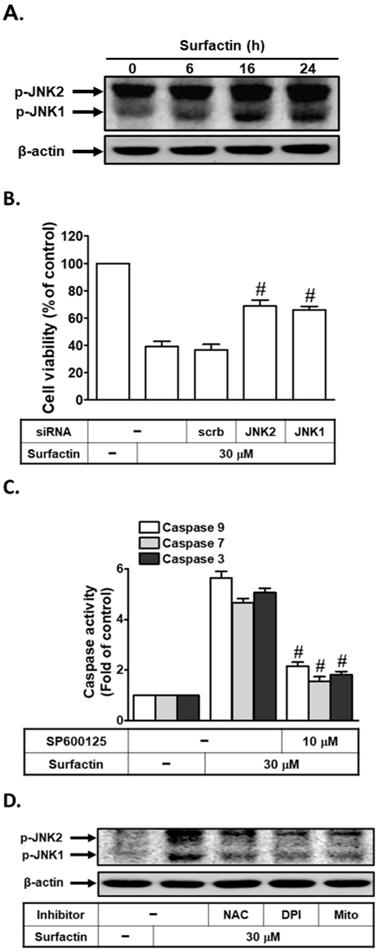
Generation of intracellular ROS is known to activate the MAPK pathway with MAPKs being relevant in apoptosis signaling [22,23]. Here, phosphorylation of JNK1/2 strongly increased after 16 h and was sustained at 24 h following exposure to surfactin (Fig. 4A). We further investigated whether JNK1/2 was critical for surfactin-induced apoptosis, cells were transfected with JNK2 or JNK1 siRNA and then analyzed for apoptosis after exposure of surfactin for 48 h. The results in Fig. 4B showed that apoptosis induced by surfactin was reduced in the presence of either JNK1 or JNK2 siRNA. In addition, we investigated the role of JNK1/2 in surfactin-induced caspase activation. As shown in Fig. 4C, surfactin-induced caspase-3, -7, and -9 activation were reduced by pretreatment with SP600125 (an anthrapyrazolone inhibitor of JNK1/2) in SCC4 cells. ROS have been shown to regulate JNK1/2 phosphorylation in various cell types [22,23]. Here, we found that surfactin-induced JNK1/2 activation was inhibited by preincubation with NAC, DPI, or MitoTEMPO (Fig. 4D). However, SP600125 pretreatment had no effects on surfactin-induced ROS generation and NADPH oxidase activation in SCC4 cells (Supplementary Fig. 2). Thus, these data suggest that surfactin induces apoptosis via the ROS/JNK1/2 signaling pathway in SCC4 cells.
ROS induces JNK1/2 activation leading to apoptosis. (A) SCC4 cells were treated with surfactin for the indicated times and then the protein levels of phospho-JNK1/2 were determined. (B) SCC4 cells were transfected with siRNA of scrambled, JNK1, or JNK2 and then treated with surfactin for 48 h. The cell viability was assayed. (C) SCC4 cells were pretreated with SP600125 for 1 h and then incubated with surfactin for 48 h. The caspase activity was analyzed. (D) SCC4 cells were pretreated with NAC, DPI, or MitoTEMPO and then incubated with surfactin for 24 h. The protein levels of phospho-JNK1/2 were determined. Data are expressed as mean±S.E.M. of three independent experiments. #P < 0.01, as compared with the cells exposed to surfactin + scrambled siRNA (B) or surfactin alone (C).
Discussion
PM exposure increases morbidity and mortality as well as causes chronic lung inflammation and cardiovascular diseases. Recently, Chu et al. proved that Taiwanese men exposed to higher concentrations of PM2.5 have an increased risk of oral cancer [2]. The seventh most common cancer in the world is OSCC which has been considered to cause detriment to human health and high mortality [24]. Surfactin is a bacterial cyclic lipopeptide generated by Bacillus subtilis [15]. Recently, surfactin has been shown to suppress cancer progression by cell cycle arrest, apoptosis, growth inhibition, and metastasis arrest [25]. The potential mechanisms by which surfactin inhibit human OSCC progression in response to PM still remain unclear. In this study, we found that surfactin decreased the viability of SCC4 and SCC25 cells by induction of apoptosis, which was associated with activation of caspase and PARP cleavage and was regulated by the mitochondrial pathway, as exemplified by mitochondrial depolarization, cytochrome c release, and down-regulation of the antiapoptotic Bcl-2 protein. We also observed that intracellular ROS production appeared essential for the activation of the mitochondrial pathway and the induction of apoptosis after exposure to surfactin. After treatment with surfactin, ROS provided a specific environment that resulted in JNK1/2-induced cell death. These data suggest a potential effect of surfactin in generating ROS and inducing mitochondria-associated apoptosis via prooxidative activity in human OSCC.
Most research in the past had explored the relationship between betel nuts, cigarettes, or alcohol consumption and oral cancer, but few studies have investigated the relationship between air pollution and oral cancer. PM is typically a representative indicator of air pollution [2,4]. It does more damage to humans than any other pollutant. The composition of PM is very complicated that includes nitrate, sulfate, ammonia, and so on. Compared to PM10, PM2.5 might cause greater and more severe adverse effects to human health [4]. PM2.5 generally penetrates the lung barrier and enters the blood system. Zhang and Li indicated that PM2.5 could induce the cell proliferation, migration, and invasion of human hepatocellular carcinoma (HCC) cell line SMMC-7721 [26]. Bacillus subtilis generates the cyclic lipopeptide surfactin. Its heptapeptide head has two negatively charged amino acid residues, and its tail consists of fatty acid residues [25]. Recently, surfactin has been shown to possess some properties including anti-cancer, anti-bacterial, and anti-viral activities [16]. Biochemical pathways of apoptosis activation have been extensively studied that can be extra- or intracellular, and caspase-dependent or caspase-independent [19]. Caspases are actively involved in inflammatory processes and apoptosis. In this study, we proved that surfactin promoted apoptosis in a caspase-dependent manner in SCC4 and SCC25 cells. Interestingly, we found that surfactin did not affect cell viability of normal human oral keratinocytes and human gingival fibroblasts. Therefore, our results directly or indirectly provide new insight into the mechanisms of surfactin-induced cancer cell death.
Oxygen-free radicals, more generally known as ROS, along with reactive nitrogen species (RNS) are well recognized to play a dual role as both deleterious and beneficial species [27]. ROS is characterized to be secondary messengers in intracellular signaling cascades, which induce and maintain the oncogenic phenotype of cancer cells; however, ROS can also induce cellular senescence and apoptosis, in turn, can function as anti-tumorigenic species [28]. The sources for ROS include NADPH oxidases, mitochondrial electron transport enzymes, xanthine oxidase, cyclooxygenase, lipoxygenase, and uncoupled nitric oxide synthase [29]. Here, we proved that ROS production induced by surfactin was decreased by pretreatment with the NADPH oxidase inhibitor, the mitochondria-targeted antioxidant, or the free radical scavenger in SCC4 cells. On the other hand, surfactin-induced mitochondrial ROS production was attenuated by pretreatment with the NADPH oxidase inhibitor, suggesting that surfactin induced mitochondrial ROS production via the activation of NADPH oxidase in these cells. Finally, we found that surfactin-induced apoptosis, caspase-3, -7, and -9 activation, and PARP cleavage were reduced by preincubation with the free radical scavenger, suggesting that increased generation of ROS was critical for surfactin-promoted apoptosis.
Mitochondria participate in a variety of important physiological and biochemical processes, such as tricarboxylic acid cycle, fatty acid metabolism and oxidative phosphorylation [30,31]. Mitochondria are indispensable for energy metabolism, apoptosis regulation and cell signaling [30]. Mitochondrial changes, including permeability transition pore opening and the collapse of membrane potential, result in the release of cytochrome c into the cytosol, which subsequently causes apoptosis by the activation of caspases [22]. In addition, the mitochondrial-mediated pathway of apoptosis is regulated by the Bcl-2 family of antiapoptotic (Bcl-2, Bcl-Xl, Bcl-1) and proapoptotic proteins (Bax, Bad, and Bak) [19]. Here, we found that surfactin induced Bad and Bax expression as well as reduced Bcl-2 expression. In addition, surfactin was also revealed to cause a mitochondrial membrane permeability transition, as determined by Western blot of the release of cytochrome c from the mitochondria into the cytosol in cells exposed to the drug. Moreover, these effects induced by surfactin were decreased by preincubation with the free radical scavenger, suggesting that surfactin induced apoptosis via the ROS-dependent mitochondrial pathway in SCC4 and SCC25 cells.
Generation of intracellular ROS is known to activate the MAPK pathway with MAPKs being relevant in apoptosis signaling [32-34]. The generic MAPK signaling pathway is shared by four distinct cascades, including the ERK1/2, JNK1/2, p38 MAPK, and ERK5 [23]. JNK1/2 pathway is reported to be associated with the cell apoptosis [35]. Indeed, we found that surfactin induced JNK1/2 activation in SCC4 cells. In addition, we found that surfactin-induced JNK1/2 activation was inhibited by pretreatment with the NADPH oxidase inhibitor, the mitochondria-targeted antioxidant, or the free radical scavenger, consistent with the results that ROS mediated JNK1/2 phosphorylation in various cell types [36,37]. Finally, we demonstrated that surfactin-induced apoptosis and caspase-3, -7, and -9 activation were reduced by JNK1 or JNK2 siRNA and SP600125, suggesting that surfactin induced apoptosis through the ROS/JNK1/2 signaling pathway in SCC4 cells.
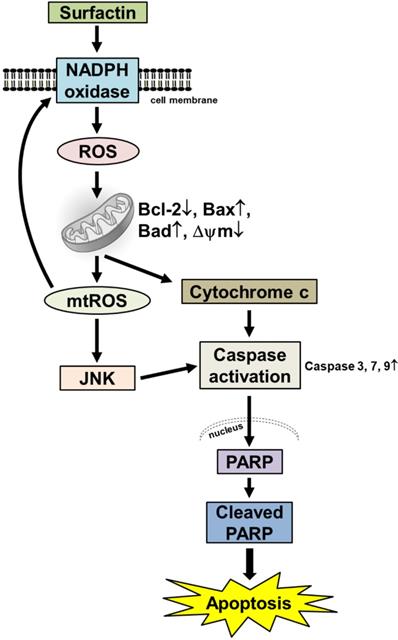
In summary, as depicted in Fig. 5, our results showed that in SCC4 and SCC25 cells, surfactin-induced apoptosis was linked to the activation of caspase and PARP cleavage mediated by the mitochondrial pathway which demonstrated by mitochondrial depolarization, cytochrome c release, and down-regulation of the antiapoptotic Bcl-2 protein. Intracellular ROS production seems to play a crucial role in the activation of the mitochondrial pathway as well as the induction of apoptosis after exposure to surfactin. Oxidative stress due to treatment with surfactin was associated with JNK1/2 activation. After treatment with surfactin, ROS generated a specific environment that lead to JNK1/2-induced cell death. These findings collectively suggest that surfactin is a potential chemotherapeutic agent for the treatment of human OSCC.
Proposed model to illustrate the anticancer activity of surfactin in SCC4 and SCC25 cells. Surfactin reduced the viability of SCC4 and SCC25 cells by induction of apoptosis. Apoptosis promoted by surfactin was associated with activation of caspase and PARP cleavage and was regulated by the mitochondrial pathway, as exemplified by mitochondrial depolarization, cytochrome c release, and down-regulation of the antiapoptotic Bcl-2 protein. Surfactin induced intracellular ROS generation. Intracellular ROS production appeared essential for the activation of the mitochondrial pathway and induction of apoptosis after exposure to surfactin. Oxidative stress due to treatment with surfactin was associated with JNK1/2 activation, as determined by JNK1/2 phosphorylation. After treatment with surfactin, ROS provided a specific environment that resulted in JNK1/2-induced cell death. These findings suggest that surfactin is a potential chemotherapeutic agent for the treatment of oral squamous cell carcinoma.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the Taipei Medical University, grant number TMU108-AE1-B10; the Taipei Medical University Hospital, grant number 109TMU-TMUH-16.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Myers D, Allen E, Essa A, Gbadamosi-Akindele M. Rapidly growing squamous cell carcinoma of the tongue. Cureus. 2020;12:e7164-7
2. Chu YH, Kao SW, Tantoh DM, Ko PC, Lan SJ, Liaw YP. Association between fine particulate matter and oral cancer among Taiwanese men. J Investig Med. 2019;67:34-8
3. Hwang J, Bae H, Choi S, Yi H, Ko B, Kim N. Impact of air pollution on breast cancer incidence and mortality: a nationwide analysis in South Korea. Sci Rep. 2020;10:5392-8
4. Zhang Z, Zhu D, Cui B, Ding R, Shi X, He P. Association between particulate matter air pollution and lung cancer. Thorax. 2020;75:85-7
5. Ko EY, Moon A. Natural Products for Chemoprevention of Breast Cancer. J Cancer Prev. 2015;20:223-31
6. Wang J, Jiang YF. Natural compounds as anticancer agents: Experimental evidence. World J Exp Med. 2012;2:45-57
7. Kornienko A, Evidente A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem Rev. 2008;108:1982-2014
8. Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768-80
9. Ulbricht CE, Chao W. Phytochemicals in the oncology setting. Curr Treat Options Oncol. 2010;11:95-106
10. Perry MC, Demeule M, Régina A, Moumdjian R, Béliveau R. Curcumin inhibits tumor growth and angiogenesis in glioblastoma xenografts. Mol Nutr Food Res. 2010;54:1192-201
11. Filippi-Chiela EC, Villodre ES, Zamin LL, Lenz G. Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PLoS One. 2011;6:e20849-61
12. Tzeng CW, Yen FL, Lin LT, Lee CW, Yen MH, Tzeng WS, Lin CC. Antihepatoma activity of Artocarpus communis is higher in fractions with high artocarpin content. ScientificWorldJournal. 2014;2014:978525-32
13. Tzeng CW, Tzeng WS, Lin LT, Lee CW, Yen FL, Lin CC. Enhanced autophagic activity of artocarpin in human hepatocellular carcinoma cells through improving its solubility by a nanoparticle system. Phytomedicine. 2016;23:528-40
14. Savitskaya MA, Onishchenko GE. Mechanisms of Apoptosis. Biochemistry (Mosc). 2015;80:1393-405
15. Park SY, Kim JH, Lee YJ, Lee SJ, Kim Y. Surfactin suppresses TPA-induced breast cancer cell invasion through the inhibition of MMP-9 expression. Int J Oncol. 2013;42:287-96
16. Cao X, Wang AH, Jiao RZ, Wang CL, Mao DZ, Yan L, Zeng B. Surfactin induces apoptosis and G2/M arrest in human breast cancer MCF-7 cells through cell cycle factor regulation. Cell Biochem Biophys. 2009;55:163-71
17. Wang CL, Liu C, Niu LL, Wang LR, Hou LH, Cao XH. Surfactin-induced apoptosis through ROS-ERS-Ca2+-ERK pathways in HepG2 cells. Cell Biochem Biophys. 2013;67:1433-9
18. Lee IT, Lin CC, Hsu CK, Wu MY, Cho RL, Yang CM. Resveratrol inhibits Staphylococcus aureus-induced TLR2/MyD88/NF-κB-dependent VCAM-1 expression in human lung epithelial cells. Clin Sci (Lond). 2014;127:375-90
19. Brentnall M, Rodriguez-Menocal L, De Guevara RL, Cepero E, Boise LH. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14:32-40
20. Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31:S170-80
21. Fleury C, Mignotte B, Vayssière JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131-41
22. Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84:581-90
23. Lee IT, Yang CM. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013;2013:791231-42
24. Oh LJ, Phan K, Kim SW, Low TH, Gupta R, Clark JR. Elective neck dissection versus observation for early-stage oral squamous cell carcinoma: Systematic review and meta-analysis. Oral Oncol. 2020;105:104661-65
25. Wu YS, Ngai SC, Goh BH, Chan KG, Lee LH, Chuah LH. Anticancer activities of surfactin and potential application of nanotechnology assisted surfactin delivery. Front Pharmacol. 2017;8:761-82
26. Zhang H, Li Z. microRNA-16 via Twist1 inhibits EMT induced by PM2.5 exposure in human hepatocellular carcinoma. Open Med. 2019;14:673-82
27. Yaacoub K, Pedeux R, Tarte K, Guillaudeux T. Role of the tumor microenvironment in regulating apoptosis and cancer progression. Cancer Lett. 2016;378:150-9
28. Bilsland J, Harper S. Caspases and neuroprotection. Curr Opin Investig Drugs. 2002;3:1745-52
29. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;2016:4350965-82
30. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44-84
31. James AM, Cochemé HM, Murphy MP. Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech Ageing Dev. 2005;126:982-6
32. Lee IS, Lim J, Gal J, Kang JC, Kim HJ, Kang BY, Choi HJ. Anti-inflammatory activity of xanthohumol involves heme oxygenase-1 induction via NRF2-ARE signaling in microglial BV2 cells. Neurochem Int. 2011;58:153-60
33. Tzeng CW, Tzeng WS, Lin LT, Lee CW, Yen MH, Yen FL, Lin CC. Artocarpus communis induces autophagic instead of apoptotic cell death in human hepatocellular carcinoma cells. Am J Chin Med. 2015;43:559-79
34. Eisele SC, Wen PY, Lee EQ. Assessment of brain tumor response: RANO and its offspring. Curr Treat Options Oncol. 2016;17:35-47
35. Yang H, Xie Y, Yang D, Ren D. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget. 2017;8:25310-22
36. Chen X, Zhang Y, Jiang S, Huang S. Maduramicin induces apoptosis through ROS-PP5-JNK pathway in skeletal myoblast cells and muscle tissue. Toxicology. 2019;424:152239-16
37. Wang H, Zhang T, Sun W, Wang Z, Zuo D, Zhou Z, Li S, Xu J, Yin F, Hua Y, Cai Z. Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2016;7:e2247-58
Author contact
![]() Corresponding author: I-Ta Lee, School of Dentistry, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan, 250 Wuxing St. Taipei 11031, Taiwan. Tel: +886-2-27361661 ext. 5162; Fax: +886-2-27362295; E-mail: itlee0128edu.tw.
Corresponding author: I-Ta Lee, School of Dentistry, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan, 250 Wuxing St. Taipei 11031, Taiwan. Tel: +886-2-27361661 ext. 5162; Fax: +886-2-27362295; E-mail: itlee0128edu.tw.