Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(15):4308-4315. doi:10.7150/jca.42849 This issue Cite
Research Paper
CDK12 promotes papillary thyroid cancer progression through regulating the c-myc/β-catenin pathway
Ning Bai, Fada Xia, Wenlong Wang, Yao Lei, Jiang Bo, Xinying Li ![]()
Department of General Surgery, Xiangya Hospital, Central South University, Changsha, China.
Received 2019-12-8; Accepted 2020-3-29; Published 2020-4-27
Abstract
Background: CDK12 is a potential therapeutic target in papillary thyroid cancer that regulates the c-myc/β-catenin pathway.
Objective: We aimed to explore the specific mechanism of CDK12 in papillary thyroid cancer and provide a new target of cancer therapy.
Methods: RT-qPCR was used to determine the CDK12 mRNA expression level. An IHC assay was performed to detect the tissue expression of CDK12. Then, we downregulated CDK12 expression in the thyroid cancer cell lines TPC-1-shCDK12 and KAT-5-shCDK12. CCK8 assays, colony formation assays, and animal xenograft models were used to evaluate the effect of CDK12 on tumorigenesis. Transwell assays and in vivo metastasis models were used to observe whether CDK12 can promote cancer metastasis. Western blotting further confirmed the mechanism of CDK12 in papillary thyroid cancer through the c-myc/β-catenin pathway.
Results: Upregulated CDK12 expression in papillary thyroid cancer promoted papillary thyroid cancer carcinogenesis in vivo, and in vitro CDK12 strengthened papillary thyroid cancer (PTC) cell migration and tumor metastasis. CDK12 promoted tumor progression by regulating c-myc/β-catenin pathway activation.
Conclusions: CDK12 affects the c-myc/β-catenin pathway to stimulate papillary thyroid cancer proliferation and metastasis. Inhibiting CDK12 might be a new method in papillary thyroid cancer therapy.
Keywords: CDK12, papillary thyroid cancer, c-myc, β-catenin, cancer progression
Introduction
According to global cancer statistics, thyroid cancer is the fifth fastest growing cancer in the world. A total of 53990 new cases of thyroid cancer were estimated in 2018, the number of female patients will reach 40900 cases, and new cases in men will reach 13090. The incidence of thyroid cancer in women is much higher than in men, and the sex ratio is almost 3:1[1]. Papillary thyroid cancer (PTC) is one subtype of thyroid cancer. PTC incidence has risen rapidly in recent years and mainly induces a high growth rate of thyroid cancer [2].Thyroid cancer pathogenesis may be induced by the unhealthy living habits [3, 4] or environmental factors [5]. Although the prognosis of PTC is usually favorable if it is diagnosed in early a stage and is treated well, some patients will still suffer lymph node metastasis, distant metastasis or other vital syndromes. Once metastasis occurs, the prognosis of PTC is poor, and patients will even die [6]. Therefore, it is urgent to reveal the detailed mechanism of PTC genesis and metastasis.
CDK12 is a member of the cyclin-dependent kinase (CDK) familythat relies on the regulatory subunit cyclin to phosphorylate the RNA polymerase II C-terminal domain toregulate cell cycle transition. CDK12 generally binds with cyclin K to maintain genomic stability by repairing DNA damage [7]. According to reports, CDK12 mutation promotes the progression of multiple cancers, such as breast cancer [8], ovarian cancer [9], and prostate cancer [10]. Jerry F. Tien et al analyzed global mRNA transcripts between normal and breast cancer cell lines with and without CDK12 amplification and found that CDK12 primarily regulates alternative last exon (ALE) splicing, which typically regulates DNA damage response activators. CDK12 modulates ALE splicing of the DNA damage response activator ATM and a DNAJB6 isoform that influences cell invasion and tumorigenesis in xenografts [8]. The deregulations of CDK12 induces tumorigenesis via DNA repair defects in ovarian cancer, such as inactivation of the homologous recombination (HR) repair pathway [11]. Inhibiting CDK12 in cancer cells without CDK12 mutations leads to gene length-dependent elongation defects, inducing cleavage maturation early, polyadenylation and loss of the expression of long (>45 kb) genes [12]. Based on the reports on mCRPC, focal tandem duplications (FTDs) associated with CDK12 loss result in highly recurrent gains at loci of genes and marked differential gene expression involved in the cell cycle and DNA replication [13]. In summary, knockdown of CDK12/CycK decreases the levels of nascent transcripts of several DDR genes [14], regulates alternative splicing [15] and interferes with effective 3'-end formation of model premRNAs [7]. Hee-Joo Choi et al found that high CDK12 expression caused by concurrent amplification ofCDK12 and HER2 in breast cancer patients through activating ErbB-PI3K-AKT or WNT-signaling cascades induced disease recurrence and poor survival [16]. This suggests that CDK12 gene amplification contributes to the pathogenesis of cancer. These findings mostly focus on the gene repair pathway of CDK12 in cancer. However, the detailed mechanism of CDK12 in papillary thyroid cancer remains largely unknown.
c-myc is a classical oncogene that can affect the progression of many cancers [17-20]. It was determined that c-myc is the downstream target of wnt/β-catenin. c-myc regulates cell proliferation, differentiation, angiogenesis, and apoptosis[21, 22]. c-Myc translation is activated by β-catenin. β-cateninis critical in many cancers and participates in a broad range of cancer processes, including stem cell self-renewal, stemness-maintaining progression, and tumorigenesis[23, 24]. The c-myc pathway is significantly correlated with poor prognosis, increased possibility of breast-to-lung metastasis and reduced overall survival [25, 26]. Herein, we performed this study to investigate the function of CDK12 and its underlying mechanism in papillary thyroid cancer progression. Our data demonstrated that CDK12 promoted cancer proliferation, migration and metastasis through the c-myc/β-catenin pathway. CDK12 may be a potential target in PTC therapy.
Method and materials
Clinical sample data
Thirty pairs of clinical samples and adjacent normal tissues were collected from patients who were diagnosed with PTC and underwent surgery at Xiangya Hospital, Central South University from 2015 to 2017, aged 22-60 years old. The sectioned cancerous tissues and paired normal mammary tissues were immediately removed and stored in RNAlater (Ambion), and the RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA) and then frozen in liquid nitrogen at -80°C. All patients provided informed consent, and this study was approved by the Medical Ethics Committee of Xiangya Hospital, Central South University.
Immunohistochemistry
According to the EnVision system (DAKO) instructions, the formalin-fixed and paraffin-embedded sections were dewaxed with xylene and graded ethanol. The sections were microwaved in citrate buffer to retrieve the antigens. Then, the sections were incubated in H2O2 solution at routine temperature for 15 min. The sections were incubated with primary antibody (CDK12, 1:100, Cell Signaling Technology) overnight at 4°C. The sections were incubated with labeled polymer-HRP anti-mouse at routine temperature for 30 min. The sections were incubated in substrate-chromogen solution for 5-30 min. The slides were dehydrated with xylene and graded ethanol. Images were taken with an optimal microscope, and the results were quantified by two pathologists. The evaluation of IHC progression followed the blind trial rules.
Cell culture and transfection
PTC cell lines (TPC-1, NPA87 and KAT-5) and the normal cell line Nthy-ori3-1 were from the Chinese Academy of Sciences (Shanghai, China). Cell lines were cultured in DMEM (Gibco, Carlsbad, CA, USA) containing 10% FBS (Gibco) and maintained in a humidified incubator at anatmosphere with 5% CO2. the stable knockdown the CDK12 expression in TPC-1 and KAT-5 cells through using Lenti-Pac™ HIV Expression Packaging Kit (GeneCopoeia) following the manufacturer's instructions.
Cell Counting Kit-8 Assay
The adherent cancer cells were lysed and 500 sh-CDK12 and sh-CTR cancer cells were seeded into a 96-well plate. The plate was incubated for 24, 48, 72, and 96 h in the incubator. Then, 10 μl of CCK-8 solution was added to each well of the plate. The plate was incubated for 1 - 4 h in the incubator. The absorbance was measured at 450 nm using a microplate reader (Bio-Tek EPOCH2, Vermont, USA).
Transwell assay
The cells were washed twice with 1x PBS and trypsinized. Then, a volume of 10% FBS in DMEM equal to the volume of trypsin was added to inactivate the trypsin. The cells were gently centrifuged. The cells were resuspended in DMEM and counted. A total of 1 x 105 cells were added into the transwell compartments. And the mitomycin was added in to the culture to avoid the effect of cancer cell's proliferation. The cells were incubated in the transwell plate at 37 °C and 5% CO2 for 24 h. The upper layer of cells was swabbed. The lower side of the insert filter was rinsed in4% formaldehyde solution for 10 min. Next, the cells were stained with 1% crystal violet for 15 min. The inserts were washed with PBS 4 times. The insert membrane was dried. The number of cells on the lower side of the filter was counted under a NIKON ECLIPSE 80imicroscope (Nikon Instruments, NY, USA). Each migration condition was tested in triplicate.
Animal xenograft model
In brief, 1×107sh-CDK12, sh-CTR TPC-1 or KAT-5 cells were subcutaneously injected into the flanks of nude mice from Charles River (Beijing, China). Each group contained 5 nude mice. Twenty-eight days later, the mice were euthanized and then the tumor volumes were analyzed. The animal research was approved by the ethical committee of Xiangya Hospital, Central South University.
RT-qPCR analysis
qRT-PCR assays were carried out with SYBR Premix Ex Taq™ (Takara, Japan) and an All-in-One™ miRNA qRT-PCR Detection Kit (GeneCopoeia) using a Bio-Rad IQTM5 Multicolor Real-Time PCR Detection System (USA).In brief, total RNA was extracted from PTC tissues and celllines. cDNA was synthesized from 1μg of total RNAin 20 µl reaction volumes using 5x PrimeScript RT Master Mix. PCR amplification was carried out with Taq DNA polymerase using 2µL of the cDNA as a template. The amplification reactions were run with 30 thermocycles of 30s at 94°C, 30 s at 55°C, and 30 s at 72°C. All primers were acquired from Sangon Biotech (Shanghai, China). Forward primer: CDK12: (5'-CTAACAGCAGAGAGCGTCACC-3'), reverse primer: CDK12 (5'-AAAGGTTTGATAACTGTGCCCA-3'). U6 was used as an internal control.
Western blot analysis
The total protein from cells was isolated with RIPA buffer and PMSF. The proteins were denatured at 100°C for 10 min and then quantified. A total of 20-30 μg of protein was loaded into the wells of the SDS-PAGE gel. The gel was run for 2 h at 100 V. The protein was transferred to the PVDF membrane for 2 h at 200 mA. The membrane was blocked in 5% skimmed milk for 1 h at room temperature. The membrane was incubated with a 1:3000 dilution of primary antibody β-catenin (Santa Cruz Biotechnology), wnt-1 (Abcam), c-myc (Abcam), CD44 (Cell Signaling Technology), slug (Cell Signaling Technology) and snail (Cell Signaling Technology), actin (Abcam) incubate the membrane and primary antibody overnight at 4°C and then exchanged the primary antibody with the secondary antibody at room temperature for 1 h.The protein bands were exposed with a ChemiImager System.
In vivo Metastasis Assay
A total of 1×105 sh-CDK12, sh-CTR TPC-1 or KAT-5 cells were injected into nude mice through the tail vein. 5 nude mice were facilitated for each group. Thirty days later, all mice were anesthetized, and the lungs were extracted. The metastatic nodules were counted by eye and then stored in 4% formaldehyde solution for further study.
Statistics
All data are reported as the mean ± SD. The paired t-test was used to evaluate the differences of the tumor tissue and paired adjacent normal tissue. The differences between two groups were determined by Student's two-tailed unpaired t-test. A p value <0.05 was regarded as statistically significant.
Results
Upregulated CDK12expression in papillary thyroid cancer
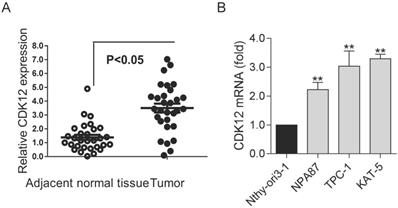
To study the role of CDK12 in PTC, we detected the expression of CDK12 in clinical PTC specimens. We found that CDK12 expression was significantly higher in tumors than in normal tissues (Figure 1A). This result indicated that CDK12 may play an important role in papillary thyroid cancer progression. Then, we detected CDK12 mRNA expression in several common thyroid cell lines. PTC cell lines (TPC-1, NPA87 and KAT-5) had prominently higher CDK12 levels than the normal cell line Nthy-ori3-1. Additionally, the CDK12 mRNA expression levels of TPC-1 and KAT-5 cells were the highest among the papillary thyroid cancer cell lines (Figure 1B). These two cancer cell lines were used for subsequent experiments.
The high expression of CDK12 in PTC: (A) Expression levels of CDK12 in 20 PTC specimens and paired adjacent normal tissues. (B) Expression levels of CDK12 by qRT-PCR in the normal thyroid cell line Nthy-ori3-1 and the TPC-1, NPA87, KAT-5 papillary thyroid cancer cell lines. β-actin was used as the internal control. A P value <0.05 was considered significant. **, P<0.01.
CDK12 promotes papillary thyroid cancer carcinogenesis in vivo and in vitro
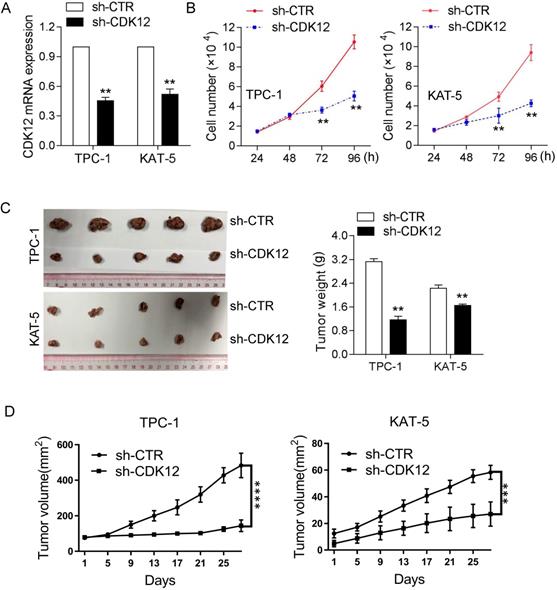
To confirm the function of CDK12 in tumorigenicity, we inhibited CDK12 mRNA expression in the TPC-1 and KAT-5 cancer cell lines (Figure 2A). The CCK8 assay results demonstrated that CDK12 downregulation inhibited cancer cell proliferation (Figure 2B). Animal research was performed to further confirm the role of CDK12 carcinogenesis in vivo. CDK12 expression was knocked down in the PTC-1 and KAT-5 cell lines and the cells were then injected into nude mice in situ. After 4 weeks, the mice were sacrificed to harvest the tumors, and the tumor volumes were measured. CDK12 inhibition resulted in tumors that were obviously smaller than those in the control group (Figure 2C-D). These results demonstrated that CDK12 promotes papillary thyroid cancer proliferation both in vivo and in vitro. Hence, CDK12 may play an important role in papillary thyroid cancer progression.
CDK12 promotes PTC progression in vivo and in vitro. (A): qRT-PCR was used to determine the mRNA expression of CDK12. TPC-1 and KAT-5 cells had effectively decreased mRNA expression of CDK12. (B): A CCK-8 assay was used to detect the cell proliferation ability of TPC-1-sh-CDK12 and KAT-5-sh-CDK12 cells. (C): Representative images of xenografts and the tumor weight statistics in nude mice. The weights of the tumors are shown in the right panel. All results are expressed as the mean±SD of three independent experiments. **, P<0.01. (D): the growth curve of xenografts, TPC-1(left), KAT-5(right), ***, P<0.001, ****, P<0.0001.
CDK12 enhancesPTCcell migration and tumor metastasis
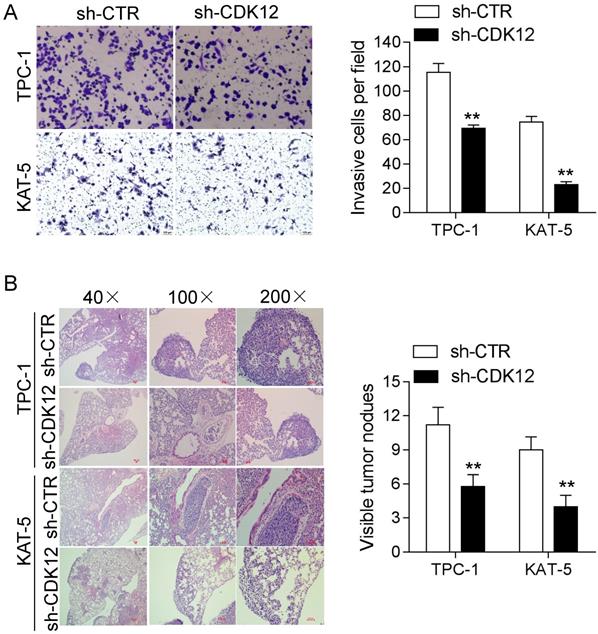
Cancer cell migration is vital intumor metastasis. Strong migration ability can accelerate the occurrence of metastasis. Therefore, we conducted a transwell assay and metastasis assay in vivo to study the mechanism of CDK12 in cancer metastasis. The transwell results showed that reduced CDK12 expression resulted in weaker migration ability compared with that of the control group (Figure 3A). The animal metastasis model also verified that the lack of CDK12 inhibited cancer metastasis, and the metastatic lung nodule loads were lower than those in the sh-CTR group (Figure 3B). In conclusion, CDK12 promotes cancer metastasis in vivo and in vitro.
CDK12 promotes PTC cell migration and tumor metastasis. (A): A transwell assay was used to analyze the migration ability in CDK12-inhibited TPC-1 and KAT-5 cell lines. Representative images of migrated cells are shown in the left panel, and the calculated results are shown in the right panel. (B): Metastasis in vivo model: representative IHC images of metastatic nodule sections are shown in the left panel, and thequantity of lung nodules is summarized in the right panel. **, P<0.01.
CDK12 promotes tumor progression by regulating c-myc/β-catenin pathway activation
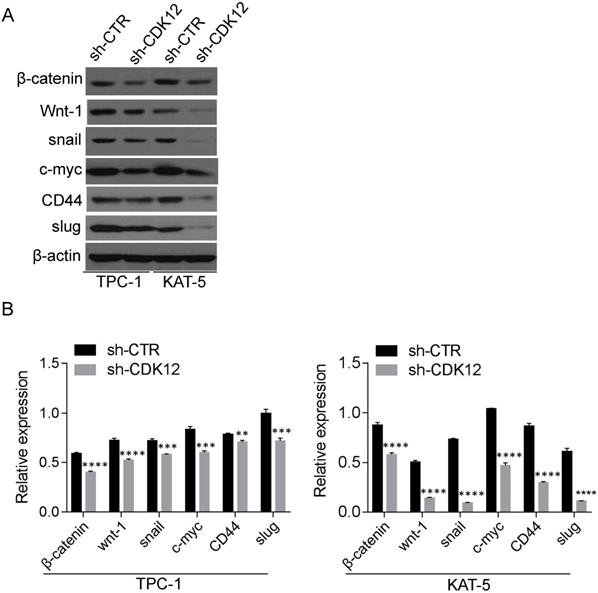
The c-myc/β-catenin pathway is a classical signaling pathway in cancer pathogenesis and is involved in cell stemness maintenance. Slug and snail were reported downstream of β-catenin and c-myc. The Western blot results revealed that CDK12 is closely related to the c-myc/β-catenin pathway (Figure 4). The proteins in the Wnt pathway, such as Wnt-1, β-catenin, c-myc, snail, and slug, were downregulated when CDK12 was knocked down. As a well-known stem cell marker, the expression of CD44 also decreased when CDK12 expression was inhibited. This indicates that CDK12 plays a vital role in the stemness of PTC cells.
CDK12 activates the c-myc/b-catenin pathway and may regulate cancer cell stemness (A): Western blotting was utilized to assess the relative expression of wnt-1, c-myc, CD44, β-catenin, slug and snail in TPC-1-sh-ctr and KAT-5-sh-CDK12 cells. Actin was used as the internal control. **, P<0.01; ***, P<0.001;****, P<0.0001.(B): the quantitative analysis of the Western blot, **, P<0.01; ***, P<0.001;****, P<0.0001.
Discussion
Although papillary thyroid cancer generally has a good prognosis, the outcome of papillary thyroid cancer metastasis is still unsatisfactory [27]. Few effective therapies can be applied for papillary thyroid cancer metastasis.
CDK12 acts as a cyclin-dependent kinase (CDK) that assembles with cyclin K to play a key role in cell cycle transition [28]. CDK12 maintains genomic stability byrepairing DNA damage. The loss of CDK12 can lead to carcinogenesis [29]. In this paper, we explored whether CDK12 is upregulated in PTC clinical specimens. The CDK12 expression in PTC cell lines further confirmed the association between CDK12 and PTC carcinogenesis. Our data demonstrated that inhibiting CDK12 expression in vivo and in vitro significantly suppressed cancer cell proliferation and tumor weights.
Metastasis is a vital challenge in cancer therapy, and it enormously threatens patient lives. Decreasing the occurrence of metastasis can effectively prolong patient survival time and reduce suffering [30]. The transwell and metastasis assay results showed that CDK12 reduced cancer cell migration and the number of lung metastatic nodules. These results indicate that CDK12 can be a potential target in PTC therapy.
The c-myc/β-catenin pathway is an important pathway in tumorigenesis. The major Wnt pathway signals depend on β-catenin. Wnt activates the receptors on the membrane surface. Then, β-catenin is activated in the cytoplasm and transferred into the nucleus. β-catenin in the nucleus regulates the expression of target genes, such as c-myc [31]. Slug and Snail are the downstream targets of c-myc. Slug and Snail regulate cell adhesion ability and play important roles in the epithelial-to-mesenchymal transition (EMT) [32-34]. Through a silicon assay, we found that CDK12 participated in the c-myc/β-catenin pathway to promote tumor progression. Western blotting confirmed that CDK12 participates in the wnt pathway to promote PTC and EMT progression. Our results help to complete the map of the c-myc/β-catenin pathway and provide a new potential target for PTC therapy.
CD44 is a well-known stemness marker. Cancer cell stemness is regarded as the cause of cancer relapse. CSCs can self-renew and maintain the ability to differentiate into other cancer types that display higher drug-resistance and malignant features [35]. The decreased expression of CD44 resulting from CDK12 inhibition revealed that CDK12 may affect the stemness of PTC cells.
In summary, CDK12 can inhibit cancer progression and metastasis by affecting cell proliferation and migration and inhibiting c-myc/β-catenin pathway expression and cancer stemness. CDK12 might become a potential biomarker or therapy target of PTC.
Conclusions
CDK12 can affect the c-myc/β-catenin pathway to stimulate papillary thyroid cancer proliferation and metastasis.CDK12 might be a new therapeutic target for papillary thyroid cancer.
Supplementary Material
Supplementary table.
Acknowledgements
This work was supported by funds from the Natural Science Foundation of Hunan Province (2019JJ40500).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30
2. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, Pacini F, Randolph GW, Sawka AM, Schlumberger M. et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26(1):1-133
3. Schmid D, Ricci C, Behrens G, Leitzmann MF. Adiposity and risk of thyroid cancer: a systematic review and meta-analysis. Obes Rev. 2015;16(12):1042-1054
4. Kitahara CM, Linet MS, Beane Freeman LE, Check DP, Church TR, Park Y, Purdue MP, Schairer C, Berrington de Gonzalez A. Cigarette smoking, alcohol intake, and thyroid cancer risk: a pooled analysis of five prospective studies in the United States. Cancer Causes Control. 2012;23(10):1615-1624
5. Dal Maso L, Bosetti C, La Vecchia C, Franceschi S. Risk factors for thyroid cancer: an epidemiological review focused on nutritional factors. Cancer Causes Control. 2009;20(1):75-86
6. Adam MA, Pura J, Goffredo P, Dinan MA, Reed SD, Scheri RP, Hyslop T, Roman SA, Sosa JA. Presence and Number of Lymph Node Metastases Are Associated With Compromised Survival for Patients Younger Than Age 45 Years With Papillary Thyroid Cancer. J Clin Oncol. 2015;33(21):2370-2375
7. Ekumi KM, Paculova H, Lenasi T, Pospichalova V, Bosken CA, Rybarikova J, Bryja V, Geyer M, Blazek D, Barboric M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic acids research. 2015;43(5):2575-2589
8. Tien JF, Mazloomian A, Cheng SG, Hughes CS, Chow CCT, Canapi LT, Oloumi A, Trigo-Gonzalez G, Bashashati A, Xu J. et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic acids research. 2017;45(11):6698-6716
9. Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. The Journal of biological chemistry. 2014;289(13):9247-9253
10. CDK12 Changes Telling in Prostate Cancer. Cancer discovery 2018, 8(9):1055
11. Ko TK, Kelly E, Pines J. CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. Journal of cell science. 2001;114(Pt 14):2591-2603
12. Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, Sharma B, Day DS, Kwiatkowski N, Pomaville M. et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nature communications. 2019;10(1):1757
13. Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N. et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell. 2018;173(7):1770-1782.e1714
14. Dubbury SJ, Boutz PL, Sharp PA. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature. 2018;564(7734):141-145
15. Chen HH, Wang YC, Fann MJ. Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Molecular and cellular biology. 2006;26(7):2736-2745
16. Choi HJ, Jin S, Cho H, Won HY, An HW, Jeong GY, Park YU, Kim HY, Park MK, Son T. et al. CDK12 drives breast tumor initiation and trastuzumab resistance via WNT and IRS1-ErbB-PI3K signaling. EMBO reports. 2019;20(10):e48058
17. Jin K, Wang S, Zhang Y, Xia M, Mo Y, Li X, Li G, Zeng Z, Xiong W, He Y. Long non-coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cellular and molecular life sciences: CMLS. 2019;76(21):4275-4289
18. Li M, Liu Y, Wei Y, Wu C, Meng H, Niu W, Zhou Y, Wang H, Wen Q, Fan S. et al. Zinc-finger protein YY1 suppresses tumor growth of human nasopharyngeal carcinoma by inactivating c-Myc-mediated microRNA-141 transcription. The Journal of biological chemistry. 2019;294(15):6172-6187
19. Liu Y, Zhao R, Wei Y, Li M, Wang H, Niu W, Zhou Y, Qiu Y, Fan S, Zhan Y. et al. BRD7 expression and c-Myc activation forms a double-negative feedback loop that controls the cell proliferation and tumor growth of nasopharyngeal carcinoma by targeting oncogenic miR-141. Journal of experimental & clinical cancer research: CR. 2018;37(1):64
20. Ye F, Gao G, Zou Y, Zheng S, Zhang L, Ou X, Xie X, Tang H. circFBXW7 Inhibits Malignant Progression by Sponging miR-197-3p and Encoding a 185-aa Protein in Triple-Negative Breast Cancer. Molecular therapy Nucleic acids. 2019;18:88-98
21. Qu A, Jiang C, Cai Y, Kim JH, Tanaka N, Ward JM, Shah YM, Gonzalez FJ. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J Hepatol. 2014;60(2):331-338
22. Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16(19):2530-2543
23. Heiser PW, Cano DA, Landsman L, Kim GE, Kench JG, Klimstra DS, Taketo MM, Biankin AV, Hebrok M. Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology. 2008;135(4):1288-1300
24. Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, Wu Y, Li X, Li X, Li G. et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Molecular cancer. 2018;17(1):45
25. Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast cancer research: BCR. 2010;12(6):213
26. Tang H, Huang X, Wang J, Yang L, Kong Y, Gao G, Zhang L, Chen ZS, Xie X. circKIF4A acts as a prognostic factor and mediator to regulate the progression of triple-negative breast cancer. Molecular cancer. 2019;18(1):23
27. Pontius LN, Youngwirth LM, Thomas SM, Scheri RP, Roman SA, Sosa JA. Lymphovascular invasion is associated with survival for papillary thyroid cancer. Endocr Relat Cancer. 2016;23(7):555-562
28. Kohoutek J, Blazek D. Cyclin K goes with Cdk12 and Cdk13. Cell division. 2012;7:12
29. Juan HC, Lin Y, Chen HR, Fann MJ. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell death and differentiation. 2016;23(6):1038-1048
30. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70(14):5649-5669
31. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691-701
32. Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116(Pt 3):499-511
33. Marin F, Nieto MA. Expression of chicken slug and snail in mesenchymal components of the developing central nervous system. Dev Dyn. 2004;230(1):144-148
34. Jiang L, Wang R, Fang L, Ge X, Chen L, Zhou M, Zhou Y, Xiong W, Hu Y, Tang X. et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics. 2019;9(9):2460-2474
35. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675-699
Author contact
![]() Corresponding author: Xinying Li, Department of General Surgery, Xiangya Hospital, Central South University; 87 Xiangya Road, Changsha, Hunan, 410008, China, E-mail: lixinyingcnedu.cn , Tel: +8613975804018
Corresponding author: Xinying Li, Department of General Surgery, Xiangya Hospital, Central South University; 87 Xiangya Road, Changsha, Hunan, 410008, China, E-mail: lixinyingcnedu.cn , Tel: +8613975804018