Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(19):5802-5811. doi:10.7150/jca.46697 This issue Cite
Research Paper
Moxidectin induces Cytostatic Autophagic Cell Death of Glioma Cells through inhibiting the AKT/mTOR Signalling Pathway
Jingjing Liu1,5#, Hongsheng Liang2#, Saadia Khilji3, Haitao Li1, Dandan Song1, Chen Chen1, Xiaoxing Wang1, Yiwei Zhang2, Ning Zhao1, Xina Li4, Aili Gao1 ![]()
1. School of Life Science, Northeast Agricultural University, Harbin, Heilongjiang, China.
2. Department of Neurosurgery, The First Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang, China.
3. Department of Cellular and Molecular Medicine, Faculty of Medicine, University of Ottawa, Ottawa, Ontario, Canada.
4. Department of Pharmacy, The First Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang, China.
5. College of Life and Health Sciences, Northeastern University, Shenyang, Liaoning, China.
#These authors contributed equally to this work.
Received 2020-4-6; Accepted 2020-7-20; Published 2020-8-3
Abstract
Moxidectin (MOX), a broad-spectrum antiparasitic drug, has been characterized as a potential anti-glioma agent. The main objective of this study was to explore autophagy induced by MOX in glioma U251 and C6 cells, and the deep underlying molecular mechanisms. In addition, the effects of autophagy on apoptosis in glioma cells were tested. Autophagy was measured by transmission electron microscopy (TEM), immunofluorescence, western blot and immunohistochemistry. Cell viability was detected with MTT and colony formation assay. The apoptosis rate was measured by flow cytometry and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL). Additonally, autophagy inhibition was achieved by using 3-Methyladenine (3-MA) and chloroquine (CQ). U251-derived xenografts were established for examination of MOX-induced autophagy on glioma in vivo. Firstly, our research found that MOX stimulated autophagy of glioma cells in a dose-dependent manner. Secondly, we found that MOX induced autophagy by inhibiting the AKT/mTOR signalling pathway. Thirdly, inhibition of autophagy could reduce apoptosis in MOX-treated glioma cells. Finally, MOX induced autophagy, and autophagy increased the apoptosis effect of MOX on U251 in vivo. In conclusion, our data provide evidence that MOX can induce autophagy in glioma cells, and autophagy could increase MOX-induced apoptosis through inhibiting the AKT/mTOR signalling pathway. These findings provided a new prospect for the application of MOX and a novel targeted therapy for the treatment of gliomas.
Keywords: Moxidectin, Glioma, Autophagy, AKT/mTOR
Introduction
Glioma is one of the most common and malignant tumors of the central nervous system, characterized by well-adapted to poorly immunogenic and hypoxic conditions [1, 2]. The current therapies mainly consist of tumour excision, adjuvant radiotherapy and chemotherapeutics. Despite the diverse strategies that have been proposed to improve the current situation, the prognosis for patients with glioma cancer still remains poor [3, 4]. It is necessary to develop new anticancer drugs and assess the molecular mechanisms of glioma cell death.
Recent studies have indicated that some macrocyclic lactones (MLs) were effective against proliferation of tumor cells [5, 6]. Kobayashi T et al. investigated the potential role of clarithromycin addition to lenalidomide and dexamethasone therapy (BiRd) in multiple myeloma [7]. Previous publications have demonstrated that ivermectin induced cytostatic autophagy by blocking the PAK1/Akt axis in Breast Cancer [8]. Meanwhile, Ivermectin inhibited angiogenesis, growth and survival of glioblastoma [9]. These findings provided insights into the anticancer efficacy of MLs, which support a preclinical rational to explore broadening the evaluation of MLs for the treatment of tumors. Among these MLs, moxidectin (MOX) belongs to the milbemycin family and is third generation macrocyclic lactone with potent endectocide activity, and is similar in structure of the avermectins [10, 11]. Specifically, MOX is a new oral treatment for human scabies which provides a solid foundation for considering its potential translation to human diseases [12]. Our previous study demonstrated that MOX had an effect on inhibiting viability of glioma cells in vitro and in vivo by inducing apoptosis and cell cycle arrest. Thus, we propose that MOX might be a potent and promising agent to combat glioma [13].
Conventional anticancer therapies primarily triggered apoptosis to promote cancer cell death [14]. However, accumulating evidence suggested that apoptosis and autophagic cell death could coexist in different chemotherapy drugs to induce cancer cell death [15, 16]. According to morphological appearance, cell death could be classified into apoptosis, autophagy, or necrosis [17]. Moreover, tumors are more autophagy-dependent than normal tissues, suggesting that there is a therapeutic window [18]. Autophagy is a self-degrading process of cellular components characterized by double-membrane autophagosomes which sequester excess or defective organelles and fuse with lysosomes or vacuoles for breakdown by resident hydrolases [19-21]. The role of autophagy in cancer is complex, and this complexity is illustrated by autophagy promoting or suppressing tumorigenesis [22-24]. Therefore, inhibiting or forcing autophagic machinery would be useful in drug cancer treatment [25]. These studies suggest autophagy lysosomal pathway is one such target that may be a promising avenue for the development of novel therapeutic strategies for treating gliomas [26].
In this study, we first detected MOX-induced autophagy in U251 and C6 cells, followed by a discussion of the the deep molecular mechanisms of autophagy. Furthermore, we explored the relationship between apoptosis and autophagy induced by MOX in vitro and vivo.
Materials and Methods
Cell lines and cell culture
Cell lines human U251 glioma and rat C6 glioma were obtained from The First Affiliated Hospital of Harbin Medical University (Harbin, China). Cells were routinely cultured in a humidified incubator at 37°C under 5% CO2 atmosphere, in Dulbecco's minimal essential medium (DMEM; HyClone, USA) supplemented with fetal bovine serum (FBS), 100 U/mL penicillin and 0.1 mg/mL streptomycin.
Reagents and chemicals
MOX was purchased from Sigma-Aldrich/Merck KGaA (Darmstadt, Germany; European Pharmacopoeia Reference Standard), and a purity of > 95%. MOX was dissolved in dimethyl sulfoxide (DMSO). FBS was provided by Zhejiang Dayhang biological technology (Hangzhou, China). Penicillin-streptomycin Solution, Annexin V-FITC Apoptosis Detection Kit was purchased from Beyotime Biotechnology (Shanghai, China). MTT, diaminobenzidine (DAB), polyvinylidene fluoride (PVDF) membranes and hematoxylin were purchased from Sigma-Aldrich/Merck KGaA. The reagents 3-methyladenine (3-MA), chloroquine (CQ) and DMSO were obtained from Sigma-Aldrich/Merck KGaA. Antibodies used in this study were against LC3B (CST, 2775S, USA), P62 (CST, 5114T, USA), AKT (CST, 9272S, USA), Phospho-AKT (Ser473) (Beyotime, AA329, China), mTOR (CST, 2972S, USA), Phospho-mTOR (S2448) (Boster, BM4840, China), p70S6K (Bioss, bs-6370R, China), Phospho-p70S6K (Ser417) (Bioss, bs-5668R, China), GSK3β (Bioss, bs-0023RR, China), Phospho-GSK3β (Bioss, bs-0028RR, China), Ki67 (Bioss, bs-23103, China), β-actin (Sigma-Aldrich, A1978, Germany), goat anti-rabbit immunoglobulin G (IgG) (H+L)-horseradish peroxidase (HRP; SunGene GmbH, LK2001, Germany) and goat anti-mouse IgG (H+L)-HRP (OriGene Technologies, ZB-2305, USA).
Transmission electron microscopy
Transmission electron microscopy (TEM) was used to analyze morphological characteristics of autophagy. U251 and C6 cells were seeded in incubation bottles for 24 h. Then cells were treated with MOX for 48 h, the untreated cells served as control group. Then the cells were harvested, washed and fixed overnight with 2.5% glutaraldehyde containing 1% tannic acid at 4°C. After washing, the cells were dehydrated using a graded series of ethanol solutions, 10 min at a time, transferred to propylene oxide and embedded in epon araldite (Polysciences, Inc., Polybed 812, USA). The ultrathin sections were observed using a JEM-100CX transmission electron microscope (Shanghai Yongming Automatic Equipments Co. Ltd., H-7650, China) and representative images were photographed and analyzed.
Plasmid transfection
MOX treatment resulted in marked autophagy induction in glioma cells, which was proved by GFP-LC3 puncta. Transfection with GFP-LC3 plasmid was obtained from The First Affiliated Hospital of Harbin Medical University. U251 and C6 cells (1×106 cells/well) were seeded in 6-well plates overnight at 37°C. Lipofectamine 2000 (Invitrogen, USA) and GFP-LC3 plasmid were added according to the manufacturer's instructions for 4 h. Then, cells were fixed with 4% paraformaldehyde (Sigma) for 30 min, washed three times with phosphate-buffered saline (PBS). The treated cells were visualized with an inverted fluorescence microscope (Feica, Germany). The cells treated with DMSO were used as control. Nuclei were stained with DAPI (Invitrogen, USA).
Western blot assay
Western blot was used to investigate the expression of autophagy-associated proteins. After treatment, U251 and C6 cells were lysed with RIPA buffer supplemented with PMSF (Beyotime, China) and quantified using the BCA protein concentration assay kit (Beyotime, China). Protein samples were separated by sodium dodecyl sulfate-polyacrylamide Gel electrophoresis (Bio-Rad, USA) and electrotransferred to a PVDF (0.22 μm). After blocking with 5% non-fat milk, the membranes were incubated with primary antibodies overnight at 4°C. The next day, the membranes were labelled with secondary antibody. Signals were detected using MiniChemi Imager (SageCreation, Beijing, China). β-actin was used as the endogenous control.
Cell viability assay
Cell viability was examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. C6 (2×104 cells/well) and U251 (1×104 cells/well) cells were seeded into a 96-well plate respectively overnight in 5% CO2 at 37℃. And the cells were pretreated with CQ (15 μM) for 1 h before being exposed to MOX. Next, Cells were incubated with 5 mg/mL MTT reagent for another 4 h at 37°C. After the medium was carefully removed, 150 μL of DMSO was added and agitated to dissolve the formazan crystals. The absorbance was recorded at 490 nm on an enzyme-linked immunosorbent assay reader (HUADONG-Y, China).
The long-term effects of MOX on tumor cell proliferation were analyzed with a colony formation assay. U251 and C6 cells were seeded into a 6-well plate. Next the cells were treated with CQ (15 μM) for 1 h before being exposed to MOX and cultured in medium for 13 days. The medium was refreshed every three days. Thereafter, the cells were washed with PBS three times, fixed with 4% paraformaldehyde and stained with 0.1% crystal violet (Sigma, USA). Quantification of colony formation was also performed using ImageJ software.
Flow cytometry
The cells were stained with Live-Dead to discriminate living cells. C6 and U251 cells (2.5×105 cells/well) were seeded into 6-well plates and pretreated with CQ (15 μM) for 1 h before being exposed to MOX. Cells were harvested, washed in ice-cold PBS, and then resuspended in PI/Annexin-V solution for apoptosis analysis according to the manufacturer's instructions. Apoptosis ratio was measured using a BD Biosciences FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The results were quantified using the Cell Quest software (BD Biosciences, USA), and apoptosis was calculated as percentage of early and late apoptotic cells.
Tumor xenograft mouse models
All animal experiments were carried out in Harbin Vic Biological Technology Development Co., Ltd., Harbin, China (Experiment number: SY-2017-Mi-027). 5-week-old female Balb/c nude mice (Beijing vitonlihua experimental animal technology co. Ltd, Beijing, China) were treated with U251 cells (2.0×106) via subcutaneous injection. When the tumor reached 70 mm3, all mice were randomized into four groups: 1) Control group, treated with 100 μL of saline; 2) CQ group, treated with 20 mg/kg/day CQ in 100 μL; 3) MOX group, treated with 20 mg/kg/day MOX in 100 μL; 4) MOX+CQ group, treated with 20 mg/kg/day CQ combined with 20 mg/kg/day MOX in 100 μL. All drugs were administered via intraperitoneal injections every day. Tumor size was measured with vernier caliper and calculated as volume (mm3) using the equation: V=0.5×length×width2. After 24 days, all mice were euthanized with ether anesthesia, and tumors were dissected and frozen in liquid nitrogen.
Immunohistochemistry
Solid tumors were removed from sacrificed mice and embedded into paraffin (Citotest, China). Paraffin-embedded tumor tissues were sectioned to 5 μm thickness and labeled with antibody LC3B, Ki67 by HRP-conjugated secondary antibody. Immunoreactivity was visualized by incubation with DAB. Hematoxylin was used for background counterstaining. Images were acquired using fluorescence microscopy (Feica, Germany) and analyzed with ImageJ software (version 2.0).
TUNEL assays
Tumor paraffin-embedded sections were stained with the TUNEL technique using an In situ Cell Death Detection Kit to evaluate the apoptotic response of tumor tissues, Fluorescein (Roche Diagnostics, Mannheim, Germany). After being deparaffinized and hydrated, slides were washed with PBS twice and incubated with proteinase K (20 μg/ml) for 25 min at 37°C. After a second round of washes, slides were incubated with TUNEL reaction mixture prepared freshly for 1 h at 37°C in a moist chamber. Images were acquired using fluorescence microscopy (Feica, Germany).
Quantitative and statistical analysis
For quantification, the intensity of bands in western blot was measured by ImageJ software (version 2.0). The values subsequent to normalizing to the loading control in the control groups were set as 1.0. LC3B, Ki67 staining intensity was measured from the number of positive cell nuclei in 25% fields using ImageJ software.
All experiments were repeated at least three times. Data are presented as the mean ± standard deviation (SD). One-factor analysis of variance (ANOVA) test was used to assess the differences among all the experiment and control or MOX groups. GraphPad Prism Package (version 5.0) and SPSS version 20.0 statistical software were used for statistical analysis. P<0.05 was considered to be indicative of a statistically significant difference.
Results
MOX stimulated autophagy of glioma cells in vitro
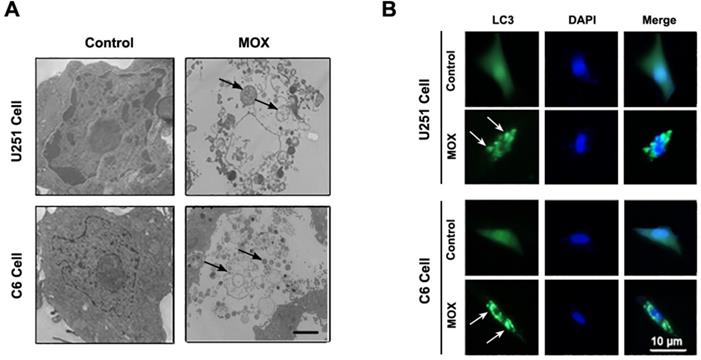
The effect of MOX on the induction cell autophagy in glioma cells was examined by TEM and GFP-LC3 transfection. As shown in Fig. 1A, TEM revealed obviously characteristic autophagosomes in U251 and C6 cells treated with MOX but not in control cells. Arrows indicated autophagosomes containing intact and degraded cellular debris. What's more, we determined the induction of autophagy by GFP-LC3 transfection. As shown in Fig. 1B, we observed abundant LC3 puncta in 48 h MOX-treated cells compared with untreated U251 and C6 cells. These data demonstrated that MOX stimulated autophagy of glioma cells in vitro.
MOX stimulated autophagy of glioma cells in vitro. (A) U251 and C6 cells were treated with MOX (0, 20 µM) for 48 h and analyzed by TEM. (B) U251 and C6 cells were transfected with GFP-LC3 plasmid then the cells were treated with DMSO (< 0.1%) or 20 µM MOX for 48 h. To determine the autophagic response, cells were inspected at 40× magnification for numbers of GFP-LC3 puncta. Scale bar, 10 µm.
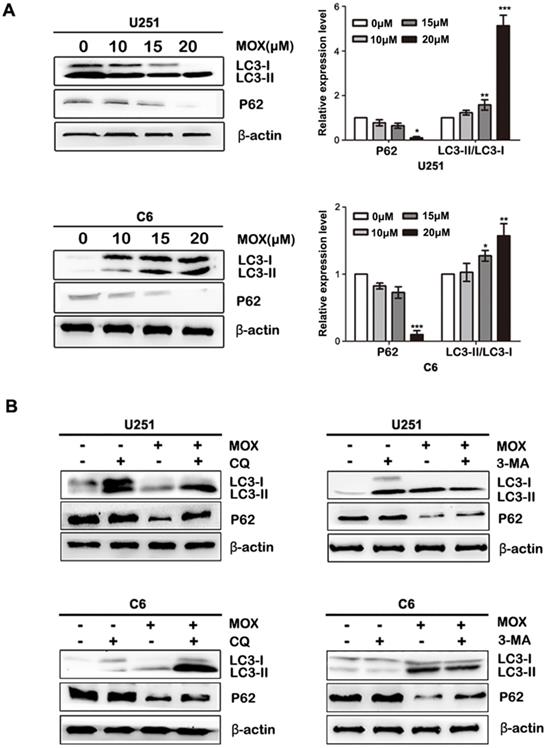
Dose-dependent effect of MOX on an autophagy-related protein LC3
In order to investigate the mechanism involved in MOX-mediated cell autophagy, the proteins related to cell autophagy were measured by western blot. As shown in Fig. 2A, LC3-II/LC3-I expression was more pronounced, and the level of P62 was decreased in U251 and C6 cells with an increased dose of MOX. Next, to explore whether MOX-induced of LC3 LC3-II/LC3-I is due to autophagy induction or the inhibition of autolysosomal function, CQ and 3-MA were used to inhibit autophagic flux. As shown in Fig. 2B, pretreatment with 3-MA resulted in the decrease of LC3-II/LC3-I compared with the MOX group in U251 and C6 cells. Meanwhile, inhibition of autophagy by using CQ, enhanced the expression of LC3-II/LC3-I compared with MOX use alone. Taken together, the result suggested that MOX could stimulate autophagy of U251 and C6 cells in a dose-dependent manner.
Dose-dependent effect of MOX on an autophagy-related protein LC3 (A) U251 and C6 cells were exposed to different concentrations (0, 10, 15, 20 µM) of MOX for 48 h. The LC3B and P62 levels were examined by western blot. (B) U251 and C6 cells were treated with 20 µM MOX in the absence or presence of CQ (15 µM) or 3-MA (5 µM) for 48 h. LC3B and P62 expressions were examined by western blot. Data were presented as the means ± SD of three independent tests. *P < 0.05, **P < 0.01, ***P < 0.001 as compared with control group. β-actin was used as an internal standard.
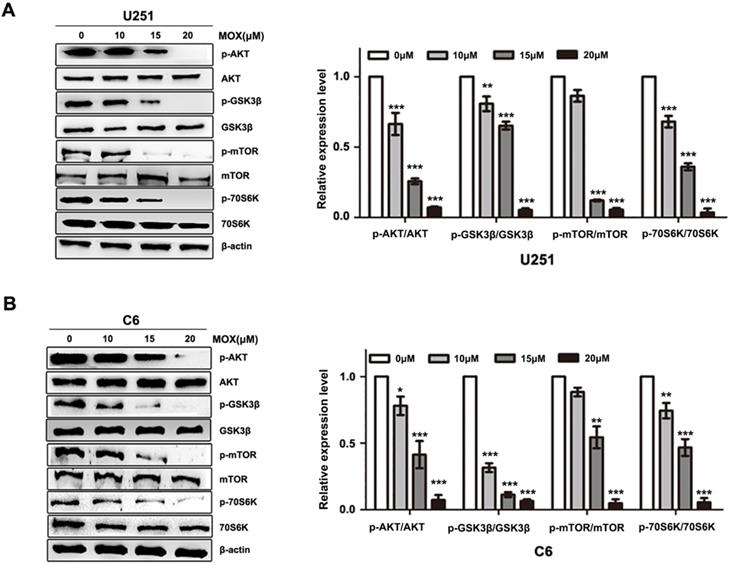
AKT/mTOR pathway is involved in MOX-induced autophagy of glioma cells
To elucidate the underlying mechanism of MOX on autophagy, AKT/mTOR pathway was investigated in MOX-induced autophagy. Compared with control group, p-AKT (Ser473) and p-mTOR (S2448) were decreased with MOX treatment in a dose-dependent manner, and the downstream protein p-p70S6K (Ser417) and p-GSK3β were reduced in U251 and C6 cells (Fig. 3A, B). These data together implied that AKT/mTOR pathway is involved in MOX-induced autophagy in U251 and C6 cells.
AKT/mTOR pathway is involved in MOX-induced autophagy of glioma cells. (A, B) U251 and C6 cells were incubated with MOX at different concentrations (0, 10, 15, 20 µM) for 48 h, the effects of MOX on the levels of AKT, p-AKT (Ser473), mTOR, p-mTOR (S2448), P70S6K, p-P70S6K (Ser417), GSK3β, p-GSK3β were examined by western blot. Data were presented as the means ± SD of three independent tests. *P < 0.05, **P < 0.01, ***P < 0.001 as compared with control group. β-actin was used as an internal standard.
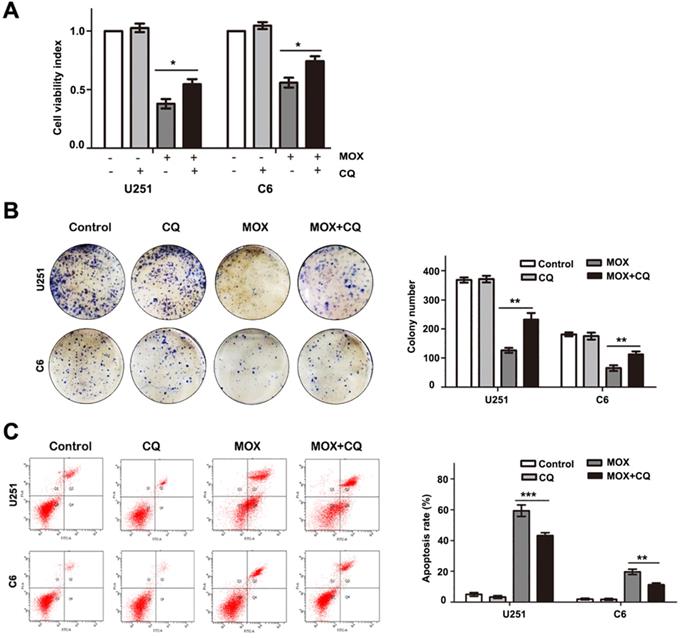
Inhibition autophagy repressed MOX-induced apoptosis in glioma cells
To determine the role of autophagy in MOX-induced cell apoptosis, we treated cells with CQ for 1 h to block autophagy before MOX treatment. Cell growth was assessed by MTT assay and colony formation analysis. As shown in Fig. 4A-B, co-treatment with CQ, the cell growth ability was increased in U251 and C6 cells. Then, flow cytometry analysis was performed using Annexin V-PI double staining. As shown in Fig. 4C, inhibition of autophagy by CQ in MOX-treated cells reduced the percentage of apoptotic cells compared with those treated with MOX alone. Specifically, the fraction of apoptotic cells reduced from 59.10±5.31% in MOX-treated U251 cells to 45.50±2.65% in co-treated cells and the apoptotic cell percentage reduced from 17.02±2.49% to 11.70%±1.67 in C6 cells. These results showed that inhibition of autophagy repressed MOX-induced apoptosis in U251 and C6 cells.
Inhibition of autophagy repressed the apoptosis effect of MOX on glioma cells. U251 cells and C6 cells were pretreated with CQ (15 µM) for 1 h before being exposed to 20 µM MOX. Cell viability was measured by MTT (A) and colony formation analysis (B). (C) The percentage of apoptotic cell was evaluated by flow cytometry after cells were incubated with MOX (20 µM) in the presence or absence of CQ (15 µM). Q2 plus Q4 areas were calculated as the apoptosis ratio. Data were presented as the means ± SD of three independent tests. **P < 0.01, ***P < 0.001 as compared with MOX group.
MOX induced autophagy in vivo
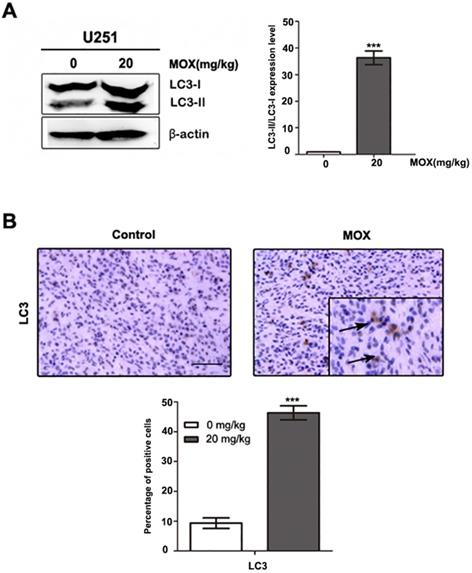
A series of therapeutic experiments were conducted in U251 cell xenograft mouse models. As shown in Fig. 5A, a massive LC3B accumulation was detected on tumor sections in MOX-treated xenografts compared with the control group. Meanwhile, we observed LC3-II expression was more pronounced in MOX-treated tumors (Fig. 5B). Together, these findings showed that MOX obviously induced autophagy in vivo.
MOX induced autophagy of glioma cell in vivo. (A) Orthotopic xenograft tissues were extracted to assess the levels of LC3-II by Western blot analysis. (B) LC3 expression in orthotopic xenografts was examined by immunohistochemical. Arrows indicate positive cells which stained brown. Scale bar, 50 µm. Data were presented as the means ± SD of three independent tests. β-actin was used as an internal standard. ***P < 0.001.
Inhibition autophagy reduced the apoptosis effect of MOX in vivo
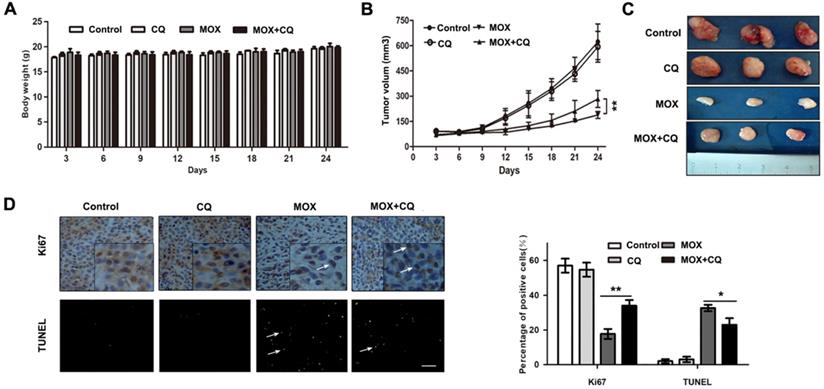
Next, to confirm the potential effect of autophagy induced by MOX on glioma tumor in vivo, an internal engraftment model was established and tested. As shown in Fig. 6A, no significant difference in the weights of the mice was observed between the experimental groups after all measured days. As shown in Fig. 6B/C, there is no significant difference in the tumor weight or volume between the control and CQ groups, but tumor weight and volume in MOX group were markedly lower than in control group. Furthermore, compared with MOX group, co-treatment of MOX and CQ was less effective in reducing the tumor size. Finally, as shown in Fig. 6D, TUNEL assay demonstrated a greater number of dead cells and an evident increase in the apoptotic proportion in MOX treated tumor tissues compared with the MOX+CQ treated tumor tissues. Immunohistochemistry was performed to label Ki 67, which was used to confirm the change in the proliferation status of the tumors. MOX+CQ xenografts displayed stronger Ki67 staining compared with that of MOX treated mice. The result demonstrated the less dead cells in MOX+CQ group compared with the MOX group. In generally, these data recapitulated the observations made in vitro and showed that autophagy increased the apoptotic effects of MOX on U251 in vivo.
Inhibition of autophagy reduced the apoptosis effect of MOX on glima cell in vivo. (A) Charting of mouse weight with time. Mice were sacrificed days after the indicated treatments, the tumor volume (B, C) were measured. (D) Immunohistochemistry staining result of Ki67 and TUNEL assay on tumor sections. Scale bar, 50 µm. Data were presented as the means ± SD of three independent tests. *P < 0.05, **P < 0.01.
Discussion
Gliomas are one of the primary brain tumors [27]. In our previous work, we proved that MOX could induce apoptotic death in glioma cells, MOX may represent a potent and promising agent to combat glioma [13]. Recent researches have demonstrated that autophagy can improve anti-cancer therapy and have an integral role in tumor maintenance. Research into autophagy is underway to better direct therapies [28, 29]. In this study, we first described autophagy phenomenon in glioma cells after MOX treatment. Second, we explored the mechanisms involved in MOX-mediated cell autophagy. Third, the relationship between cell apoptosis and autophagy in glioma cells treated with MOX was discussed.
Autophagy is a key mechanism for maintaining cell homeostasis by adjusting cell metabolism to nutrient supply and removing damaged organelles [30]. Research has demonstrated that chemotherapy or radiotherapy could activate autophagy [31]. For example, Liu R et al. showed that itraconazole inducted autophagy in glioblastoma cells [32], and Zhao WY et al. demonstrated that isogambogenic acid inhibited the growth of glioma through activating autophagy [33]. Conversion of LC3-I into LC3-II is widely used as a marker for autophagosome formation. LC3-I is the soluble form while LC3-II is the autophagosome membrane bound form [34]. P62 is incorporated in completed autophagosomes and degraded in autolysosomes [35]. In this study, we found MOX could induce autophagy in U251 and C6 cells by TEM, GFP-LC3 transient transfection and western blot analysis. In addition, Li CG et al. proved that 3-MA inhibited the initial step of autophagy, and CQ inhibited autophagosome-lysosome fusion and degradation steps in glioblastoma Cells [36]. Therefore, CQ and 3-MA were used as autophagy inhibitors to investigate MOX-induced up regulation of LC3B because of autophagy induction. Thus, we show that MOX induced autophagy in U251 and C6 cells.
Furthermore, we explored the signalling pathways regulating autophagy. The well characterized AKT/mTOR pathway is an attractive therapeutic target that contributes to the initiation and maintenance of cancer [37]. AKT signalling has an important role in the regulation of cell proliferation, angiogenesis, migration and invasion. Chronologically, AKT can stimulate p-mTOR that is an upstream protein [38-40]. Cell growth is partly regulated through mTOR [41, 42]. In our study, p-AKT, p-mTOR, p-p70S6K and p-GSK3β were decreased in U251 and C6 cells after MOX treatment. These results further indicated that MOX induced autophagy in U251 and C6 cells through the AKT/mTOR signalling pathway.
Some studies indicated that autophagy induced by therapeutic interventions can cause the death of cancer cells that are resistant to apoptosis [43-46]. However, the relationship between autophagy and apoptosis in cancers is complex. In our previous study, MOX could induce cell apoptosis of glioma cells. Thus, the relationship of apoptosis with autophagy is deemed necessary for investigation. The autophagy flux of glioma was inhibited by CQ which could decrease lysosomal function [47, 48]. The results indicated that the effect of autophagy inhibitor involved in autophagy and apoptosis. The inhibition of autophagy increased the cell ability measured by MTT and colony formation assay, and reduced apoptosis measured by flow cytometry in glioma cells. These findings suggested that autophagy played a significant role in MOX-induced cell death and that inhibition autophagy observably reduced the anti-tumor effects of MOX.
At the same time, the mechanisms of MOX in mouse xenograft models were performed. Previous study showed that MOX resulted in inhibition of the tumor growth without overall gross toxicity. The selected dose of the MOX injections was 20 mg/kg, and MOX could reduce the tumor mass of U251 xenografts [13]. Western blot and immunohistochemistry analysis confirmed the increase in LC3B following MOX treatment. These results demonstrated that MOX can induce autophagy of glioma in vivo. It is reported that CQ can be regarded as autophagy inhibitor in vivo [49, 50]. In follow-up experiment, we selected the dose of CQ injections as 20 mg/kg, where it did not exert a significant inhibitory effect on tumor growth. In this study, it was found that the intraperitoneal injection of MOX prominently inhibited the growth of glioma cells compared with MOX+CQ group and neither showed any abnormality in behavior nor significant major organ-related toxicity in vivo. As a result, we recapitulated the observations made in vitro and showed that autophagy enhanced the anti-glioma treatment effects of MOX. Further studies in glioma cancer animal models as well as in human clinical trials are necessary.
In conclusion, our results demonstrated that MOX can induce autophagy in glioma cells while exploring some of the underlying molecular mechanisms. Moreover, we stated that the AKT/mTOR signalling pathway contributed to MOX-induced autophagy in glioma cells. Apoptosis was an important cause of death of glioma cells, while autophagy induced by MOX promoted apoptosis. This study provided a new prospect for the application of MOX and modulating autophagy may represent an effective strategy for the treatment of glioma.
Acknowledgements
The authors would like to thank Qiao Li from the University of Ottawa (ON, Canada) for assisting in the preparation of the manuscript. This work was supported by a grant from the National Natural Science Foundation of China [No. 81201723 and 81501050].
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institutional and/or national research committee (Harbin Vic Biological Technology Development Co., Ltd., Harbin, China). The protocol was approved by the management and welfare of experimental animal ethics committee, Animal Experiment of Harbin Vic Biological Technology Development Co., Ltd., Harbin, China.
Authorship
Aili Gao performed conception and design, experiments performing, data analysis. Jingjing Liu contributed to experiments performing. Hongsheng Liang, Dandan Song and Chen Chen performed data analysis. Xiaoxing Wang, Yiwei Zhang, Ning Zhao and Xina Li contributed to conception and design, data analysis, manuscript writing. Haitao Li and Saadia Khilji contributed to financial support, and final approval of manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wen PY, Kesari S. Malignant gliomas in adults. New Eng J Med. 2008;359:492-507
2. Louis DN, Ohgaki H, Wiestler OD. et al. The WHO classification of tumors of the central nervous system. Acta Neuropathol. 2017;114:97-109
3. Stupp R, Mason WP, van den Bent MJ. et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New Eng J Med. 2005;352:987-996
4. Furnari FB, Fenton T, Bachoo RM. et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Gene Dev. 2007;21:2683-2710
5. Korystov YN, Ermakova NV, Kublik LN. et al. Avermectins inhibit multidrug resistance of tumor cells. Eur J Pharmacol. 2004;493:57-64
6. Mani T, Bourguinat C, Keller K. et al. Interaction of macrocyclic lactones with a Dirofilaria immitis P-glycoprotein. Int J Parasitol. 2016;46:631-640
7. Kobayashi T, Miura M, Abumiya M. et al. The potential role of clarithromycin addition to lenalidomide and dexamethasone therapy (BiRd) in multiple myeloma. Ann Hematol. 2018;97:1097-1099
8. Dou QH, Chen HN, Wang K. et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016;76:4457-4469
9. Liu YY, Fang SS, Sun QS. et al. Anthelmintic drug ivermectin inhibits angiogenesis, growth and survival of glioblastoma through inducing mitochondrial dysfunction and oxidative stress. Biochem Bioph Res Commun. 2016;480:415-421
10. Cotreau MM, Warren S, Ryan JL. et al. The antiparasitic moxidectin: safety, tolerability, and pharmacokinetics in humans. J Clin Pharmacol. 2003;43:1108-1115
11. Prichard R, Menez C, Lespine A. Moxidectin and the avermectins: Consanguinity but not identity. Int J Parasitol Drugs Drug Resist. 2012;2:134-152
12. Mounsey KE, Bernigaud C, Chosidow O. et al. Prospects for Moxidectin as a New Oral Treatment for Human Scabies. PLOS Negl Trop Dis. 2016;10:1-8
13. Song DD, Liang HS, Qu B. et al. Moxidectin inhibits glioma cells proliferation by inducing G0/G1 cell cycle arrest and apoptosis in vitro and in vivo. Oncol Rep. 2018;40:1348-1358
14. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178-194
15. De Bruin EC, Medema JP. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat Rev. 2008;34:737-749
16. Shin SY, Lee KS, Choi YK. et al. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis. 2013;34:2080-2089
17. Gu JJ, Zhang JH, Chen HJ. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Oncol Lett. 2009;12:5015-5022
18. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res. 2015;21:5037-5046
19. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signl. 2014;20:460-473
20. Mizushima N. Autophagy: process and function. Gen Dev. 2007;21:2861-2873
21. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell biol. 2007;9:1102-1109
22. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999-2010
23. He CC, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67-93
24. Fimia GM, Piacentini M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci. 2010;67:1581-1588
25. Giansanti V, Torriglia A, Scovassi AI. Conversation between apoptosis and autophagy: Is it your turn or mine? Apoptosis. 2011;16:321-333
26. Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S. et al. The autophagic tumor stroma model of cancer or battery-operated tumor growth: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297-4306
27. Kruchko C, Ostrom QT, Gittleman H. et al. The CBTRUS story: providing accurate population based statistics on brain and other central nervous system tumors for everyone. Neuro-Oncology. 2018;20:295-298
28. Bush NAO, Chang SM, Berger MS. Current and future strategies for treatment of glioma. Neurosure Rev. 2017;40:1-14
29. Moretti L, Yang ES, Kim KW. et al. Autophagy signaling in cancer and its potential as novel target to improve anti-cancer therapy. Drug Resist Update. 2007;10:135-143
30. Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell. 2011;147:728-741
31. Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25:37-45
32. Liu R, Li J, Zhang T. et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy involvement of abnormal cholesterol trafficking. Autophagy. 2014;10:1241-1255
33. Zhao WY, Peng F, Shu MT. et al. Isogambogenic acid inhibits the growth of glioma through activation of the AMPK-mTOR pathway. Cell Physiol Biochem. 2017;44:1381-1395
34. Giatromanolaki A, Sivridis E, Mitrakas A. et al. Autophagy and lysosomal related protein expression patterns in human glioblastoma. Cancer Biol Ther. 2014;15:1468-1478
35. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672-4678
36. Li CG, Liu YH, Liu HL. et al. Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol Biochem. 2015;35:1303-1316
37. LoPiccolo J, Blumenthal GM, Bernstein WB. et al. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist Update. 2008;11:32-50
38. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253-262
39. Dunlop EA, Tee AR. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121-129
40. Hennessy BT, Smith DL, Ram PT. et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988-1004
41. Liu R, Li JY, Zhang T. et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy involvement of abnormal cholesterol trafficking. Autophagy. 2014;10:1241-1255
42. Shinojima N, Yokoyama T, Kondo Y. et al. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635-637
43. Bursch W, Ellinger A, Gerner C. et al. Programmed Cell Death (PCD) Apoptosis, Autophagic PCD, or Others? Ann N Y Acad Sci. 2000;926:1-12
44. Chi KH, Wang YS, Huang YC. et al. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget. 2016;7:58075-58088
45. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891-2906
46. Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70-77
47. Choi JH, Yoon JS, Won YW. et al. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. Apmis. 2012;120:597-604
48. Kimura T, Takabatake Y, Takahashi A. et al. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013;73:3-7
49. Wang F, Tang JY, Li PC. et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell Physiol Biochem. 2018;45:54-66
50. Yuan G, Yan SF, Xue H. et al. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J Biol Chem. 2014;289:10607-10619
Author contact
![]() Corresponding author: Aili Gao, School of Life Science, Northeast Agricultural University, 600 Changjiang Road, Xiangfang District, Harbin, 150030, P.R. China. Tel.: 86-131-9950-0261; E-mail: gaoailiedu.cn.
Corresponding author: Aili Gao, School of Life Science, Northeast Agricultural University, 600 Changjiang Road, Xiangfang District, Harbin, 150030, P.R. China. Tel.: 86-131-9950-0261; E-mail: gaoailiedu.cn.