Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(10):2887-2920. doi:10.7150/jca.41324 This issue Cite
Review
Evidence for immortality and autonomy in animal cancer models is often not provided, which causes confusion on key issues of cancer biology
Xixi Dou1 ![]() , Pingzhen Tong2
, Pingzhen Tong2 ![]() , Hai Huang3
, Hai Huang3 ![]() , Lucas Zellmer4, Yan He5, Qingwen Jia1, Daizhou Zhang1, Jiang Peng6, Chenguang Wang6, Ningzhi Xu7, Dezhong Joshua Liao2
, Lucas Zellmer4, Yan He5, Qingwen Jia1, Daizhou Zhang1, Jiang Peng6, Chenguang Wang6, Ningzhi Xu7, Dezhong Joshua Liao2 ![]()
1. Shandong Provincial Key Laboratory of Transmucosal and Transdermal Drug Delivery, Shandong Freda Pharmaceutical Group Co., Ltd., Jinan 250101, Shandong Province, P.R. China.
2. Department of Pathology, The Second Hospital of Guizhou University of Traditional Chinese Medicine, Guiyang 550001, Guizhou Province, P.R. China.
3. Center for Clinical Laboratories, The Affiliated Hospital of Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China.
4. Masonic Cancer Center, University of Minnesota, 435 E. River Road, Minneapolis, MN 55455, USA.
5. Key Lab of Endemic and Ethnic Diseases of The Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China.
6. Department of Orthopaedics, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, P.R. China.
7. Tianjin LIPOGEN Gene Technology Ltd., #238 Baidi Road, Nankai District, Tianjin 300192, P.R. China.
8. Laboratory of Cell and Molecular Biology & State Key Laboratory of Molecular Oncology, National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences, Beijing 100021, P.R. China.
Received 2019-10-20; Accepted 2020-2-8; Published 2020-3-4
Abstract
Modern research into carcinogenesis has undergone three phases. Surgeons and pathologists started the first phase roughly 250 years ago, establishing morphological traits of tumors for pathologic diagnosis, and setting immortality and autonomy as indispensable criteria for neoplasms. A century ago, medical doctors, biologists and chemists started to enhance “experimental cancer research” by establishing many animal models of chemical-induced carcinogenesis for studies of cellular mechanisms. In this second phase, the two-hit theory and stepwise carcinogenesis of “initiation-promotion” or “initiation-promotion-progression” were established, with an illustrious finding that outgrowths induced in animals depend on the inducers, and thus are not authentically neoplastic, until late stages. The last 40 years are the third incarnation, molecular biologists have gradually dominated the carcinogenesis research fraternity and have established numerous genetically-modified animal models of carcinogenesis. However, evidence has not been provided for immortality and autonomy of the lesions from most of these models. Probably, many lesions had already been collected from animals for analyses of molecular mechanisms of “cancer” before the lesions became autonomous. We herein review the monumental work of many predecessors to reinforce that evidence for immortality and autonomy is essential for confirming a neoplastic nature. We extrapolate that immortality and autonomy are established early during sporadic human carcinogenesis, unlike the late establishment in most animal models. It is imperative to resume many forerunners' work by determining the genetic bases for initiation, promotion and progression, the genetic bases for immortality and autonomy, and which animal models are, in fact, good for identifying such genetic bases.
Keywords: Transgenic, cancer, carcinogenesis, immortality, autonomy, cancer stem cells, senescence.
Introduction
Cancer research has been going on for 2,700 years [1], but in our opinion systematic studies of cancer had not been expedited until the 1760s when several historical events occurred that greatly accelerated the research: First, in 1761 John Hill published his observation of cancers in the nasal cavity of snuff users, which is the first report on a connection between chemicals and human cancer [2]. Second, in 1775 Pott reported his “chirurgical” observation of cancers in several body sites and identified cancer in the scrotal skin of British chimney sweeps [3]. Because chimney sweeps in other European countries did not have this cancer, it was suspected that British chimney sweeps, unlike those in other countries, did not bathe as a matter of honor, which allowed carcinogenic hydrocarbons from soot to be retained on the scrotal skin. Ensuing requirement of bathing at least once a week, recommended by Hill, significantly prevented this cancer occurrence. This is the first documented success in cancer prevention, as described by Sell [4]. Third, according to Triolo's comprehensive review [5], the French Royal Academy of Surgery in 1772 and the Lyon Academy of Science in 1733 offered prizes for original essays on the question of “what is cancer”, which drove a national-wide search for the nature and definition of cancer. Fourth, the latter prize mentioned above was awarded to the surgeon-chemist Bernard Peyrilhe for his work on the inoculation of dogs with human-derived cancer fluid, which was published in 1774 as the first documented animal experiment on cancer in the literature [5], to our knowledge.
One of the results from the last 250 or so years of extensive research on cancer is the so enormous size of the literature that often becomes a tribulation for researchers. Actually, even in 1955, Alexander Haddow (1907-1976) had pointed this out, writing that “the mere abundance of the data …presents a growing problem, towards which there are two extreme types of reaction: first, that of the happy researcher who is content to ignore the original literature, and to rely upon others for his information; and secondly, the reaction of those whom the literature totally enslaves” [6]. Few of today's researchers peruse the ancient literature due to their many tiers of stress, such as dwindling funding and increasing difficulty in obtaining tenured faculty positions, besides the colossal volume of the literature to read. As a repercussion, cancer research has manifested a discontinuous growth, just like cancer itself that is a discontinuous growth from its normal parental cell, meaning that few of today's cancer students know and address the questions raised by their predecessors.
We write this perspective article to review some seminal findings by different trailblazers in “carcinogenesis research”, which is herein defined as the study 1) on the procedures that convert a normal cell to a malignant one and then to more malignant states, and 2) on the mechanisms underlying these steps. Cancer's clinical quarter will not be touched upon to avoid digression. The idiom of “tumorigenesis”, which is of broader scope as it also covers the formation of benign tumors, is used sometimes, partly because many animal models produce both benign and malignant lesions.
In our opinion, modern research on tumorigenesis has undergone three phases. The first one began in the late 18th century and went through the entire 19th century [5, 7, 8]. The cancer research fraternity in this phase was dominated by surgeons, pathologists, and anatomists. They established autopsy and biopsy as the routine pathology practice, which led to the establishment of the morphological traits of neoplasms [5, 9] and some theoretical achievements, such as the supposition by Virchow (1821-1902) that cancer resulted from chronic irritation [10-13], mainly inflammation [7, 8, 14]. The second phase had its inception roughly at the beginning of the 20th century and was coined as an epoch of “experimental cancer research” by prominent cancer pathologists James Ewing (1866-1943), according to Cardiff and Kenney [15], and Harold Leroy Stewart (1908-1998) [9]. In this incarnation, medical doctors, biologists, and chemists established and characterized many animal models of chemical-induced tumorigenesis [16]. Using these models, they established the two-hit theory [17-21], mutation theory [22-24], clonal evolution theory [25-28], as well as the multi-stage [29-32], i.e. initiation-promotion [33-38] or initiation-promotion-progression [39-44], models of carcinogenesis. Animal models of irradiation-induced carcinogenesis emerged during this period as well [45].
Starting about 40 years ago, molecular biologists, many lacking strict training and clinical experience in surgical pathology or oncology, have gradually replaced medical doctors and traditional biologists to now dominate the fraternity of carcinogenesis research [46, 47], thus moving “experimental cancer research” into a new phase. In this latest incarnation, molecular biologists have established numerous genetically manipulated animal models of carcinogenesis and in vitro systems of neoplastic transformation of normal cells which have led us to deeper mechanisms of how genes regulate behaviors of normal and neoplastic cells. We now enjoy enormous amounts of information and great details on molecular signaling pathways for almost all physiological functions and pathological alterations in the human body. However, few of the genetic animal models established so far address the traditional multiple stages of “initiation-promotion-progression” [41], leaving those mavens who are familiar with their predecessors' work to wonder how to couple the stepwise biological changes observed previously with the molecular alterations seen in these genetic models. Moreover, few of the publications reporting these genetic models provide material evidence for immortality and autonomy of the resulting lesions. To warrant this statement, we encourage readers to search published reports of these genetic models for “immortal”, “autonomous”, or similar keywords, to see how many of them describe these properties of the resulting lesions. By reviewing the work of many forerunners, most being preeminent cancer pathologists, we attempt in this essay to reinforce immortality and autonomy as the cardinal, yet long-neglected, criteria to qualify outgrowths as neoplasms.
Many chemical-induced tumors in animals remain dependent on that chemical until late stages
To our knowledge, the first experimentally induced tumors in animals were reported, in the German literature, by Ledoux-Lebard in 1885 [48], who, according to Triolo [7], observed epithelioma in the lungs of the rabbits injected with a mixture of sweet almond oil and croton oil. In 1900, Brosch induced atypical epithelial growths in the crushed skin of a guinea pig with applications of a xylol-paraffin solution [49]. As described by Davis [50, 51] and Vasiliev [52], in 1906, Fischer showed that subcutaneous injections of Scarlet Red into the ears of rabbits induced papilloma, which regressed upon discontinuation of the injections but reappeared with further injections [53]. According to Davis [50, 51], these phenomena were confirmed by Helmholz in 1907 and by Werner in 1908. Between 1914 and 1924, Katsusaburo Yamagiwa (1863-1930), after he left Virchow and returned to Japan [54, 55], induced papilloma and papillocarcinoma in rabbits' ears by painting the ears with coal tar; metastases were seen in lymph nodes in some cases. However, the tumors regressed upon cessation of the tar-painting but recurred quickly if the painting resumed [56, 57]. Yamagiwa thus concluded that “carcinomas do not develop as carcinomas from the beginning, and do not always continue as carcinomas” [57]. This “do not always continue” is the first statement in the literature, to our knowledge, stating that induced cancer can disappear spontaneously. During 1930s and 1940s, Peyton Rous (1879-1970), a Nobel laureate, confirmed the regression of the lesions upon tar discontinuation and their quick reappearance upon tar repainting [38, 58, 59]. Actually, according to Rous, Des Ligneris had already confirmed in 1930 that,“…a second period of tarring brings out warts sooner than the first” [58]. Realizing that the reversible lesions could not be authentic neoplasms, Rous described them as warts, which are hyperplastic lesions, and wrote in 1940 that, “…it will be seen that the tar warts of rabbits are tumors by all of the standard criteria except two. They have no capacity for independent growth like that exhibited by most (but not all) classical tumors; and the changes in their cells may conceivably be reversible since they often become smaller and vanish” [58]. The two unmet criteria in Rous' observations, i.e. “no capacity for independent growth” and being “reversible”, are later referred to as “autonomy” and “immortality” in the literature. Rous further wrote that, “…in the current definition of a tumor no allowance is made for neoplasms which depend upon favoring factors for existence, and it cannot be used to rule them out” [58]. Here, the lesion inducer is dubbed as “favoring factors”.
Chronic treatment of rats with 7,12-diemthylbenz(a)anthracene (DMBA) can induce mammary tumors, but sustenance of the tumors requires continuation of DMBA [60-63]. Continuous feeding of rats with 3-methylcholanthrene could induce palpable mammary tumors as early as the 20th day from the start of the feeding [64]. Painting the skin of C57 brown mice with 3-methylcholanthrene could also induce palpable tumors as early as the 31st day [65], but 15 of the 22 induced skin tumors regressed completely upon cessation of the inducer and only three of the persisting 15 evolved to histological malignancies [65]. Similarly, a large number of papilloma could be induced by painting the skin of albino mice with 3:4-benzyprene, but the tumors actually sloughed off and only a few progressed to carcinomas [66]. After having studied successive stages of carcinogenesis [67-70], Rusch wrote in 1950 that carcinogenesis generally consisted of “induction, reversibility and progression” [71], which clearly points out that lesions can be immortal and autonomous only at a late stage. The typical inducer-dependency until a late stage can be exemplified by the skin carcinogenesis model presented by Berenblum in 1947 [34], which, in Haddow's words, “proceeds from the normal epithelium first to an early non-specific hyperplasia, second to a specific pre-neoplastic hyperplasia, and then to the emergence of papillomata, and how later stages can be recognized in the progressive growth of such papillomata, their conversion into carcinoma, and the uncontrolled growth of the latter…This general sequence takes place equally well whether exposure to the carcinogen is continued or not” [72].
Hormone-induced tumors in animals are inducer-dependent until late stages
As comprehensively reviewed by Cardiff and N Kenney [15], breast cancer has been known to be regulated by female hormones since 1896 when Beatson reported the regression and recurrence of a breast lump in a 33-year-old woman following removal of her ovaries [73]; other similar cases were also reported in the following years [74, 75]. Lathrop and Loeb also reported similar findings in spayed mice in 1916 [76]. Chronic treatment of rats and mice with estrogens can induce cancers in the bladder and mammary glands and benign tumors in the pituitary and testes [77-92], and can also induce uterine tumors [93]. The ACI strain (August strain crossed with Copenhagen strain, also called AxC) of rats may be more susceptible than other strains to the induction of the mammary and pituitary tumors [78, 94], but we once found that about one-fourth of the females lacked one side of the uterus and ovary (DJ Liao's unpublished data), suggesting that the ACI strain may bear a recessive mutation in a relevant but not yet identified gene. Treatment of mice with estrogen, or with both estrogen and androgen, can induce benign and malignant tumors in the cervix and vagina; these malignant tumors are transplantable to other mice treated with the hormones [95-101]. Administration of androgens to rats can induce prostate [102-106] and uterine [107, 108] cancers. Concomitant treatment of rats with estrogen and androgen can induce mammary and prostate cancers much more quickly than treatment with androgen or estrogen alone [102-106, 109-114] and can induce uterine leiomyomas as well [115]. Administration of estrogen to hamsters can induce malignant renal tumors with abdominal metastases [116-121], while administration of both estrogen and androgen to hamsters can induce malignant tumors in the kidneys and induce benign and malignant tumors in the uterus, in the skin, and in the epididymal tail and adjacent ductus deferens [102, 122, 123]. Moreover, gonadal and gonadotrophic hormones have also been shown to possess the ability to induce endocrine cell tumors in ovaries or testes [90, 124-127]. Transplantation of the ovary into the spleen can cause neoplasia of the ovary as well, because it eliminates the feedback control regulating hormonal synthesis in the ovary and provides the ovary with unrequited stimulus of pituitary hormones [128].
Estrogen-induced mammary cancer, as well as pituitary and testicular tumors, have been known since the 1930s to regress partially or completely upon withdrawal of the hormone, and the tumors can sustain themselves without estrogen treatment only at very late stages [77, 87, 100, 129-146] (and DJ Liao's personal experience). Initially, estrogen-induced pituitary tumors can be transplanted only to animals treated with estrogens, but not to the untreated animals, evincing their dependency on an excessive amount of estrogen [77]. However, they can eventually evolve to estrogen-independency [147, 148]. The Nobel laureate Charles B. Huggins (1901-1997) had shown in both animal studies and human clinics that castration or treatment with estrogens could cause regression of prostate cancer at certain stages, signifying that this cancer is hormone-dependent until a late stage [149-151]. In the words of Jacob Furth (1896-1979), a renowned pathologist [152-154], “this (prostate) tumor is an example of a growth in man with a spectrum ranging from conditioned to highly autonomous type. The cases of Huggins that were controlled by castration (that is, removal of sources of androgens) may be regarded as dependent; those which partially or temporarily regressed after castration or estrogen treatment, as partially dependent; those not influenced by such therapy, as autonomous” [155]. Estrogen-induced renal tumors in hamsters, including their abdominal metastases, will regress upon cessation of the estrogen treatment unless the tumors are at very advanced stages [156-159] (and DJ Liao's empirical knowledge). Initially, these renal tumors are transplantable only to those hamsters that are treated with estrogen, connoting that the tumors still depend on an excessive amount of estrogen, but autonomy can eventually be achieved by manipulation of the estrogen in the recipient animals [119, 159, 160]. The hormone dependency seen in all of these studies is the rationale behind the anti-hormone treatments of hormone-dependent cancers [161-163].
Treatment of mice with iodine-131 (I-131) or other anti-thyroid drugs can induce pituitary tumors that secrete thyroid stimulating hormone (TSH) [77, 164-168] because the drugs damage the thyroid and thus decrease the levels of thyroid hormones. This, in turn, stimulates proliferation of TSH secretory cells in the pituitary [169-173]. By the same principle, partial thyroidectomy of rats and mice can cause pituitary adenomas as well [174-179]. The tumors can be transplanted [180]; initially only to those mice treated with thiouracil or other goitrogenic compounds that induce TSH and then gradually to normal mice [181], which again shows the trajectory of “initial dependence and then autonomy”.
Thyroid neoplasms can be induced in mice by treatment with thiouracil or other goitrogenic compounds [174, 177, 178, 182-192] or with I-131 [171, 193-196] as a sequel of a high amount of TSH secreted from the pituitary. These thyroid tumors are TSH-dependent but often metastasize to lymph nodes [155] and the lungs [155, 174, 185], although the tumor cells can be converted to TSH-independence via continuous subpassage in culture [155, 174, 177, 178, 183]. The hormone dependency of thyroid tumors, including adenocarcinomas, also occurs in a Zebrafish colony as feeding the fish with salt that contains iodine causes regression of the tumors [197]. In 1953, Furth wrote that “conditions can be created whereby uncontrolled proliferation of one cell type is obtained, resulting in a tumor-like growth. Manipulations attaining this need not involve any intrinsic alteration in cells causing them to behave as cancer cells. Whether or not such tumors and the similar human metastasizing thyroid adenomas are considered neoplastic depends on the definition of a neoplasm. In our terminology such thyroid tumors are conditioned neoplasms. In the course of subpassage in thiouracil-treated mice the dependent growths give rise to autonomous growths which possess individual features of their own and can be grafted on normal mice. Thyroid adenomas induced by TSH-secreting pituitary tumors are indistinguishable from those induced by thiouracil” [155]. Here, Furth used “tumor-like growth”, “behave as cancer cells”, and “conditional neoplasms” to express his reservation in considering the induced pituitary and thyroid tumors, even the spontaneous human thyroid tumors, as authentic; despite their ability to metastasize. In his punditry, “dependent tumors are those in which apparently normal cells proliferate in an altered host; autonomous tumors are those in which permanently altered cells proliferate in normal hosts” [155], although, based on our training in human pathology, we opine that dependent “tumor cells” are not normal but are hyperplastic.
Some tumors from genetically manipulated animals are inducer-dependent as well
The c-myc gene or a k-ras mutant can induce malignant tumors in many lines of transgenic mice, as we have shown or reviewed before [139,198-205]. However, many of the tumors have been shown to regress upon turning off the transgene and can be sustained without the expression of the transgene only at advanced stages, although, once they have regressed, they can be quickly re-induced by reactivation of the transgene [205-223]. Xmrk, c-myc, mutant k-ras, or SV40 large T oncogene can also induce liver cancer in transgenic Zebrafish, and again, the tumors will regress after inactivation of the transgene [224-230]. Conversely, inactivation of the tumor suppressor gene p53 via conditional knockout can beget tumor formation, but reactivation of the p53 leads to regression of the tumors [228, 229, 231-235]. This phenomenon of “regression upon inducer withdrawal and quick repopulation upon reintroduction of the inducer” is a full reflection of the same phenomenon seen in the chemical- or hormone-induced carcinogenesis described above, and has become a rationale for targeting therapy in cancer [236-238]. Our contemporaries in the third phase of carcinogenesis research consider “regression upon inducer withdrawal” as “oncogene addiction” and “tumor dormancy” as the reason for “the tumor repopulation upon reintroduction of the inducer” [206, 210, 213, 216-218, 236-239], but, peculiarly, without mentioning the same phenomenon observed by our predecessors.
Spontaneous regression of human neoplasms occurs but is rare
In humans, spontaneous regression or remission of a neoplasm is extremely rare, but it is recurrently shown in case reports [240-255] with a frequency varying between 1/60,000 and 1/140,000 cases [242, 256-258]. Malignant melanoma may have the highest rate of spontaneous regression [259-265]. Ever since its first case reported in 1866, as reviewed by Kalialis [266], it has been reported that 10-50% of cutaneous malignant melanoma cases show partial or complete regression without treatment [267, 268], including 0.23% of the metastatic cases [267]. High rates of spontaneous regression have also been reported for indolent histologic subtypes of non-Hodgkin's lymphoma, varying between 10% and 20% in selected series, as reviewed by Drobyski and Qazi [269]. Pediatric neuroblastoma is another malignancy with a high frequency of spontaneous regression, especially those cases categorized into stage IV-S [270,271]. Other types of cancer often showing spontaneous regression include renal cell carcinoma, choriocarcinoma, lymphoid malignancies, etc. [240-242, 272-275].
Some sporadic tumors in animals and plants also regress spontaneously in a seasonal manner
Spontaneous regression also occurs in animal tumors, such as in mice [276]. Mention should be made of tumors in some species of fish and amphibians that often regress spontaneously in a seasonal or temperature-sensitive manner [277-285]. The ambient temperatures in some seasons may be hostile for the tumor-inciting micropathogens to grow, and thus fewer tumors occur, but it remains obscure why overt tumors in these cold-blooded creatures disappear in these seasons. The fact that the fish or amphibians themselves live well while the tumors are sloughed off suggests that the tumors require a different microenvironment to sustain their autonomous lives. Similarly, it has also been known for almost a century that some plants will not develop tumors at some hot ambient temperatures [286], albeit both the plants and the tumor-inciting micropathogens can grow happily at those temperatures [286-290]. Whether overt tumors in these plants will regress at a hostile temperature remains unknown.
Immortality and autonomy had already become indispensable criteria for neoplasms a century ago
The studies described above on chemical- or hormone-induced outgrowths are among the earliest ones that point out the problem of “inducer-dependency” and set immortality and autonomy as criteria for neoplasms. Actually, as reviewed by Triolo in 1965 [5], research on human cancers in the 19th century had already led researchers, mainly surgeons and pathologists, to a theory that, “cancer cells are autonomous, endow themselves with the power of an independent existence, and divert their entire resources into an unlimited capacity for growth.” This theory finally entered into a rudimentary form and was given as a formal introduction of cancer by J. George Adami in 1901 [291] and, according to Rous [58], as a cancer definition in some German pathology textbooks published in the 1910s.
Furth [155] and Ewing [292] considered that all tumors should be in some form of autonomy. Haddow wrote in the 1947 that, “…we now know that, while constitutional and genetic factors can greatly influence susceptibility to cancer, and many even determine the site of its spontaneous occurrence, the disease is one of the individual cells as a separate organism and with no relation to the needs of the body as a whole. It is this which gives cancer its unique position in pathology, accounts for its intractable nature, and explains its growth, in Paget's words 'irrespective of the maintenance of the rest of the body, discordant from its normal type, and with no seeming purpose' (Paget, 1853)” [72]. The quoted words of Paget had already, in 1853, pointed out the tumors' autonomous nature. Indeed, according to Haddow [72] and Knauss et al [293], a cancer has long been regarded as a new race or new strain of organism, which is another way of describing autonomy dating back to 1897 by David Hansemann, 1903 by G. Hauser (Beitr. Path. Anat., 1903; 33, 1), and 1926 by Menetrier. Many other former pundits also described carcinogenesis as an atavistic procedure, further pointing out that the resulting “new race of organism” is evolutionarily-lower than its host animal [294-301].
Immortality betokens that a tumor can survive as a “newly developed independent organism” [58] that parasitizes the host [294, 295] and forever maintains its life by continuous replication of its cells [296, 302, 303]. As adduced by Paget in 1889 [304], “as Langenbeck says, every single cancer cell must be regarded as an organism, alive and capable of development.” Harry Greene (1904-1969), a preeminent surgical pathologist at Yale University, elaborated on the autonomy by writing in 1951 that, “…the definition of a tumor as an autonomous growth has enjoyed persistent popularity in textbooks of pathology. In such definitions the adjective 'autonomous' is employed to express the idea of independence with respect to two different particulars. One of these relates to freedom from the laws restraining and coordinating normal tissue growth, and the other concerns release from the necessity of a continued stimulus” [305]. According to Furth's translation [155], in an article written in German from 1951, Bungeler considered that a dependence seen in a large variety of human outgrowths indicates that the outgrowths are not true tumors and, more critically, there is no transition between the dependent and autonomous outgrowths. This “no transition” means that whether an outgrowth is autonomous or not is a black-and-white demarcation between neoplasms and non-neoplasms. Describing human cancer's properties, Emmanuel Farber (1918-2014), a superlative cancer pathologist, also accentuated autonomy as a cornerstone of cancer biology [306]. Notwithstanding, it still needs to be pointed out that autonomy of tumor cells may be achieved via non-autonomous mechanisms, e.g. various interactions with other cell types [307-309].
Mutation and inauthenticity may explain some cases of spontaneous regression
Since malignant tumors keep randomly mutating, theoretically some mutations may be good ones that direct the cells to differentiation or facilitate clearance of tumor cells by immune cells, such as mutation of the FBXW7 gene [310]. Conversely, some mutations may be deleterious, killing the cells by themselves or by working with other harmful single nucleotide polymorphisms (SNPs), since about 12% of the SNPs are harmful in the human genome [311], especially in Europeans [312]. Actually, since the most common genetic changes found in tumors are large chromosomal deletions [313,314], severe genomic damage may lead to the loss of those genes required for cell survival. Moreover, some pernicious mutants may undergo mutation again, back to the wild type or to a better version, which may have reverse evolution as its essence [315] and cause differentiation of the cells. This so-called “back mutation” or “reverse mutation” is occasionally discerned in drosophila [316], as well as in some human genetic diseases [317-320] and in some cancers treated with chemotherapeutic agents [321-323].
Inauthenticity of the tumors may be another reason for spontaneous regression. For instance, it was often reported in the 1970s-1980s that hepatomas and hepatocellular carcinomas in women chronically using estrogen-rich oral contraceptives regressed upon termination of the contraceptive use [324-330], which substantiates the human relevance of estrogen-induced hepatomas in rodents reported in the 1950s-1960s [331-333]. As another example, low-grade lymphomas can result from infection by Helicobacter pylori (HP). These tumors are basically curable by eradication of the bacteria with antibiotic treatment [334-339] but, if left untreated, some of them will progress and become incurable, as reviewed by Park and Koo [340]. Similarly, Chronic HTLV-I (human T cell lymphotropic virus type I) infection may spawn adult T cell leukemia or lymphoma, but the neoplasm can be well controlled or even cured by antiviral treatment against HTLV-1 [341-343]. To us, these properties of these estrogen-, bacterium-, or virus-caused outgrowths resemble those induced in many animal models described above, and thus are not authentically neoplastic at their early time point although their diagnoses meet pathological criteria for neoplasms and they, if left untreated, may eventually evolve to genuine neoplasms. Or, we can take a non-pathological definition of cancer proposed by Robert Axelrod, who majored in political science but became a prominent cancer ecologist [344], that incipient cancer cells might just have been partly transformed, and not yet fully malignant, thus requiring collaboration with each other for survival and for collective presentation of a cancer phenotype [345].
Using very strict criteria, there may not be pure spontaneous regression or remission of cancer in humans, because it is unlikely that patients will do absolutely nothing for their illness. Some patients' self-management towards the neoplasm may actually be effective, although their doctors may not realize it. The patients may have experienced severe infection, especially a febrile one, since an infection or fever may be an effective cancer remedy partly by enhancing the immune attack on the cancer cells [256, 346-367], as we have reviewed before [296, 368]. Moreover, regression may occur via an unknown mechanism, such as via spontaneous epigenetic or genetic changes leading to a full differentiation of the tumor cells [369-371] or increased stimulation of immune function by the tumor cells [372, 373].
Tissue culture and transplantation were once used to determine immortality and autonomy
Even over a century ago, whether or not a patient's tumor was immortal and autonomous had been a concern of, and thus had often been tested by, surgical pathologists, because they had realized that morphological traits should not be the solitary criterion, and the tumor's behavior should also be considered, for an infallible diagnosis of cancer. The tests had been conducted, ever since 1901 [374,375], mainly with culture of surgically removed tumor tissues or with transplantation of the tissues to animals, the two modern techniques aforetime. Actually, a technique involving both transplantation and culture was done by inoculating tumor cells into a fertile egg and then hatching it [376-386], which is the parentage of some modern chick embryo assays for cancer research [387-399] such as the chick heart invasion assay [400-405]. The rationale for using tissue culture is that neoplastic cells are immortal and can self-renew to forever maintain themselves as a “new organism” by incessant cell division. Even after the patient has died, the “organism” can be maintained as cell lines, embodied by the Hela cell line established in 1951 from cervical cancer of the late patient Henrietta Lacks [406].
Human tissue transplantation to animals, started by Peyrilhe in 1773 with cancer fluid [5] and by Hanau in 1889 with solid tissue [407], has been overwhelmingly used in cancer research, as extensively reviewed even many decades ago [407-422]. Mention should be made of the studies over a century ago that involved tumor transplantations to humans [423-425], with the heroic trial by Senn who inoculated himself with pieces of cancerous lymph nodes [7, 425]. Moreover, as reviewed by Triolo [7], transplantation of animal tumors to other animals have also been performed since in 1860s [426-430]. The rationale for this approach is to use tumor cells' behaviors, mainly autonomy, to determine its authenticity. As shown in table 1, transplantation of animal tumors can generally be divided into five categories [305, 431], i.e. 1) autologous transplantation, or transfer back elsewhere in the same animal; 2) homologous I transplantation, or transfer to a tumor-bearing animal of the same species; 3) homologous II transplantation, or transfer to a normal animal of the same species; 4) heterologous I transplantation, or transfer to a tumor-bearing animal of a different species; and 5) heterologous II transplantation, or transfer to a normal animal of a different species.
Five common types of tumor tissue transplantation.
| Name | Definition |
|---|---|
| Autologous | transfer back elsewhere in the same animal |
| Homologous I | transfer to a tumor-bearing animal of the same species |
| Homologous II | transfer to a normal animal of the same species |
| Heterologous I | transfer to a tumor-bearing animal of a different species |
| Heterologous II | transfer to a normal animal of a different species |
A seminal finding by Greene in the 1940s, among his many other findings [305, 431-438], is that some cancers are not transplantable to normal animals but are transplantable to the animals that bear a spontaneous tumor, especially one of the same tissue origin [305, 431]. For instance, the Brown-Pearce rabbit tumor typically does not grow in normal C3H mice but it grows rapidly in those bearing spontaneous tumors [433], and a Rous chicken sarcoma grows well subcutaneously in tumor-bearing C3H mice but not in normal C3H mice [305]. These results led Greene to a conclusion that the factors affecting the take of transplanted tumors “are constitutional in distribution and are not localized at the site of the primary growth” [432]. However, lymphoblastic leukemia and lymphosarcoma are graftable to every normal genetically compatible host but do not produce tumors in the anterior chamber of an eye of an alien host, showing a difference from other tumors [305]. The difference between normal and tumor-bearing hosts in response to a tumor graft suggests that tumor-bearing animals possess some factors affecting the graft's survival. A plausible interpretation is that the spontaneous tumor preexisting in the host has already suppressed the host's immune function that is supposed to reject the graft. Studies of these inhibitory effects have later been extended to the interaction between normal cells and tumor cells not only in vivo but also in vitro [439-448], as has been reviewed by us [449], by Rubin [450-453], by Aktipis [454,455] and by Thomas et al [456-458] from different slants. For instance, it has been shown that normal cells suppress the growth of adjacent tumor cells in culture [459] and in skin grafts on mice [460]. Unfortunately, identifying these tumor or host factors has largely been neglected, although it is important since manipulation of these factors may be helpful in curing cancer.
Another trailblazing finding by Greene et al. in the 1940s is that the tumors that are capable of metastasizing are heterologously transplantable, as they can grow in the brain or the anterior chamber of an eye of animals of a different species, whereas tumors that are still incapable of metastasizing cannot [431, 435, 437, 438]. Based on these observations, Greene concluded that only those lesions which can metastasize are fully autonomous and can be regarded as cancers, whereas those which do not possess this ability are still conditionally autonomous and thus should not be regarded as malignancy [435, 437]. Although in pathology textbooks metastasis is not a canon for diagnosis of a malignancy, it is the only reliable yardstick to distinguish malignant neoplasms from benign ones [461]. Considering that even today, compared with Green's epoch, in the surgical pathology service we still do not have a simpler or more reliable approach to determine whether a primary tumor removed from a patient has encompassed the ability to metastasize, it is a pity that Greene's simple but reliable test has not been used in clinical service until now, probably due partly to an ethical concern on the eye graft.
Most animal models have not yet been tested for the trajectory of “induction, reversibility, and progression”
Many animal models of carcinogenesis induced by chemicals or hormones have not yet been determined for the inducer-dependency. Even worse, except the several models described in an above section, like the ones described by Sanchenz-Garcia's group [220,462], the vast majority of genetically manipulated animal models have not yet been tested either. This severe defect is presumably ascribable to two reasons: first, probably many molecular biologists have not realized that immortality and autonomy are prerequisite criteria for neoplasms. Second, the genetic manipulation in many, probably most, of these animal models is not set in a “turn-on/turn-off” mode, and thus does not allow researchers to control the target gene to determine whether or not the lesions are inducer-dependent. Moreover, for the induction of visceral tumors, like the N-nitrosobis(2-hydroxypropyl)amine-instigated lung tumors [463], the determination is more difficult as it requires sacrifice of the animals. We surmise that most of the undetermined animal models may also show an inducer dependency until a late stage, with their carcinogenic procedures following the aforementioned trajectory of “induction, reversibility, and progression” described by Rusch [71]. Considering that the lesions wrought by c-myc and mutant k-ras, the two most potent oncogenes, already manifest such dependency, other genetically manipulated models will likely show this trajectory as well. Notwithstanding, this conjecture needs to be substantiated by studying untested animal models, especially the new ones to be established in the future using, for example, a conditional transgenic or knockout approach.
Loss of allegiance to the host's body is the essence of neoplastic cells' immortality and autonomy
Sporadic tumors can be derived only from those cell types that are renewable, i.e. have a lifelong ability to replicate, because mutation needs to be perpetuated by at least one round of DNA replication and to be passed to filial cells via cellular divisions [155, 449]. That permanence becomes possible because the fitness testing of cells is usually conducted after the mutation is made permanently heritable [464]. We tag those highly renewable cell types as “anabolic” for their great susceptibility to cancers and those that have lost their replicative ability in adulthood, such as neurons and cardiac myocytes, as “catabolic” for their role in the development of type 2 diabetes [465]. Even for those renewable cells, it will take about one-fourth to one-third of the lifespan to complete the procedure of sporadic carcinogenesis, which is about 20-30 years for human beings [306, 371], although it could take 50 years by others' estimation [19]. Therefore, the aforementioned tumors induced by 3-methylcholanthrene in just 3-4 weeks cannot be authentically neoplastic [64, 65], since the lifespan of experimental mice and rats is three years or longer [205], although their counterparts in the wild live much shorter lives [24]. Indeed, we are not aware of any rodent model in which a sporadic cancer can be induced in a period less than a few months, except those genetic models in which the genetic manipulation has already been effective during an embryonic stage, thus mimicking a pediatric (but not a sporadic) carcinogenesis, as to be expanded upon later.
All cell types in an evolutionarily complex animal have a physiological total number. For renewable cell types, if the cell number is decreased for some reason, the body will trigger cell proliferation to restore the physiological number. Conversely, if the number is higher than normal, as seen in over-regeneration that often happens following a regeneration procedure, the body will goad some of the cells into apoptosis to avoid cell redundancy [296, 302, 303, 466-469]. This is because apoptosis evolves as a specific mechanism to eliminate useless, redundant cells from the tissue or organ [302, 303, 466, 468], but not as a demise mechanism triggered by compensatory proliferation as thought by some peers [470-473]. Indeed, compensatory proliferation is regeneration and does not aim to engender excessive, i.e. hyperplastic, cells, although it usually does mildly because of a slight overproduction of cells. Killing excessive cells via apoptosis can be implemented in an evolutionarily complex animal because all cells have allegiance to the animal's body, as we described before [296, 302, 303, 466-468], or “conform with the law of organisms”, as put by Rous in 1941 [58]. This allegiance as the “law of organisms” allows the host's body to require some renewable cells to sacrifice their lives for the body's ultimate interest. An instructive example is that white blood cells are often put on the frontier by the host's body to fight against infectious micropathogens and die in the battle, so that the host as a whole can survive [466, 474]. However, sometimes some renewable cells, such as select bone marrow cells, epidermal keratinocytes, and mucal cells in the gastric-intestinal tract, have lost their altruism, usually due to acquisition of tumor-driving mutations that make the cells egocentric. These selfish cells want to survive stress such as micropathogen infection, over-regeneration-trigged apoptosis, etc., and become independent of the body, i.e. become autonomous. Reiterated, this loss of loyalty to the host's body is the essence of, or the reason for, autonomy of some cells. “Fail to conform with the law of organisms” as said by Rous [58], or “become autonomous” as outlined by Ewing to be the pathological concept of a tumor [475], was set as “the signature of a genuine neoplasm” by Borst in 1903 [476] and has, until today, been a salient feature of benign and malignant tumors.
In addition to apoptosis, an accelerated aging procedure leading to senescent death may be an additional mechanism for elimination of the excessive, i.e. hyperplastic, cells in the early lesions of animal models; although studies on the mechanism for the inducer-dependency have hardly been extended to this type of cell death. We define cell death via aging as “senescent death” [302], because normal cells have their lifespans [477-482] and ever since it was first observed in 1965, this senescent phenomenon has immediately been linked to aging [483, 484]. Indeed, a host of studies have shown that aging and senescence are highly interrelated [24, 484-497], although senescence itself is defined as a permanent growth arrest that does not necessarily lead to death of the cell [487, 491-493,498]. Senescent death is also an evolutionarily developed demise program, but unlike apoptosis, it aims to eliminate those aged, although still useful, cells [302, 303, 466].
Hyperplasia is the responsive type whereas neoplasia is the intrinsic type of growth
Hyperplastic and neoplastic cells differ starkly in not only their cell death pattern but also their growth pattern. Leslie Foulds (1902-1974) split growth rate into “the responsive” and “the intrinsic” components, with the total growth of the cells being the sum of the two [155]. He wrote in 1953 that, “all cells which can give rise to cancer possess the ability to multiply at a given rate, provided the environmental conditions are constant. They also have the capacity to respond to nutritional and hormonal growth factors, temperature, pH, etc. The intrinsic growth rate of normal cells is in general low; their responsive growth rate is high. The cancerous change goes with acquisition of a greater intrinsic growth rate and diminished responsiveness; the more malignant a cell, the greater the intrinsic and the less the responsive growth” [155]. In today's language of cancer research, “the responsive growth” is the regenerative type of cell proliferation that is controlled by the host's body [466] and dwindles away during carcinogenesis, whereas “the intrinsic growth” is the autonomous proliferation that is controlled by the cells themselves and is strengthened during carcinogenesis. Hyperplastic cells are still loyal to the host's body and thus their growth belongs to the “responsive” type.
Autonomy is manifested not only as uncontrolled replication but also as uncontrolled function
Although a neoplastic nature is defined as “uncontrolled replication” attributed to the gain of intrinsic replicative ability, in reality there are some tumors that do not actually kill patients by expansive cell proliferation but, instead, by their uncontrolled functions [499]. Examples include some endocrine tumors, such as some islet-cell carcinomas that secrete insulin [500] and pheochromocytomas that secrete catecholamines such as adrenalin [501]. As the most salient feature of these tumors, the patient's body has lost its control over the tumor's functions. While the tumor is still small without invasion or metastasis, a virulently high level of the hormone it secreted may have already killed the patient. Keloid scar, which is not classified as tumor in pathology textbooks but show neoplastic features such as recurrence and incurability, may be an example of uncontrolled function of benign lesion [502-504], as its fibroblasts constantly produce collagen. Moreover, uncontrolled function may sometimes show as uncontrolled metabolisms, embodied by such as cachexia-incurring cancers that elicit high metabolic rates to cannibalize many cells of the patient for energy. Therefore, disloyalty to the host's body can be manifested mainly as the loss of the host's control over the tumor's functions or metabolisms, and not predominantly as the loss of the control over the tumor's cell proliferation, as Markert [499] and Pitot [505] had already pointed out in 1968.
Animal models can generally be dichotomized
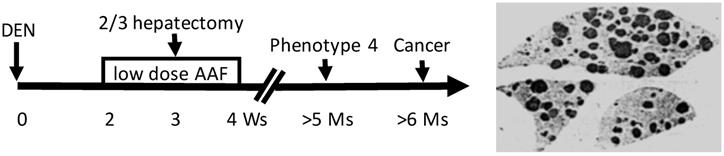
Animal models established since the 1900s have evolved using, as the inducer, a single agent to using a complex regimen or manipulations. Nevertheless, we try to split all animal models into two groups, based on whether or not the inducer is a potent genotoxic agent, although there are many intermediate models in which the inducer is a combination of both genotoxic and non-genotoxic agents [506]. In one group wherein the inducers are potent in causing mutations, mutation(s) responsible for the initiation occur early. A prime example is the Solt-Farber's “resistant hepatocyte” model of hepatocarcinogenesis in the rat (Fig. 1) [507, 508], or our modified version of it in which the promoting agent 2-acetylaminofluorene is routed via gavage instead of by feeding ad libitum [509-512]. Carcinogenesis in this group follows a trajectory of “initiation-promotion” or “initiation-promotion-progression”, as detailed by Farber [39-41, 513, 514]. It is clear that the genes and their mutations responsible for initiation are not those responsible for immortality and autonomy. This can be discerned in the Solt-Farber model wherein spontaneous proliferation, which reflects immortality and autonomy, occurs only in the lesions coined by Farber as “phenotype 4” that appear months after the establishment of initiated cells and after the completion of the carcinogenic regimen (Fig. 1) [514].
The Solt-Farber's “resistant hepatocyte” model of liver carcinogenesis in the rat. A toxic dose of diethylnitrosamine (DEN) will 1) cause liver necrosis and 2) create initiated hepatocytes. Two weeks later, when the liver has recovered from the necrosis, the rat will be given a low dose of 2-acetylaminofluorene (AAF) for two weeks, function of which is to inhibit proliferation, so-called mitoinhibition, of hepatocytes, but the initiated cells are resistant to this inhibition. In the middle of AAF treatment, hepatectomy will be performed to remove two-thirds of the liver, which provides a strong impetus for regeneration. Because normal hepatocytes are mitoinhibited, all regeneration pressure is imposed onto the initiated cells, driving them to proliferate robustly and form nodules. The image at the left shows these nodules visualized by immunohistochemical staining of the P form of glutathione S transferase, a marker for the nodular cells, in the three remaining lobes of the liver four weeks post cessation of AAF treatment [201,852]. These nodules will regress afterwards but some new focal cells, which can proliferate spontaneously and are coined by Farber as “phenotype 4”, will later develop from some of the nodules [507,508]. One or several of these phenotype-4 lesions will eventually progress to overt cancers.
The other group of animal models uses non-mutagenic agents as the inducers, which in the literature are often dubbed as “epigenetic carcinogens or agents” [515-520], “nongenotoxic carcinogens” [519, 520], or “cocarcinogens” [34, 521-523]. In our opinion, carcinogenesis in this group often incepts with promotion, but not with initiation, unlike that in the aforesaid group. This is because the nongenotoxic inducer in this group kindles proliferation of normal cells without incurring mutation(s) or even epigenetic aberration(s) to establish initiated cells in the early incarnation, and therefore the early proliferative lesions are not of initiated cells, meaning that initiation with some genetic changes, and the subsequent neoplastic transformation, occur much later in this group of models than in the above one. Alternatively, initiation in this group of animal models might not involve mutations, as considered by some investigators [307, 524-531].
Unlike in animal models, immortality and autonomy may occur early in most human tumors
When are immortality and autonomy established during a lengthy tumorigenesis in humans? It is an enthralling brainteaser, so far without an answer [532]. For several reasons we infer that in most cases they occur at an early time point (Fig. 2). First, spontaneous regression of tumors is rare, and thus nearly all tumors, many of which are diagnosed at early stages, are considered immortal. Second, in our pathology service and in the literature [533], we occasionally encounter very tiny malignant tumors in patients. Albeit the small tumor had already been surgically extirpated or considered cured, some patients still died of its metastasis years later [534], which substantiates the malignant authenticity of the small primary tumor. Third, autopsies of humans that died of various causes found about 3-27% of the bodies had an occult pituitary adenoma [535-539] (and DJ Liao's empirical knowledge), and magnetic resonance imaging of normal human volunteers found this tumor in about 10% of normal persons [540]. Similarly, it has been known since 1934 that a large number of men over 40 years of age have occult prostate adenomas or adenocarcinomas, although many of the lesions do not develop to clinical cancer before the men die from other reasons [541-546]. A much higher incidence of occult tumor occurs in the thyroid, since one early report showed that 49.5% of 821 clinically normal people contained nodules, 17 of which were histologically malignant [547,548]. Similarly, unselected autopsies of children before three months of age also found neuroblastomas in the adrenals at a frequency 40-50 times higher than the reported incidence of this tumor [549]. Fourth, as summarized by Blagosklonny [532] and Kolquist [550], even many premalignant lesions in humans show immortal traits, such as elevated expression or activity of telomerase. Nevertheless, more tangible proof for the speculative early-establishment of immortality and autonomy is still needed.
Illustration of a speculative difference at the time point for the establishment of immortality and autonomy between the tumorigenesis in most animal models and that in most human situations. In humans, immortality (Immort.) and autonomy (Auto.) may occur at a very early time point, thus establishing small lesions as genuinely benign or malignant neoplasms. In contrast, tumorigenesis in most animal models is a stepwise procedure of initiation, promotion and, in some cases, progression as well. Initiated cells are still mortal and thus are not neoplastic. Immortality and autonomy in animal models occur at late promotion or at the progression.
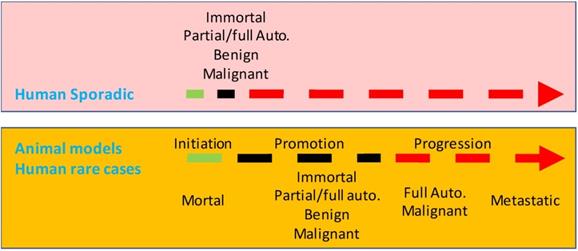
In humans, tumor-promoting momentum is much weaker, including the impetus provided by those relatively potent promoters such as cigarette smoking or chronic viral hepatitis, compared with that provided in various animal models. Therefore, human lesions grow and progress much more slowly, allowing immortality and autonomy to occur much earlier with respect to the size of the lesions, and allowing the neoplastic transformation to occur as the result of some relevant mutation(s), long before the patients feel something wrong and go to see their doctors. This is partly because a lengthier course allows accumulation of more haphazardly-occurring mutations, including the one(s) required for immortality and autonomy, if we accept the notion that tumors, especially cancers, occur as repercussions of mutations that have cell-autonomous modes of action [29-31, 525, 526, 551-558].
The inducer-dependency of the early tumors in most, if not all, of animal models indicates a very late establishment of immortality and autonomy, which collides with the perceivable early-establishment of immortality and autonomy in human lesions described above. In other words, few, if at all, animal models established so far reflect the tumorigenic course in most human situations (Fig. 2). Fortunately, in some rare human situations, immortality and autonomy are likely to be established in a late stage. For example, familial colorectal polyps that will sooner or later progress to cancer are developed due to inherited mutations in some genes, like the APC (adenomatous polyposis coli) gene [559-563]. The constant presence of the mutation serves as a lasting coercion on colorectal mucal cells, keeping them in an unremitting state of proliferation to form polyps. These polyps are considered in pathology as premalignant lesions, pursuant to their morphology and to the fact that cancer likely ensues. Notwithstanding, we are curious about whether the polyps would regress if we have a way to correct the mutation, since spontaneous regression not only of polyps but also of small colorectal adenomas has been well recognized by pathologists [564-566]. Probably, from the point of immortality and autonomy, the still-mortal polyps are “preneoplastic”, an elegant jargon used by Haddow [72] and Rubin [567], or are “precursor lesions”, another good appellation used by Farber [568]. Other embodiments of the late establishment of immortality and autonomy in human outgrowths include the abovementioned curable hepatomas and hepatocellular carcinomas caused by chronic use of oral contraceptives [324-330], and lymphomas or leukemia caused by HP [334-340] or HTLV-1 [341-343]. Some thyroid tumors may also be mortal and may not evolve to authentic neoplasms in the patients' lifetime [569], which dovetails with Forth's opinion in 1953 [155].
What do the two genetic hits do in carcinogenesis, and is a third hit needed?
Tumorigenesis may sometimes occur via only “one-hit” [570-572], but “two hits” are usually required [17-20]. Although the “two hits” are still ill-defined, sometimes as two genetic alterations but some other times as “initiation” and “promotion”, the concept accepts the century-old ideas that carcinogenesis results from genetic alterations and that cancer cells owe their properties to mutations [450, 573-576]. We mingle the two different “two hits” definitions together and consider that the first genetic hit is for creation of initiated cells that differ from their surrounding cells in response to promoting environment (Fig. 3). According to Farber [39-42, 513, 514, 568, 577-582], in most cases promoting agents cause “mitoinhibition”, i.e. inhibition of mitosis or proliferation, of normal cells, whereas initiated cells are resistant to this inhibition (Fig. 1) [200]. Actually, a condition disfavoring cell growth in cell culture, such as a lower serum concentration or a cell confluence situation, is an impetus to drive neoplastic transformation as well [313]. Therefore, in a promoting environment, probably also in humans [313], only initiated cells can robustly proliferate to form lesions, especially when many of their adjacent normal cells die and the organ or tissue has a strong demand for regeneration [200, 201, 583]. This “mitoinhibition” theory conforms with the hypothesis of Rozhok and DeGregori that cancer occurs more often in old age [453, 584], because normal cells in the elderly, compared with their counterparts in the young, have less proliferative capacity, thus being more “mitoinhibited” and providing the spontaneously-occurring initiated cells with a stronger promoting momentum [24]. The molecular mechanisms of promotion via mitoinhibition still remain enshrouded. We extrapolate, with trepidation as sans evidence, that mitoinhibited normal cells promote proliferation of initiated cells in part via a mechanism similar to that used by senescent cells to promote carcinogenesis of their adjacent cells, since senescence is a state of permanent growth arrest [487, 491-493, 498], i.e. “permanent mitoinhibition”. This mechanism is coined as SASP (senescence-associated secretory phenotype) [585-587], and its effect on carcinogenesis has been extensively reviewed in the literature [487, 588-596].
Illustration of our three-hit hypothesis. Coupling the traditional two-hit principle with the initiation-promotion theory leads us to a supposition that the first genetic hit establishes initiated cells that are still mortal and non-autonomous, whereas the second hit creates immortality and autonomy, thus establishing neoplastic cells, either benign or malignant. Since formation of benign neoplasms also requires two genetic hits, we extrapolate that, in some animal models and probably also in many human situations, establishment of malignant morphologies and behaviors requires a third hit on the relevant gene(s).
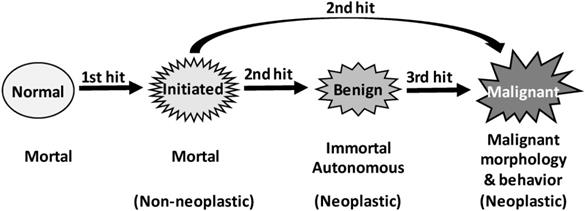
In Rubin's punditry, the cells of skin papilloma produced in the aforementioned animal models that regress upon withdrawal of the inducer are initiated [451], which connotes that initiated cells are not immortal. Indeed, in Farber's “mitoinhibition model” of hepatocarcinogenesis described above, most initiated cells in the focal lesions eventually die of apoptosis [39-42, 513, 514, 568, 577-582]. In our meditation, the second hit converts initiated-cells into a neoplastic state, benign or malignant, by rendering the cells immortal and autonomous (Fig. 3). This second hit occurs in a later promotion stage of the “initiation-promotion” models or in the progression stage of the “initiation-promotion-progression” models. In sporadic carcinogenesis in humans, initiated cells may also exist, although they are technically difficult to identify. Nevertheless, “preneoplastic” cells in humans may have already experienced the first hit, while “pre-cancerous cells” may have also experienced the second hit.
In some carcinogenic procedures wherein a malignancy does not require a benign lesion as a precursor and thus a second hit is sufficient, the mutation(s) responsible for immortality and autonomy may also be responsible for malignant morphologies and behaviors (Fig. 3). However, in other animal models and in human situations, the mutations responsible for establishing immortality and autonomy may not be the ones responsible for establishing malignant morphologies and behaviors, since benign neoplasms have also experienced the second hit. Therefore, in these situations a third genetic hit may be required to establish malignant morphologies and behaviors (Fig. 3). Of course, malignant neoplasms continue to evolve via many subsequent hits to be more and more heterogeneous and heinous.
An old, but still unanswered, question is how many mutations are needed for completing a carcinogenesis
The target or targets of the abovementioned two or three genetic hits remain unknown to us. Initiation created by the first hit likely involves only one or several genes, since initiated cells are morphologically indistinguishable from uninitiated ones [40, 42, 514, 597]. Immortality and autonomy created by the second hit may involve only one or several genes as well, since many benign tumor cells, such as uterine leiomyoma cells, are quite similar to their normal counterparts in cellular morphology. Therefore, it is not surprising that acquisition of immortality does not require genetic instability, and cancer cells can be created and sustained without gross genetic changes [598-600], although instability and gross mutations can occur even at an early time point of carcinogenesis [601]. The inference that only one or several genes are involved is also supported by the fact that immortalization of a mortal cell to establish a cell line has been proven to be easy, especially in vitro [139, 602-608]. For instance, targeting both the p16ink4 and c-myc genes can immortalize human mammary epithelial cells invitro [598], and the IgEGF and SV40T bi-transgenes can immortalize murine cells [609]. Actually, immortalization is easier when the cell has a small-rodent parentage. Simply knocking out the p53 gene alone can immortalize mouse hepatocytes [609], and even ectopic expression of a 3'-untranslational region of a gene without expression of the protein [610] can immortalize rat embryonic cells. A so-called “3T3 protocol”, mainly transferring 3x103 cells from a flask to another every three days, had been established almost six decades ago as an effective procedure to immortalize mouse cells, especially embryonic ones [611-613]. This simplicity is presumably because small-rodent cells have their telomerase constantly “on” and have only a single barrier to immortalization controlled by the RB (retinoblastoma protein) pathway [602, 614, 615].
The third hit, if it is needed, may also require only a small number of genes, in our opinion, since the second hit can do both, i.e. can immortalize the cells and confer malignant morphology and behavior upon the cells. Therefore, the sum of the two or three hits may be congruent with the estimation by Hahn and Weinberg that five alterations are required for converting human cells to malignant phenotype [616], or by Armitage and Doll in 1954 [31] and by Vogestein in 1993 [617], that carcinogenesis requires only six or seven mutations. Fluid cancers such as leukemia may require even fewer and thus may be relatively easier to cure, generally speaking, as we inferred before [449]. A caveat is that different hits in different cases may involve different genes, especially for the third hit that is responsible for cellular and histological morphologies and behaviors that can vary greatly among different cases of the same cancer type. This variation makes the sum of “initiator genes”, “immortalizer genes”, or “malignant morphology responsible genes” large, and the sum of all three even larger, which is a major reason why there have been a huge number of genes found to be cancer-relevant.
Actually, whether mutation is needed or not for tumor formation is still debatable
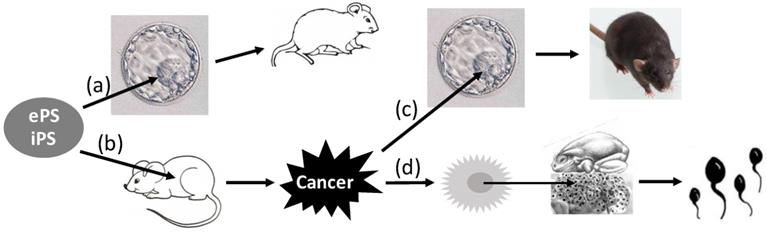
Although it has become “Tumor 101” that tumors are caused by variegated genetic alterations, collectively coined herein as “mutations”, there has always been a theory considering that mutations are not necessarily required, which, since it was broached by Rous in 1947 [531], has continued receiving supportive laboratory data [307, 525-530, 618-621]. Some peers even consider that heterogeneity of cancer cells may not necessarily be related to the increases in mutations either [622]. A major piece of evidence supporting this non-mutation theory is from Stevens' reports in the 1960s, which showed that transplantation of germinal stem cells from early male mouse embryos of the 129 strain to testicles of adult mice led to the development of teratoma or teratocarcinoma (Fig. 4) [623-625]. As reviewed by Buta, Bustamante-Marin, Damjanov, Arechaga, Blum, Sell, Martin, and Pierce [4, 626-639], many other researchers also reported later that early embryonic cells, including those of human origin [630], placed into several extrauterine sites of adult animals could develop to teratoma or teratocarcinoma [629, 640-644]. A slew of studies in the past decade have extended these findings by showing that induced pluripotent stem cells transplanted to animals can develop to teratoma or teratocarcinoma as well (Fig. 4) [626, 645-650]. Considering that extrauterine sites should not be mutagenic, these observations support the non-mutation theory. Moreover, this tumorigenesis involving embryonic or induced pluripotent stem cells can be minimized or prevented by different manipulations [626, 647, 648, 650-652]. Conversely, teratocarcinoma cells injected into the blastocyst can be incorporated into the developing embryos, and the organs or tissues of the mice developing from such embryos contain cells from both the blastocyst and the cancer (Fig. 4) [653-661]. More convincingly, injection of the nuclei isolated from the Lucké renal cancer cells of the frog origin into enucleated frog eggs allows the eggs to hatch out tadpoles that are normal without any trace of cancer (Fig. 4) [285, 662-668]. Similar conversion back to a normal state by an embryonic microenvironment has also been shown for a few other cancer cell types [448, 669-673, 673, 674]. For instance, with models of chick embryo and Zebrafish embryo as well as with intrauterine injection approach in mice, many studies have shown that human malignant melanoma cells in an embryonic microenvironment do not develop to tumors but, instead, differentiate to neural-crest-like cells [675-678]. Some cells of squamous cell carcinomas have also been observed to differentiate into mature keratinized cells as squamous pearls [679]. Therefore, as pointed out by Pierce in 1974 [632], the concept of “once a cancer cell, always a cancer cell” may not always be correct. Because mutations are unlikely to disappear by themselves [632], development of mature tissues from cancer cells lends color to the non-mutation theory.
Reversion of pluripotent cells between normal and cancers. Embryonic (e) or induced (i) pluripotent stem (PS) cells introduced into the blastocyst in the uterus can develop to live animals (a). However, if the cells are transplanted to extrauterine sites of adult animals, they will likely develop to teratomas or teratocarcinomas (b). If teratocarcinoma cells are inoculated into the blastocyst, they will be incorporated into the developing embryo, and the tissues of the animal developed from the embryo will be chimeric, i.e. containing cells from both the embryo and the cancer (c). Moreover, if inoculation of the nuclei isolated from the Kucké renal cancer cells of the frog origin into enucleated frog eggs, the eggs can hatch out live tadpoles with all tissues normal (d).
It is worthy of mentioning that the abovementioned experiments with teratocarcinoma cells have a much better version in plant tumor systems, as thoroughly reviewed by Braun four decades ago [680]. It has been shown in several plant species that teratomas can be reverted to normal plant cells and that tumor cells grafted to another plant can develop to a normal plant that can bloom and produce seeds, and the seeds can germinate and grow to normal plants [20, 681-687].
Another major piece of evidence invigorating the non-mutation theory comes from the studies showing the reversibility of transformed cells back to the normal in cell culture [446, 453, 620, 688-691] and in animals or humans [197, 370, 692-695], with or without induction by chemicals [696-704]. Actually, this reversion has received attention for almost a century [446,688,689], in part because direction of cancer cells to differentiation is a tantalizing strategy for cancer therapy. This reversibility is more clearly discerned in tumors of some species of fish and amphibians that are seasonal or temperature-sensitive, as aforementioned [277-285]. However, the reversion does not necessarily indicate that the transformed cells were not initially transformed by mutations. It could be that the genetic alterations the transformed cells bear cannot prevent the reversion elicited by other genetic mutations or by some epigenetic changes. Or, alternatively, the extrinsic factors that cause the reversion can circumvent or override the initial mutations responsible for the neoplastic morphology and behavior (Fig. 5).
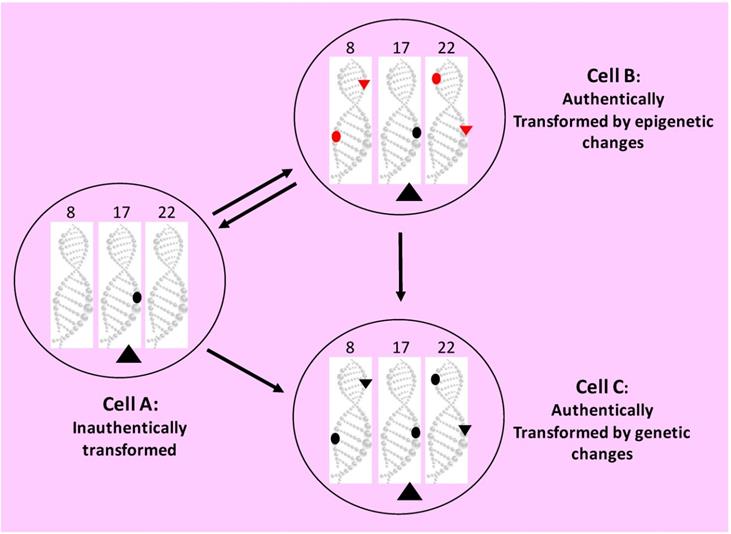
Depiction of the “coercion hypothesis”. Our manipulation, say as a transfected cDNA (the large black triangle in the cytoplasm of Cell A) or as a transgene or a gene-knockout on the chromosome 17 (black dot in Cell A), coerces the primary cell into incessantly replicating and manifesting transformed morphology or behavior, such as colony formation in agar. The relentless proliferation will eventually lead to spontaneous occurrence of the epigenetic (red dots on the DNA of chromosomes 8 and 22 in Cell B) or genetic (black dots on the DNA of chromosomes 8 and 22 in Cell C) alterations that establish immortality and autonomy, making the cell truly neoplastic in behavior. Continuous proliferation will also cause spontaneous occurrence of the epigenetic (small red triangles on the DNA of chromosomes 8 and 22 in Cell B) or genetic (small black triangles on the DNA of chromosomes 8 and 22 in Cell C) alterations that establish neoplastic morphology. This is to say that immortality and autonomy as “the behavior aspect” of neoplastic property, as well as “the morphology aspect” of neoplastic property, may sometimes be controlled separately by different sets of epigenetic or genetic alterations, i.e. different sets of “hits”. Moreover, the cell authentically transformed via epigenetic mechanisms (Cell B) may initially be reversible back to the normal, but later it will likely develop such genetic alterations that make the cell lose the reversibility and progress into the state of Cell C. If our manipulation is made in a controllable manner and is withdrawn early, the primary cell (Cell A) will no longer manifest the transformed morphology and behavior and will undergo senescent death (if the cell is in a culture dish) or both senescent death and apoptosis (if the cell is in a live animal). However, the truly transformed cells (Cells B and C) may retain their neoplastic properties sustained by the epigenetic or genetic alterations, unless some extrinsic factors (such as a chemical) cause the cells to circumvent or override the epigenetic or genetic alterations and make the cells reverse back to the normal state with or without retaining the alterations. In other words, neoplastic morphology and behavior incurred by our manipulation, an extrinsic factor, are inauthentic, but those caused intrinsic epigenetic or genetic alteration(s) are authentic.
Epigenetic alterations certainly make a considerable contribution to the formation and progression of tumors [705]. What we still wonder is whether these changes alone, without involvement of genetic mutations, are sufficient for the development of authentic neoplasms that are immortal, autonomous and irreversible (i.e. without undergoing spontaneous regression) and progress continuously towards more and more diabolical states, as seen in most cancer patients.
Are immortality, autonomy, and transformation extricable from one another, and which occurs first?
In 1983 Land et al showed that embryonic fibroblasts expressing a ras mutant could form colonies in soft agar [706], which was shown by Freedman in 1974 as an insignia of a transformed state [139, 707]. However, the transformed cells could not grow constantly and were still mortal, and their immortalization required concomitant expression of the c-myc gene or a viral oncogene [706]. Similarly, expression of the SV40 large T antigen in mouse embryonic fibroblasts goads the cells into forming colonies in soft agar, but most of the cells eventually die [708,709]. Primary cells concomitantly expressing a CDK4 gene and a ras mutant can form colonies in agar and develop to invasive tumors in animals, but the cells still cannot grow indefinitely in culture [710]. All these data and others [711] strongly suggest that neoplastic transformation can occur before, and thus can be extricated from, immortalization, which is braced by the observations that telomerase itself is capable of prodding primary cells into growing in agar and in animals, independent from immortalization [532, 712] and transformation [371, 713]. However, this seems to collide with the two-stage model that is sometimes perceived as that the “initiation” immortalizes normal cells whereas the “promotion” transforms the immortalized cells [370, 371]. Newbold, Reddel, and some other cancer wizards also consider that immortality is an early and prerequisite step of transformation [477-481, 714].
While the above discrepancy still awaits an answer, a related question is raised as to whether autonomy can also be extricated from immortality, although this segregation collides with the facts that human cancers rarely regress spontaneously and that no human tumor shows this segregation. A keloid scar may be the only tumor-like lesion in the human we know that seems to show this extrication, as it shows functional autonomy by constant collagen production without showing clear immortality of its fibroblasts [502-504]. Some animal models seem to show this extrication as well: epithelial cells have been shown to be evasive, disseminating, and able to enter into the bloodstream before they form primary tumors [715]; and mammary epithelial cells can be manipulated to metastasize and colonize in the lungs before they are malignantly transformed [716, 717]. Nevertheless, such separation is not discerned in some tests with traditional approaches involving chemical carcinogens, such as the “Syrian hamster embryo cell transformation assay” [718, 719].
In our opinion, if an in vitro study shows the above extrications, more-tenable proofs for the neoplastic state are needed. This is to say that once the cells are shown to be capable of forming colonies in soft agar, they need to be tested for immortality before we can announce that they have been transformed. Unfortunately, many published studies of in vitro transformation do not show this additional evidence corroborating the immortal nature of the cells.
It is worth ruminating about why we can only induce several tumors in an animal
There is an obvious discrepancy between neoplastic transformation in cell culture and tumor formation in animals, which has been baffling us for a long time [200, 720]: in vitro transformation assay usually results in a large number of colonies in soft agar. Because each colony develops from a single transformed cell, the appearance of many colonies means that many cells have been transformed; this in turn means that the transformation assay is very effective, although it is a short-time procedure. However, in most genetically modified animal models, each animal develops only several tumors in its lifetime, albeit the target organ or tissue of the animal, say the liver or the five pairs of mammary glands, have trillions of cells bearing the same genetic modification. For instance, only 1 of 10 mammary glands in a c-myc transgenic mouse develops a tumor [721, 722] and only 4 or 5 of 100 pancreatic islets develop β-cell tumors in a SV40-LT-transgenc mouse [723]. Actually, most chemical-carcinogenesis models produce only one to several tumors per animal, to our knowledge. If evaluated with the number of tumors per animal as the criterion, the only plausible conclusion is that the efficacy of our in vivo transformation approach is negligible, as trillions of the targeted cells fail to be neoplastic. Researchers are usually content with the high percentage, sometimes 100%, of the manipulated animals that develop tumors, and do not ask why the remaining trillions of cells in the target organ or tissue of the animal, which received the same manipulation simultaneously, do not evolve to overt tumors.
Many animal models are overpraised, due to neglect of the immortality issue
As aforesaid, evidence for immortality and autonomy has not been provided for the lesions resulting from the vast majority of genetically-modified animal models of carcinogenesis. More correctly, the time point has not been determined at which lesions in these animal models enter into the immortal and autonomous state. This is an uncomfortable but undeniable flaw of the relevant studies, although we should have been content with the profuse information provided by these models on the functions and underlying mechanisms of the genes manipulated. It is possible that many peers have already harvested the “cancers” from the animals for the mechanistic analyses before the lesions, probably large in size, have evolved to genuine neoplasms; likely due to the unawareness of the importance of the immortality and autonomy issue. In 1948, Greene emphasized that, “the problem of cancer is primarily a problem of behavior. A pathologist who examines tumor tissue under the microscope may observe significant details of form and structure, but he can never determine its malignancy from its appearance alone; only by its behavior in the living body can malignant tissue be unmistakably identified. Of two tumors with cells that look exactly alike, one may remain static or even disappear while the other inexorably spreads and kills the patient. Unfortunately many kinds and conditions of tissue which are not malignant bear a remarkable resemblance to cancer” [431]. Because animal lesions, even if they are large in size, have not yet established immortality and autonomy, they can be cured easily, simply by withdrawal of the inducer or by chirurgic extirpation. This contrasts with the fact that most human cancers are not curable, at least not so easily. Moreover, “malignant” tumors in most animal models do not metastasize within the lifespan of the animals, whereas most human cancers will metastasize if untreated. Indeed, an early estimation has shown that fewer than 30% of genetically modified animal models of carcinogenesis produce metastases [724], although there are several models of mammary carcinogenesis showing metastases [725, 726]. Some tumors from animal models can metastasize, but the metastases may still be inducer-dependent, embodied by the aforementioned metastases of the TSH-instigated thyroid tumors [155, 181, 185] and of the estrogen-induced renal tumors in hamsters [156-158]. In a nutshell, the mortality and non-autonomy, the inducer-dependency, and the inability to metastasize are telltale evidence that many animal cancers are easy to cure and thus are disarming, which starkly contrasts with most human cancers. Moreover, among the species differences is that most spontaneous malignant lesions are mesodermal-originated sarcomas in mice but are epithelial-originated cancers in humans [727-729].
We still have no way of directly transforming cells in vitro and in vivo
In the above sections we have described five phenomena that dissent from, and may threaten the bedrock of, the orthodox doctrine of how cancer develops: 1) transformation, immortality, and autonomy can be segregated from one another in the lab, and which one occurs first depends on the experimental design, although immortalization occurs first in the two-stage model. 2) In vitro transformed cells may still be mortal and reversible back to the normal; 3) formation of tumors may not necessarily involve mutations; 4) outgrowths induced in animals are inducer-dependent until advanced stages; and 5) neoplastic transformation in animal models may be perceived to have a negligibly low efficacy because each animal develops very few tumors after a long latency. All of these phenomena deliver us a single message: although numerous alterations have been identified in a huge number of genes, none of our in vitro or in vivo manipulations are able to directly cause the epigenetic or genetic alteration(s) required for the establishment of immortality and autonomy (Fig. 5). In animal models, there so far has not been any evidence showing that turning on or off a gene, which has been technically feasible for decades, can quickly turn on or off immortalized or autonomous features of the targeted primary cells. In cell culture, none of our manipulations of physical, chemical, or biological factors can quickly immortalize and transform primary human or animal cells. The targeted cells show immortal and transformed features only weeks later in the culture or months later in the animal, obviously as the events secondary to our manipulations. Exceptions do exist peculiarly, as some plant cells can be transformed after only 34-48 hours of manipulation [287, 730, 731]; with a few more days of manipulation creating more aggressive cells [680, 732-739]. Nevertheless, to our knowledge mouse or rat models require 6-9 months for the tumor induction, which is about 1/6-1/4 of the mouse or rat lifespan; although in some rare cases very potent chemical carcinogens might induce tumors in 3-4 months, i.e. about 1/12-1/9 of the lifespan. Considering that the early tumors are still inducer-dependent, it is likely that the actual latency for the occurrence of authentic neoplasm is much longer.
We extrapolate that all of our manipulations to transform animal or human cells may just coerce the cells into incessantly replicating, sustaining the cells' life, and manifesting neoplastic morphology and/or behavior without actually transforming the cells (Fig. 5). Therefore, when the coercion ends, the “transformed” cells either return back to the normal or undergo apoptosis as they are redundant [740-742]. For instance, expression of the SV40 large T antigen in primary cells can confer additional 20-30 population doublings upon the cells, during which some cells are immortalized spontaneously [743]. In animal models, the cells of the duress-sustaining lesions still have allegiance to the animal's body. This preserved allegiance, which bespeaks the non-autonomous nature and categorizes the lesions into hyperplasia (although showing malignant morphology), is the reason for why the cells regress via apoptosis so that the host organ or tissue does not possess redundant cells [203, 296, 303, 466-469,474]. Actually, the lesions may show higher rates of apoptosis, and probably also senescent death, than their host tissue because of their hyperplastic nature, although the inducers may suppress the apoptosis and senescent death as components of their coercive mechanisms. In cell culture, because mortal cells in dishes are no longer under the control of the animal's body and thus do not need to care about the cell redundancy issue, the cells die of only senescent death, and not of apoptosis [302, 468]. For this reason, the manipulations in most in vitro transformation assays are made in a perpetual manner, such as being made as stably-expressing cell clones, to prevent the loss of the coercer. Actually, some techniques of conditional immortalization and/or transformation [744-749], along with many conditionally immortalized cell lines [743, 750-759], like the temperature-controlled ones [756, 760], have been widely established to make it feasible to turn on or off the coercer gene. There are even transgenic animals established to facilitate the establishment of such conditionally immortalized cell lines [755]. Some of these conditionally immortalized cells can form colonies in soft agar when the coercer is turned on, but no colony is formed when it is turned off [756, 759, 761]. Obviously, the “conditional” means reversable, implying that the immortality or transformation occurs simply under the duress of the immortalizer or the transforming gene, and not due to the relevant epigenetic or genetic alterations.
In all in vitro and in vivo models, the epigenetic or genetic alteration(s) for immortality and autonomy occur only spontaneously in a random and stochastic manner during constant cell replication caused by the duress (Fig. 5). Because of this manner, it occurs only to several cells in animals at an early-enough time point that leaves the cells with a sufficient time to evolve to overt tumors. This late establishment of immortality and autonomy betokens that primary cells put more guards on the genes responsible for immortality and autonomy as the second defensive line, compared to the guards on the genes relevant to the initiation as the first defensive line, meaning that the first hit is easy, but the second hit is difficult. Until now, no exogenous agent, a chemical, irradiation, biological factor, or any other, has been identified that can break through this second defensive line of cells, and we have no idea on which genes are involved. Actually, a question that tantalizes us is whether such genes really exist. What a lengthy promotive period in all animal models established so far tells us is that breaking through this second defensive line can only be made by currently-unknown intrinsic factor(s). Fortunately, our manipulations as extrinsic factors can accelerate the breaking-through by sustaining the cells' life; accelerating cell replication, damaging DNA, and/or inhibiting DNA repair.
Regardless of its mechanism, the reversibility of in vitro transformation and the inducer-dependency of animal lesions remind us that we cannot consider cells or lesions neoplastic based solely on their morphology and behavior. Diagnosis of outgrowths induced in animals should not solely rest on the pathological morphology and formidable behavior, although the relentless proliferation begot by the duress will one day lead to the epigenetic or genetic alterations for true neoplastic transformation. This “coercion hypothesis” (Fig. 5), proposed by us a few years ago [720] and recently [47, 303, 466] on the essence of animal models of carcinogenesis, deserves experimental testing.
We still lack a good strategy to determine molecular pathways leading normal cells to cancers
The genes mediating the two or three genetic hits described above remain unknown. One of the reasons is that we have been encountering a logical plight for decades regarding our research strategies and approaches, as repeatedly pointed out by us before [47, 296, 720, 762]: the results from the approaches we used, such as genetic engineering, can only tell us that certain manipulations or alterations, like concomitant overexpression of the c-myc and a k-ras mutant, and the ensuing cascades of molecular changes, can eventually cause neoplastic transformation or tumor formation. However, we still do not know whether cells in humans or in untreated animals really do spontaneously develop to neoplasms because of the abnormalities of these genes and via these cascades of molecular changes. In an analogy, we have built the highway Interstate-95 (I-95) and know that Mr. Trump can go from New York City (NYC) to Washington DC by taking it, but we do not actually know whether this is indeed the path, but not one of the others, he took. If we still cannot find a way to break this impasse, our attempt to learn why and how some cells in humans become neoplastic will continue to be prodigal financially and in effort. This is because we will continue to identify (more correctly, to create) many more molecular pathways leading normal cells to neoplasms, besides the many pathways already known or created [763], while we remain unable to hold any particular pathway(s) accountable for sporadic carcinogenesis in humans. Restated, we are creating, but not identifying or discovering, pathways, such as by creating otherwise non-existing transgenic or knockout mice, and surmise that these man-made paths are the carcinogenic procedures occurring in patients' bodies. In another analogy, we already have many paths leading from NYC to DC but will endlessly build many more while remaining unable to know which path(s) Mr. Trump took or will take. Probably, we have been upending things or putting the cart before the horse in our research.
Neglecting immortality causes confusion on aging-caused cell death in outgrowths
Normal animal cells undergo aging and eventually die of it [465, 474, 764-774], and so do cells in overt outgrowths from many animal models, more often in morphologically-benign than in morphologically-malignant ones [769, 771-773]. This type of cell death has established “cancer cell senescence” as a popular research bailiwick [474], although many relevant studies do not involve lesions from animals but, peculiarly, use cancer cell lines that are immortal. Because its essence is “dies from aging”, the “cancer cell senescence” concept is illogical and collides with the immortality and autonomy criteria for a neoplastic state, since it connotes that immortal cells still undergo aging and eventually die of it. In our logic, neoplastic cells are immortal and thus cannot undergo aging and eventually die from it, whereas cells that can undergo aging and die from it cannot be regarded as neoplastic no matter how much their morphology and behavior resemble those of neoplastic cells. In all those lesions that are morphologically malignant and even metastatic but are still inducer-dependent, such as the TSH-dependent thyroid tumors and their metastases [155, 174, 185], cells may undergo aging and die via senescent death because they have not yet become authentically neoplastic.
Neglecting immortality causes confusion on cancer stem cells
As Sell has pointed out before [4], in the mid-1990s there were two important concepts on stem cell theory of cancer reemerging simultaneously in the literature. One is the hypothesis that cancers arise from normal stem cells in the organ or tissue, which somehow had gone awry and lost the ability to differentiate while having acquired the ability to proliferate indefinitely [635]. This theory actually appeared in the 19th century [775] and reemerged in the literature in the 1960s and 1970s [4, 499, 632-636, 776, 777]. It developed from the concept that cancers originated from embryonic cells with stemness. As reviewed by Triolo [5], this concept was first suggested by Virchow's teacher Johannes Müller in 1838, involved the work of Boll, Cohnheim, Durante, and many others, and had become popular as the “blastema theory” in the 19th century. As reviewed by Trosko et al [778-783], this initial concept annotates cancer stem cells (CSCs) as those organ- or tissue-specific stem cells that somehow go amiss, likely due to some epigenetic and/or genetic aberrations [781,784], and gradually evolve to cancers [4, 634-636, 764, 785-787]. Sometimes these abnormal stem cells are also called “transformed stem cells [788]”, “cancer progenitor cells [789]”, or “cancer-initiating cells [782, 790]”. According to a denomination of carcinogenic mechanisms, in a renewable cell type a stem cell that has gone awry may stop differentiation, in Sell's words, “showing maturation arrest” [635], during an embryonic stage or during a tissue regenerative procedure, and continues proliferating to form a neoplasm, as we described before [449, 720]. Actually, this “stop-differentiation” mechanism is presumably a reason why nonrenewable cell types still develop childhood neoplasms: pediatric tumorigenesis, incurred by such reasons as carriage of certain germ-line mutations or in utero exposure to a carcinogen [791], had already incepted during an embryonic stage when the cells still had their replicative ability [449]. For this reason, we have suggested that molecular biologists should be wary of using those DNA elements that are activated during an embryonic stage [199, 205], such as the Mist-1 promoter [792, 793], as the promoters to drive transgenes. This is because the resulting transgenic animal models may show stop of differentiation and thus mimic only the formation of childhood cancer, whereas most cancers in humans are sporadic [199, 205]. For example, both female and male transgenic mice expressing the MMTV-PyV middle T antigen develop mammary tumors at a very young age [794], in contrast to humans.
The other concept is that cancer is maintained by a small fraction of the cancer cells in the tumor mass that have the property of stem cells [795-797]. In Clarke's words, “…a subset of cancer cells within some tumors, the so-called cancer stem cells, may drive the growth and metastasis of these tumors” [798]. More detailed by Chiodi, “in many types of cancers a subset of cells shows peculiar characteristics, such as the ability to induce tumors when engrafted into host animals, self-renew and being immortal, and give rise to a differentiated progeny. These cells have been defined as CSCs or tumor initiating cells [799]”. Similarly, in the words of Weinberg's group, “the CSC hypothesis posits the existence of subpopulations of neoplastic cells within a tumor that exhibit an elevated ability to seed new tumors upon experimental implantation in appropriate animal hosts” [800]. They also say that, “evidence is accumulating that both normal and fully neoplastic cell populations harbor subpopulations of stem cells (SC) that can both self-renew and spawn more differentiated progeny” [801]. This CSC definition, which is used in most recent CSC publications [518, 785, 802-806], was derived from some findings in the 1990s that many leukemia cells showed different degrees of differentiation, and a small subset of them have stem cell properties with a great potency to populate to a tumor mass when transplanted into animals [795-798, 807]. Actually, many much-earlier studies, started by Furth and Kahn in 1937 [808], have already shown that single cancer cells in late progression stages were highly transplantable and could grow rapidly in recipient animals [809-813], as reviewed by George Klein six decades ago [408].
The two CSC concepts described above, one about cancer-origin and the other about cancer-maintenance mechanism, are unrelated and are both correct and clear, as pointed out by Visvader [814]. However, the number of the studies on the second concept has been soaring in the past 20 years, which unfortunately sets this concept as the orthodox CSC definition, admixes the two unrelated concepts together, and makes many researchers confused. For instance, CSCs are described as “cancer-initiated cells” in both concepts [782, 790, 799]. Indeed, the CSC definition in the literature of the past 20 years has remained erratic or, in the words of Dumont et al [815, 816], “fuzzy and evolving”, partly because it has never been lucid in distinguishing CSCs on the one hand from normal stem cells in embryos or in adult organs and on the other hand from the vast majority of cancer cells [817].
The “subset”, “subpopulation”, or similar words used in the second CSC concept about a cancer maintenance mechanism hint slyly, in a tacit manner, that except for a tiny fraction, the vast majority of cancer cells are not immortal and are not able to self-renew, which obviously collides with the definition of neoplasm in all pathology textbooks published since the 1900s. Benign and malignant cells relentlessly undergo symmetrical binary fission, just like bacterial cells that unremittingly divide to maintain their strains, although some aging research wizards consider that bacterial cells also undergo asymmetrical division and undergo aging as well [818-820], likely for maintaining their vitality [821]. Moreover, malignant cells are highly plastic and can differentiate to various cell types [807,822]. For instance, quite different types of cancer, even pre-cancer lesions [823], of the epithelial origin [824-834] manifest bone histology, or osseous metaplasia in pathological phraseology. Therefore, a strong pluripotency should not be used to dichotomize cancer cells into CSC and non-CSC groups. It is true that in cancers many cells die at a much higher rate than others due to various stressors, such as insufficient oxygen or nutrient nourishment, or overly severe genetic damage [554, 555, 835]. The opposite is also true that in a cancer mass some cells' ability of self-renewal via symmetrical division is much more potent than that of the others. However, these quantitative differences simply reflect the well-known heterogeneity of malignant cells [453, 836-840], which is largely ascribed to the stemness of some cells [841] and the great genetic variation among most cells [835, 842], and should not be used to split cancer cells into CSC and non-CSC groups either. More critically, “ability of self-renewal vs inability of self-renewal” is actually “immortality vs mortality”, which is a black-and-white demarcation between the neoplastic and non-neoplastic states and thus should not be used again as the demarcation between CSCs and non-CSCs. We would like to quote Paget's words in 1889 again: “as Langenbeck says, every single cancer cell must be regarded as an organism, alive and capable of development” [304]; obviously, both Paget and Langenbeck used “every single” to extend the “self-renewal” ability to all cancer cells.
We proffer that, since CSCs in the second definition differ from other cancer cells only quantitatively in such as the competency of self-renewal, metastasis, therapy-resistance, etc., clear quantitative parameters in these vicious behaviors should be established to separate those highly-competent cells from their less-competent counterparts, just like the establishment of the normal ranges for blood pressure, blood sugar, etc. Identification of biomarkers for these cells, as performed by many cancer researchers now, is part of this line of work. Once these quantitative parameters have been set as criteria, these abominable cells can be more easily defined, identified and studied for their behaviors in the quarters of chemotherapy, metastasis, patients' prognosis, etc., without calling them CSCs. For example, it is unnecessary to call “CD44(+)/CD24(-) and ALDH1(+)” breast cancer cells as CSC [843-846], since “CD44(+)/CD24(-) and ALDH1(+)” defines them more specifically and clearly than the equivocal “CSC”. Meanwhile, returning the CSC concept to the cancer-origin hypothesis will be helpful to solve the current snafu in this realm of cancer research.
Mention should be made of some publications which prefer not to provide a pellucid CSC definition, like the ones by Sarkar [847-849], but, instead, prefer to sway between different definitions. In this type of even more baffling definitions, a CSC could be a normal stem cell that eventually evolves to a cancerous one, the benign stage towards a cancerous cell, or the earliest form of a cancerous cell. While some peers, such as Reya et al [850,851], seem to consider both cancer-origin and cancer-maintenance types of cells as CSCs, some others, such as Chaffer and Weinberg [801], sometimes consider that those cancer-maintaining CSCs, but not all cancer cells of the cancer mass, may be derived from tissue-specific normal stem cells in a stepwise manner. These latter cancer pundits elegantly incorporate the hypothesis on cancer-maintenance mechanism into the notion of normal-stem-cell origin of cancer, but it still indirectly suggests that only CSCs, and not the vast majority of cancer cells in the tumor mass, can self-renew, and thus still denies immortality as a sine qua non for a neoplastic state.
In our long-term cogitation on the hypothesis of CSC as “a”, if not “the”, mechanism for cancer maintenance, an intriguing question occurs to us that whether benign tumors also utilize their “benign tumor stem cells (BTSC)” for their maintenance. As implicated in some of the above sections, benign tumor cells are also immortal and thus have acquired self-renewal ability, although they may be too differentiated to be distinguishable from their normal counterparts in cellular morphology, with uterine leiomyoma as an epitome. Is the continuing expansion of a benign tumor mass also ascribable to a tiny subset of BTSCs, but not to the vast majority of the tumor cells? Otherwise, why does a cancer have to be maintained by a tiny subset of cells?
Concluding remarks
Over a century ago, pathologists had set immortality and autonomy as indispensable canons for neoplasms, including the benign ones. It has been known ever since the 1900s that many overgrowths induced in animals are inducer-dependent until very late stages. Unfortunately, indubitable evidence for immortality and autonomy has not been provided for the lesions resulting from most genetically modified animal models of carcinogenesis at the time when the lesions are collected from the animals. Although these lesions have indeed provided us with a profusion of information on the functions and underlying mechanisms of the manipulated genes, especially on the quarters of cell proliferation and death, the lack of this needed evidence still makes us apprehensive, as much confusion on the behaviors of these animal lesions may be caused by their neoplastic inauthenticity. There are five lines of phenomena that dissent from the orthodox doctrine of cancer theory but should not be neglected, i.e. 1) the extrication of one from another among transformation, immortality, and autonomy; 2) the reversibility of in vitro transformation; 3) the irrelevance of mutation to cancer formation and cancer cell heterogeneity in some experimental systems; 4) the inducer-dependency of outgrowths induced in animals; and 5) the insinuation of a poor in vivo transformation efficacy by a low tumor yield in each animal of most model systems after a long latency. Probably, our in vitro and in vivo manipulations cannot mimic most situations of human cancer formation in which mutations are likely required for early establishment of immortality and autonomy. We should, with numerous genetic models created, embark on the following research quests: 1) What is the genomic basis for immortality or autonomy? 2) What is the genomic basis for initiation, promotion, or progression, or for the first, second, or third genetic hit outlined herein? 3) How are immortality and autonomy linked to initiation, promotion, and progression? 4) Which are good animal models for us to use in tackling the above three tasks? Concerns raised in this essay are obviously provocative but deserve reconsideration by cancer researchers, especially those at the pinnacle of cancer research.
Abbreviations
APC: adenomatous polyposis coli; ACI: August strain crossed with Copenhagen strain; CSC: cancer stem cell; DC: Washington DC; DMBA: 7,12-diemthylbenz(a)anthracene; HTLV-1: human T cell lymphotropic virus type I; HP: helicobacter pylori; MMTV: mouse mammary tumor virus long terminal repeat; RB: retinoblastoma protein; NYC: New York City; SASP: senescence-associated secretory phenotype; SD: senescent death; TSH: thyroid stimulating hormone.
Acknowledgements
We would like to thank Dr. Fred Bogott at Austin Medical Center, Mayo Clinic in Austin, Minnesota, USA, for his excellent English editing of this manuscript.
Author Contributions
XD drafted the manuscript. YH prepared the table and figures and participated in discussion. LZ revised the manuscript and performed English editing. QJ, DZ, NX and CW participated in discussions. PT, HH and DJL formulated the concepts. DJL finalized the manuscript.
Consent for publication
All authors agree on the publication.
Funding
This work was supported by grants from the National Natural Science Foundation of China to D. Joshua Liao (grant No. 81660501) and Hai Huang (Grant No. 81460364 and 81760429), as well as by Guizhou Provincial Innovative Talents Team of China to Hai Huang (Grant No. 2019-5610).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Faguet GB. A brief history of cancer: age-old milestones underlying our current knowledge database. Int J Cancer. 2015;136:2022-2036
2. Redmond DE Jr. Tobacco and cancer: the first clinical report, 1761. N Engl J Med. 1970;282:18-23
3. Pott P (1775). Chirurgical observations relative to the cataract, the polypus of the nose, the cancer of the scrotum, the different kinds of ruptures, and the mortification of the toes and feet. Natl Cancer Inst Monogr (Reprinted). 1963;10:7-13
4. Sell S. On the stem cell origin of cancer. Am J Pathol. 2010;176:2584-494
5. Triolo VA. Nineteenth Century Foundations Of Cancer Research Advances In Tumor Pathology, Nomenclature, And Theories Of Oncogenesis. Cancer Res. 1965;25:75-106
6. Haddow A. Neoplastic diseases (cancer). Annu Rev Med. 1955;6:153-186
7. Triolo VA. Nineteenth century foundations of cancer research. Origins of experimental research. Cancer res. 1964;24:4-27
8. No Author. Retrospect: 1889. Br Med J. 1889;2:1433-1471
9. Stewart HL. The cancer investigator. Cancer Res. 1959;19:804-818
10. Schmidt A, Weber OF. In memoriam of Rudolf virchow: a historical retrospective including aspects of inflammation, infection and neoplasia. Contrib Microbiol. 2006;13:1-15
11. Cuddihy J. Rudolf Ludwig Carl Virchow. Cancer Cells. 1991;3:110-112
12. Morton LT. Rudolf Ludwig Carl Virchow (1821-1902): bibliography. J Med Biogr. 1993;1:46-47
13. Wullstein HL, Hellmer L. Rudolf Ludwig Carl Virchow (1821 to 1902). Arch Otolaryngol. 1970;92:299-301
14. Power D. Some effects of chronic irritation upon living tissues, being first steps in a rational study of cancer. Br Med J 893, 2: 830-834.
15. Cardiff RD, Kenney N. Mouse mammary tumor biology: a short history. Adv Cancer Res. 2007;98:53-116
16. Shimkin MB, Triolo VA. History of chemical carcinogenesis: some prospective remarks. Prog Exp Tumor Res. 1969;11:1-20
17. Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157-162
18. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820-823
19. Loeb LA, Harris CC. Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res. 2008;68:6863-6872
20. Turgeon R, Wood HN, Braun AC. Studies on the recovery of crown gall tumor cells. Proc Natl Acad Sci U S A. 1976;73:3562-3564
21. Ashley DJ. The two "hit" and multiple "hit" theories of carcinogenesis. Br J Cancer. 1969;23:313-328
22. Nordling CO. Evidence regarding the multiple mutation theory of the cancer-inducing mechanism. Acta Genet Stat Med. 1955;5:93-104
23. Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68-72
24. Rozhok AI, DeGregori J. The evolution of lifespan and age-dependent cancer risk. Trends Cancer. 2016;2:552-560
25. Nowell P. HD: A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497
26. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85-109
27. Nowell PC. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut. 1962;8:65-66
28. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-28
29. Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. 1954. Int J Epidemiol. 2004;33:1174-1179
30. Armitage P, Doll R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 1957;11:161-169
31. Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1-12
32. Ito N, Hasegawa R, Imaida K, Hirose M, Asamoto M, Shirai T. Concepts in multistage carcinogenesis. Crit Rev Oncol Hematol. 1995;21:105-133
33. Berenblum I, Shubik P. A new, quantitative, approach to the study of the stages of chemical cartinogenesis in the mouse's skin. Br J Cancer. 1947;1:383-391
34. Berenblum I. Cocarcinogenesis. Br Med Bull. 1947;4:343-345
35. Berenblum I, Trainin N. Possible two-stage mechanism in experimental leukemogenesis. Science. 1960;132:40-41
36. Berenblum I. Established principles and unresolved problems in carcinogenesis. J Natl Cancer Inst. 1978;60:723-726
37. Berenblum I. New ideas on the biological stages of carcinogenesis. Acta Unio Int Contra Cancrum. 1954;10:21-24
38. Friedewald WF, Rous P. The Initiating And Promoting Elements In Tumor Production: An Analysis Of The Effects Of Tar, Benzpyrene, And Methylcholanthrene On Rabbit Skin. J Exp Med. 1944;80:101-126
39. Farber E. Carcinogenesis-cellular evolution as a unifying thread: Presidential address. Cancer Res. 1973;33:2537-2550
40. Farber E, Cameron R. The sequential analysis of cancer development. Adv Cancer Res. 1980;31:125-226
41. Farber E. Cancer development and its natural history. A cancer prevention perspective. Cancer. 1988;62:1676-1679
42. Farber E. Hepatocyte proliferation in stepwise development of experimental liver cell cancer. Dig Dis Sci. 1991;36:973-978
43. Pitot HC. Adventures in hepatocarcinogenesis. Annu Rev Pathol. 2007;2:1-29
44. Pitot HC, Hikita H, Dragan Y, Sargent L, Haas M. Review article: the stages of gastrointestinal carcinogenesis-application of rodent models to human disease. Aliment Pharmacol Ther. 2000;14(Suppl 1):153-160
45. Furth J, Tullis JL. Carcinogenesis by radioactive substances. Cancer Res. 1956;16:5-21
46. Fais S. A nonmainstream approach against cancer. J Enzyme Inhib Med Chem. 2016;31:882-889
47. He Y, Yuan C, Chen L, Liu Y, Zhou H, Xu N. et al. While it is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action. Int J Med Sci. 2018;15:309-322
48. Ledoux-Lebard A. Le cancer, maladie parasitaire. Arch Gen Med. 1885;155:423
49. Brosch A. Theoretische und experimentelle untersuchungen zur pathogenesis und histogenesis der malignen geschwulste. Arch Path Anat Physiol. 1900;162:32-84
50. Davis JS. III. The Effect of Scarlet Red, in Various Combinations, upon the Epitheliation of Granulating Surfaces. Ann Surg. 1910;51:40-51
51. Davis JS. X. A Further Note on the Clinical Use of Scarlet Red and its Component, Amidoazotoluol, in Stimulating the Epitheliation of Granulating Surfaces. Ann Surg. 1911;53:702-719
52. vasiliev JM, Cheung AB. Evolution of epithelial proliferation induced by scarlet red in the skin of normal and carcinogen-treated rabbits. Br J Cancer. 1962;16:238-245
53. Fischer B. Die experimentelle erzeugung atypischer epithelwucherungen und die entstehung bosartiger geschwulste. Munch Med Woschschr. 1906;53:2041-2047
54. No Authors Available. Katsusaburo Yamagiwa (1863-1930). CA Cancer J Clin. 1977;27:172-173
55. Hino O. The 90-year anniversary of Katsusaburo Yamagiwa's carcinogenesis. Proceedings of the 1st International Awaji Liver Symposium. December 1, 2005. Awaji Island, Japan. Oncology. 2007;72(Suppl 1):1-140
56. Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of carcinoma. CA Cancer J Clin. 1977;27:174-181
57. Fujiki H. Gist of Dr. Katsusaburo Yamagiwa's papers entitled "Experimental study on the pathogenesis of epithelial tumors" (I to VI reports). Cancer Sci. 2014;105:143-149
58. Rous P, Kidd JG. Conditional Neoplasms And Subthreshold Neoplastic States: A Study Of The Tar Tumors Of Rabbits. J Exp Med. 1941;73:365-390
59. Friedewald WF, Rous P. The pathogenesis of deferred cancer; a study of the after-effects of methylcholanthrene upon rabbit skin. J Exp Med. 1950;91:459-484
60. Haslam SZ, Bern HA. Histopathogenesis of 7,12-diemthylbenz(a)anthracene-induced rat mammary tumors. Proc Natl Acad Sci U S A. 1977;74:4020-4024
61. McGuire WL, Chamness GC, Costlow ME, Shepherd RE. Hormone dependence in breast cancer. Metabolism. 1974;23:75-100
62. Mobbs BG. Uptake of (3H)oestradiol by dimethylbenzanthracene-induced rat mammary tumours regressing spontaneously or after ovariectomy. J Endocrinol. 1969;44:463-464
63. Young S, Cowan DM. Spontaneous regression of induced mammary tumours in rats. Br J Cancer. 1963;17:85-89
64. Huggins C, Briziarelli G, Sutton H Jr. Rapid induction of mammary carcinoma in the rat and the influence of hormones on the tumors. J Exp Med. 1959;109:25-42
65. Mider GB, Morton JJ. Skin tumors following a single application of methylcholanthrene in C57 brown mice. Am J Pathol. 1939;15:299-302
66. Cabot S, Shear N, Shear MJ, Perrault A. Studies in carcinogenesis: XI. Development of skin tumors in mice painted with 3:4-benzpyrene and creosote oil fractions. Am J Pathol. 1940;16:301-312
67. Rusch HP. A theory on carcinogenesis. Med Bull (Ann Arbor). 1956;22:501-516
68. Rusch HP. Carcinogenesis; a facet of living processes. Cancer Res. 1954;14:407-417
69. Rusch HP, Kline BE. Influence of interrupted carcinogenic treatment on tumor formation. Proc Soc Exp Biol Med. 1948;69:90-95
70. Rusch HP, Kline BE. Further evidence for successive stages in the formation of neoplasms. Arch Pathol (Chic ). 1946;42:445-454
71. Rusch HP. Stages in cancer research. Tex Rep Biol Med. 1950;8:207-214
72. Haddow A. Mode of action of chemical carcinogens. Br Med Bull. 1947;4:331-342
73. Beatson GT. On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Trans Med Chir Soc Edinb. 1896;15:153-179
74. Beatson GT. The Treatment of Operable and Inoperable Carcinoma of the Mamma. Glasgow Med J. 1910;74:161-165
75. Beatson GT. The Treatment of Inoperable Carcinoma of the Female Mamma. Glasgow Med J. 1911;76:81-87
76. Lathrop AE, Loeb L. Further investigations on the origin of tumors in mice. III. On the part played by internal secretion in the spontaneous development of tumors. J Cancer Res. 1916;1:1-19
77. Anderson E, Clifton KH, Furth J, Gadsden EL. Autonomous mammotropic pituitary tumors in mice; their somatotropic features and responsiveness to estrogens. Cancer Res. 1956;16:600-607
78. Colletti JA, Leland-Wavrin KM, Kurz SG, Hickman MP, Seiler NL, Samanas NB. et al. Validation of six genetic determinants of susceptibility to estrogen-induced mammary cancer in the rat and assessment of their relevance to breast cancer risk in humans. G3 (Bethesda ). 2014;4:1385-1394
79. Dennison KL, Samanas NB, Harenda QE, Hickman MP, Seiler NL, Ding L. et al. Development and characterization of a novel rat model of estrogen-induced mammary cancer. Endocr Relat Cancer. 2015;22:239-248
80. Dunning WF, Curtis MR, Segaloff A. Strain differences in response to diethylstilbestrol and the induction of mammary gland, adrenal and bladder cancer in the rat. J Mich State Med Soc. 1948;47:305-315
81. Dunning WF, Curtis MR, Madsen ME. Diethylstilbestrol-induced mammary gland and bladder cancer in reciprocal F1 hybrids between two inbred lines of rats. Acta Unio Int Contra Cancrum. 1951;7:238-245
82. Dunning WF, Curtis MR, Segaloff A. Strain differences in response to estrone and the induction of mammary gland, adrenal, and bladder cancer in rats. Cancer Res. 1953;13:147-152
83. Fels E. [Pituitary tumors produced by a synthetic estrogen]. Rev Soc Argent Biol. 1950;26:38-43
84. Gardner WU, Strong LC. Strain-limited Development of Tumors of the Pituitary Gland in Mice Receiving Estrogens. Yale J Biol Med. 1940;12:543-548
85. Gardner WU. Studies on steroid hormones in experimental carcinogenesis. Recent Prog Horm Res. 1947;1:217-259
86. Gardner WU. Steroid hormones in the induction of cancer. Cancer Res. 1947;7:37-52
87. Gardner WU. Studies on ovarian and pituitary tumorigenesis. J Natl Cancer Inst. 1954;15:693-709
88. Richardson FL. Incidence of mammary and pituitary tumors in hybrid mice treated with stilbestrol for varying periods. J Natl Cancer Inst. 1957;18:813-829
89. Sabatino ME, Petiti JP, Sosa L, V, Perez PA, Gutierrez S, Leimgruber C. et al. Evidence of cellular senescence during the development of estrogen-induced pituitary tumors. Endocr Relat Cancer. 2015;22:299-317
90. Twombly GH, Meisel D, Stout AP. Leydig-cell tumors induced experimentally in the rat. Cancer. 1949;2:884-892
91. Zondek B. Hypophyseal tumors induced by estrogenic hormone. Am J Cancer. 1938;33:555-559
92. Zondek B. Oestrogens and tumour genesis. Acta radiol. 1947;28:433-450
93. Ferrigno M, Gardner WU. Unusual neoplastic lesions of the uterine horns of estrogen-treated mice. J Natl Cancer Inst. 1956;17:601-613
94. Schaffer BS, Lachel CM, Pennington KL, Murrin CR, Strecker TE, Tochacek M. et al. Genetic bases of estrogen-induced tumorigenesis in the rat: mapping of loci controlling susceptibility to mammary cancer in a Brown Norway x ACI intercross. Cancer Res. 2006;66:7793-7800
95. Gardner WU. Sensitivity of the vagina to estrogen: genetic and transmitted differences. Ann N Y Acad Sci. 1959;83:145-159
96. Gardner WU. Experimental induction of uterine cervical and vaginal cancer in mice. Cancer Res. 1959;19:170-176
97. Gardner WU. Carcinoma of the uterine cervix and upper vagina: induction under experimental conditions in mice. Ann N Y Acad Sci. 1959;75:543-564
98. Gardner WU, Allen E. Malignant and Non-malignant Uterine and Vaginal Lesions in Mice Receiving Estrogens and Estrogens and Androgens Simultaneously. Yale J Biol Med. 1939;12:213-234
99. Pan SC, Gardner WU. Carcinomas of the uterine cervix and vagina in estrogen- and androgen-treated hybrid mice. Cancer Res. 1948;8:337-345
100. Gardner WU. Hormonal aspects of experimental tumorigenesis. Adv Cancer Res. 1953;1:173-232
101. Gardner WU. Uterine cervical and vaginal cancers in experimental animals. Acta Unio Int Contra Cancrum. 1961;17:905-909
102. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80:175-189
103. Noble RL. The development of prostatic adenocarcinoma in Nb rats following prolonged sex hormone administration. Cancer Res. 1977;37:1929-1933
104. Noble RL. Sex steroids as a cause of adenocarcinoma of the dorsal prostate in Nb rats, and their influence on the growth of transplants. Oncology. 1977;34:138-141
105. Noble RL. Production of Nb rat carcinoma of the dorsal prostate and response of estrogen-dependent transplants to sex hormones and tamoxifen. Cancer Res. 1980;40:3547-3550
106. Noble RL. Prostate carcinoma of the Nb rat in relation to hormones. Int Rev Exp Pathol. 1982;23:113-159
107. van Nie R. Biological aspects of the genesis of uterine tumours induced in mice by testosterone. Jaarb Kankeronderz Kankerbestrijd Ned. 1964;14:247-252
108. van NIE, Benedetti EL, Muhlbock O. A carcinogenic action of testosterone, provoking uterine tumours in mice. Nature. 1961;192:1303
109. Liao DZ, Pantazis CG, Hou X, Li SA. Promotion of estrogen-induced mammary gland carcinogenesis by androgen in the male Noble rat: probable mediation by steroid receptors. Carcinogenesis. 1998;19:2173-2180
110. Xie B, Tsao SW, Wong YC. Induction of high incidence of mammary tumour in female Noble rats with a combination of 17beta-oestradiol and testosterone. Carcinogenesis. 1999;20:1069-1078
111. Xie B, Tsao SW, Wong YC. Sex hormone-induced mammary carcinogenesis in female noble rats: the role of androgens. Carcinogenesis. 1999;20:1597-1606
112. Xie B, Tsao SW, Wong YC. Sex hormone-induced mammary carcinogenesis in female Noble rats: expression of TGF-beta1 and its receptors, TGF-alpha, and EGF-R in mammary carcinogenesis. Breast Cancer Res Treat. 1999;58:227-239
113. Xie B, Tsao SW, Wong YC. Sex hormone-induced mammary carcinogenesis in the female Noble rats: expression of bcl-2 and bax in hormonal mammary carcinogenesis. Breast Cancer Res Treat. 2000;61:45-57
114. Liao DJ, Dickson RB. Steroid hormone-growth factor interactions in proliferative controls of the mammary gland and breast cancer: a rapidly evolving perspective. J Steroid Biochem Mol Biol. 2002;80:135-136
115. Zhang J, Sun Y, Liu Y, Sun Y, Liao DJ. Synergistic effects of androgen and estrogen on the mouse uterus and mammary gland. Oncol Rep. 2004;12:709-716
116. Kirkman H, Bacon RL. Malignant renal tumors in male hamsters (Cricetus auratus) treated with estrogen. Cancer Res. 1950;10:122-124
117. Kirkman H, Bacon RL. Estrogen-induced tumors of the kidney. II. Effect of dose, administration, type of estrogen, and age on the induction of renal tumors in intact male golden hamsters. J Natl Cancer Inst. 1952;13:757-771
118. Kirkman H, Bacon RL. Estrogen-induced tumors of the kidney. I. Incidence of renal tumors in intact and gonadectomized male golden hamsters treated with diethylstilbestrol. J Natl Cancer Inst. 1952;13:745-755
119. Kirkman H, Algard FT. Autonomous variants of an androgen/estrogen-induced and -dependent ductus deferens leiomyosarcoma of the Syrian hamster. Cancer Res. 1970;30:35-40
120. Liao DZ, Hou X, Bai S, Li SA, Li JJ. Unusual deregulation of cell cycle components in early and frank estrogen-induced renal neoplasias in the Syrian hamster. Carcinogenesis. 2000;21:2167-2173
121. Matthews VS, Kirkman H, BACON RL. Kidney damage in the golden hamster following chronic administration of diethylstilbestrol and sesame oil. Proc Soc Exp Biol Med. 1947;66:195
122. Kirkman H, Algard FT. Characteristics of an androgen-estrogen-induced uterine smooth muscle cell tumor of the Syrian hamster. Cancer Res. 1970;30:794-800
123. Kirkman H. Hormone-related tumors in Syrian hamsters. Prog Exp Tumor Res. 1972;16:201-240
124. Furth J. Transplantability of induced granulosa cell tumors and of luteoma in mice; secondary effects of these growths. Proc Soc Exp Biol Med. 1946;61:212-214
125. Li MH, Gardner WU. Further studies on the pathogenesis of ovarian tumors in mice. Cancer Res. 1949;9:35-41
126. Peckham BM, Greene RR. Experimentally produced granulosa-cell tumors in rats. Cancer Res. 1952;12:25-29
127. Peckham BM, Greene RR. Experimentally produced granulosa-cell tumors in rabbits. Cancer Res. 1952;12:654-656
128. Biskind GR, Biskind MS. Experimental ovarian tumors in rats. Am J Clin Pathol. 1949;19:501-521
129. Burrows H. Carcinoma mammae occurring in a male mouse under continued treatment with oestrin. Am J Cancer. 1935;24:613-616
130. Cutts JH. Estrogen-induced breast cancer in the rat. Proc Can Cancer Conf. 1966;6:50-68
131. Cutts JH, Froude GC. Regression of estrone-induced mammary tumors in the rat. Cancer Res. 1968;28:2413-2418
132. Cutts JH. Unusual response to androgen of estrogen-dependent mammary tumors. J Natl Cancer Inst. 1969;42:485-488
133. Cutts JH. Enzyme activities in regressing estrone-induced mammary tumors of the rat. Cancer Res. 1973;33:1235-1237
134. Geschickter CF, Lewis D, Hartman CG. Tumors of the breast related to the oestrin hormone. Am J Cancer. 1934;21:828-859
135. Huggins C. Endocrine-induced regression of cancers. Cancer Res. 1967;27:1925-1930
136. Huggins C. Endocrine-induced regression of cancers. Am J Surg. 1978;136:233-238
137. Mceuen CS. Occurrence of cancer in rats treated with oestrone. Am J Cancer. 1938;34:184-195
138. Noble RL, Cutts JH. Mammary tumors of the rat: a review. Cancer Res. 1959;19:1125-1139
139. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57-67
140. Noble RL, Hochachka BC, King D. Spontaneous and estrogen-produced tumors in Nb rats and their behavior after transplantation. Cancer Res. 1975;35:766-780
141. Gardner WU. Steroids in experimental carcinogenesis. Cancer. 1957;10:726-730
142. Andervont HB, Canter HY, Shimkin MB. Effect of discontinued estrogenic stimulation upon the development and growth of testicular tumors in mice. J Natl Cancer Inst. 1957;18:1-39
143. Andervont HB, Shimkin MB, Canter HY. Some factors involved in the induction or growth of testicular tumors in BALB/c mice. J Natl Cancer Inst. 1960;25:1083-1096
144. Andervont HB, Shimkin MB, Canter HY. The growth of estrogen-induced interstitial-cell testicular tumors in BALB/c mice. J Natl Cancer Inst. 1960;24:1219-1237
145. Andervont HB, Shimkin MB, Canter HY. Testicular tumors in mice after removal of stilbestrol-cholesterol pellets. Acta Unio Int Contra Cancrum. 1961;17:105-112
146. Furth J. Discussion of problems related to hormonal factors in initiating and maintaining tumor growth. Cancer Res. 1957;17:454-463
147. Gardner WU. Hormonal imbalances in tumorigenesis. Cancer Res. 1948;8:397-411
148. Dunning WF. Response of some isologously transplanted rat neoplasms to steroids. Ann N Y Acad Sci. 1958;76:696-704
149. Huggins C. Prostatic cancer treated by orchiectomy; the five year results. J Am Med Assoc. 1946;131:576-581
150. Huggins C, BERGENSTAL DM. Inhibition of human mammary and prostatic cancers by adrenalectomy. Cancer Res. 1952;12:134-141
151. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;168:9-12
152. Angevine DM. Significant events in the life of Jacob Furth. Cancer Res. 1966;26:351-356
153. Weinhouse S, Furth JJ. Jacob Furth - September 20, 1896-July 23, 1979. Biogr Mem Natl Acad Sci. 1992;62:167-197
154. Furth J. The making and missing of discoveries: an autobiographical essay. Cancer Res. 1976;36:871-880
155. Furth J. Conditioned and autonomous neoplasms: a review. Cancer Res. 1953;13:477-492
156. Bloom HJ, Baker WH, Dukes CE, Mitchley BC. Hormone-dependent tumours of the kidney. II. Effect of endocrine ablation procedures on the transplanted oestrogen-induced renal tumour of the Syrian hamster. Br J Cancer. 1963;17:646-656
157. Bloom HJ, Dukes CE, Mitchley BC. Hormone-dependent tumours of the kidney. I. The oestrogen-induced renal tumour of the Syrian hamster. Hormone treatment and possible relationship to carcinoma of the kidney in man. Br J Cancer. 1963;17:611-645
158. Kirkman H. Steroid tumorigenesis. Cancer. 1957;10:757-764
159. Kirkman H. Autonomous derivatives of estrogen-induced renal carcinomas and spontaneous renal tumors in the Syrian hamster. Cancer Res. 1974;34:2728-2744
160. Kirkman H, Chesterman FC. Additional data on transplanted tumours of the golden hamster. Prog Exp Tumor Res. 1972;16:580-621
161. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321:288-300
162. Sharma D, Kumar S, Narasimhan B. Estrogen alpha receptor antagonists for the treatment of breast cancer: a review. Chem Cent J. 2018;12:107
163. Peddi PF. Hormone receptor positive breast cancer: state of the art. Curr Opin Obstet Gynecol. 2018;30:51-54
164. Upton AC, Furth J. Induction of pituitary tumors by means of ionizing irradiation. Proc Soc Exp Biol Med. 1953;84:255-257
165. Gorbman A. Tumorous growths in the pituitary and trachea following radiotoxic dosages of I131. Proc Soc Exp Biol Med. 1949;71:237-240
166. Gorbman A. Pituitary tumors in rodents following changes in thyroid function: a review. Cancer Res. 1956;16:99-105
167. Gorbman A. Factors influencing development of hypophyseal tumors in mice after treatment with radioactive iodine. Proc Soc Exp Biol Med. 1952;80:538-540
168. Furth J, Clifton KH. Experimental pituitary tumors and the role of pituitary hormones in tumorigenesis of the breast and thyroid. Cancer. 1957;10:842-853
169. Dent JN, Gadsden EL, Furth J. On the relation between thyroid depression and pituitary tumor induction in mice. Cancer Res. 1955;15:70-75
170. Dent JN, Gadsden EL, Furth J. Further studies on induction and growth of thyrotropic pituitary tumors in mice. Cancer Res. 1956;16:171-174
171. Furth J, Burnett WT Jr. Hormone-secreting transplantable neoplasms of the pituitary induced by I131. Proc Soc Exp Biol Med. 1951;78:222-224
172. Furth J, Gadsden EL, Burnett WT Jr. Autonomous transplantable pituitary tumors arising in growths dependent on absence of the thyroid gland. Proc Soc Exp Biol Med. 1952;80:4-7
173. Furth J, Burnett WT Jr, Gadsden EL. Quantitative relationship between thyroid function and growth of pituitary tumors secreting TSH. Cancer Res. 1953;13:298-307
174. Bielschowsky F. Chronic iodine deficiency as cause of neoplasia in thyroid and pituitary of aged rats. Br J Cancer. 1953;7:203-213
175. Bielschowsky F, HALL WH. Carcinogenesis in the thyroidectomized rat. Br J Cancer. 1953;7:358-366
176. Bielschowsky F. Functional acidophilic tumours of the pituitary of the rat. Br J Cancer. 1954;8:154-160
177. Bielschowsky F. Neoplasia and internal environment. Br J Cancer. 1955;9:80-116
178. Bielschowsky F, Horning ES. Aspects of endocrine carcinogenesis. Br Med Bull. 1958;14:106-115
179. Furth J, Dent JN, Burnett WT Jr, Gadsden EL. The mechanism of induction and the characteristics of pituitary tumors induced by thyroidectomy. J Clin Endocrinol Metab. 1955;15:81-97
180. Halmi NS, Gude WD. The morphogenesis of pituitary tumors induced by radiothyroidectomy in the mouse and the effects of their transplantation on the pituitary body of the host. Am J Pathol. 1954;30:403-419
181. Bielschowsky F. The role of thyroxine deficiency in the formation of experimental tumours of the thyroid. Br J Cancer. 1949;3:547-9
182. Bielschowsky F, Griesbach WE. Studies on experimental goitre; the transplant-ability of experimental thyroid tumours of the rat. Br J Cancer. 1949;3:541-6
183. Bielschowsky F. Hormonal factors in neoplasia of the thyroid. Acta Unio Int Contra Cancrum. 1960;16:133-137
184. Money WL, Godwin JT, Rawson RW. The experimental production of thyroid tumors in the rat by the administration of sodium-5-iodo-2-thiouracil. Cancer. 1957;10:690-697
185. Morris HP, Dalton AJ, Green CD. Malignant thyroid tumors occurring in the mouse after prolonged hormonal imbalance during the ingestion of thiouracil. J Clin Endocrinol Metab. 1951;11:1281-1295
186. Morris HP, Green CD. The role of thiouracil in the induction, growth, and transplantability of mouse thyroid tumors. Science. 1951;114:44-46
187. Morris HP. Experimental thyroid tumors. Brookhaven Symp Biol. 1955;7:192-218
188. Purves HD, Griesbach WE. Studies on experimental goitre; thyroid carcinomata in rats treated with thiourea. Br J Exp Pathol. 1946;27:294-297
189. Purves HD, Griesbach WE. Studies on experimental goitre; thyroid tumours in rats treated with thiourea. Br J Exp Pathol. 1947;28:46-53
190. Dalton AJ, Morris HP, Striebich MJ, Dubnik CS. Histologic changes in strain C mice following long-term ingestion of thiouracil. J Natl Cancer Inst. 1950;11:391-413
191. Dalton AJ, Morris HP, Dubnik C. Change in the thyroid and other organs in mice receiving thiouracil. Fed Proc. 1946;5:219-230
192. Morris HP, Green CD, Dalton AJ. The effect of ingestion of thiouracil on strain C mice. J Natl Cancer Inst. 1951;11:805-815
193. Goldberg RC, Lindsay S, Nichols CW Jr, Chaikoff IL. Induction of neoplasms in thyroid glands of rats by subtotal thyroidectomy and by the injection of one microcurie of I-131. Cancer Res. 1964;24:35-43
194. Goldberg RC, Chaikoff IL. Induction of thyroid cancer in the rat by radioactive iodine. AMA Arch Pathol. 1952;53:22-28
195. Goldberg RC, Chaikoff IL. Development of thyroid neoplasms in the rat following a single injection of radioactive iodine. Proc Soc Exp Biol Med. 1951;76:563-566
196. Furth J. Thyroid-pituitary tumorigenesis. J Natl Cancer Inst. 1954;15:687-691
197. Murray KN, Wolf JC, Spagnoli ST, Lains D, Budrow N, Kent ML. Reversibility of Proliferative Thyroid Lesions Induced by Iodine Deficiency in a Laboratory Zebrafish Colony. Zebrafish. 2018;15:558-565
198. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143-164
199. Liao JD, Adsay NV, Khannani F, Grignon D, Thakur A, Sarkar FH. Histological complexities of pancreatic lesions from transgenic mouse models are consistent with biological and morphological heterogeneity of human pancreatic cancer. Histol Histopathol. 2007;22:661-676
200. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13:1677-1693
201. Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11:615-626
202. Liao DJ, Natarajan G, Deming SL, Jamerson MH, Johnson M, Chepko G. et al. Cell cycle basis for the onset and progression of c-Myc-induced, TGFalpha-enhanced mouse mammary gland carcinogenesis. Oncogene. 2000;19:1307-1317
203. Liao DJ, Dickson RB. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab Invest. 2003;83:1437-1449
204. Liao DJ, Wang Y, Wu J, Adsay NV, Grignon D, Khanani F. et al. Characterization of pancreatic lesions from MT-tgfalpha, Ela-myc and MT-tgfalpha/Ela-myc single and double transgenic mice. J Carcinog. 2006;5:DOI 10.1186/1477-3163-5-19
205. Ma Y, Jia Y, Chen L, Ezeogu L, Yu B, Xu N. et al. Weaknesses and Pitfalls of Using Mice and Rats in Cancer Chemoprevention Studies. J Cancer. 2015;6:1058-1065
206. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313-317
207. D'Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE. et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235-239
208. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199-207
209. Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ. et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249-3262
210. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S. et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112-1117
211. Tilli MT, Furth PA. Conditional mouse models demonstrate oncogene-dependent differences in tumor maintenance and recurrence. Breast Cancer Res. 2003;5:202-205
212. Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J. et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One. 2008;3:e2125-doi 10.1371/journal.pone.0002125
213. Shachaf CM, Felsher DW. Rehabilitation of cancer through oncogene inactivation. Trends Mol Med. 2005;11:316-321
214. Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O. et al. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 2008;68:5132-5142
215. Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577-586
216. Felsher DW. Tumor dormancy: death and resurrection of cancer as seen through transgenic mouse models. Cell Cycle. 2006;5:1808-1811
217. Felsher DW. Tumor dormancy and oncogene addiction. APMIS. 2008;116:629-637
218. Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer. 2010;1:597-604
219. Hengstler JG, Bockamp EO, Hermes M, Brulport M, Bauer A, Schormann W. et al. Oncogene-blocking therapies: new insights from conditional mouse tumor models. Curr Cancer Drug Targets. 2006;6:603-612
220. Martin-Lorenzo A, Gonzalez-Herrero I, Rodriguez-Hernandez G, Garcia-Ramirez I, Vicente-Duenas C, Sanchez-Garcia I. Early epigenetic cancer decisions. Biol Chem. 2014;395:1315-1320
221. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-846
222. Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N. et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468-472
223. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57-60
224. Nguyen AT, Emelyanov A, Koh CH, Spitsbergen JM, Parinov S, Gong Z. An inducible kras(V12) transgenic zebrafish model for liver tumorigenesis and chemical drug screening. Dis Model Mech. 2012;5:63-72
225. Uchiyama K, Watanabe D, Hayasaka M, Hanaoka K. A novel imprinted transgene located near a repetitive element that exhibits allelic imbalance in DNA methylation during early development. Dev Growth Differ. 2014;56:653-668
226. Zheng W, Li Z, Nguyen AT, Li C, Emelyanov A, Gong Z. Xmrk, kras and myc transgenic zebrafish liver cancer models share molecular signatures with subsets of human hepatocellular carcinoma. PLoS One. 2014;9:e91179-doi 10.1371/journal.pone.0091179
227. Sun L, Nguyen AT, Spitsbergen JM, Gong Z. Myc-induced liver tumors in transgenic zebrafish can regress in tp53 null mutation. PLoS One. 2015;10:e0117249
228. Anders K, Kershaw O, Larue L, Gruber AD, Blankenstein T. The immune system prevents recurrence of transplanted but not autochthonous antigenic tumors after oncogene inactivation therapy. Int J Cancer. 2017;141:2551-2561
229. Dolezal JM, Wang H, Kulkarni S, Jackson L, Lu J, Ranganathan S. et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J Biol Chem. 2017;292:10068-10086
230. Li H, Lu JW, Huo X, Li Y, Li Z, Gong Z. Effects of sex hormones on liver tumor progression and regression in Myc/xmrk double oncogene transgenic zebrafish. Gen Comp Endocrinol. 2019;277:112-121
231. Tonelli C, Morelli MJ, Sabo A, Verrecchia A, Rotta L, Capra T. et al. Genome-wide analysis of p53-regulated transcription in Myc-driven lymphomas. Oncogene. 2017;36:2921-2929
232. Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323-1334
233. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L. et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661-665
234. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-660
235. Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T. et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893-904
236. Weinstein IB. Cancer. Addiction to oncogenes-the Achilles heal of cancer. Science. 2002;297:63-64
237. Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction-a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3:448-457
238. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077-3080
239. Shachaf CM, Felsher DW. Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res. 2005;65:4471-4474
240. Challis GB, Stam HJ. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol. 1990;29:545-550
241. Everson TC. SPONTANEOUS REGRESSION OF CANCER. Ann N Y Acad Sci. 1964;114:721-735
242. Everson TC. Spontaneous regression of cancer. Prog Clin Cancer. 1967;3:79-95
243. Kleef R, Jonas WB, Knogler W, Stenzinger W. Fever, cancer incidence and spontaneous remissions. Neuroimmunomodulation. 2001;9:55-64
244. Stephenson HE Jr. Spontaneous regression of cancer evaluated by computerized data. Natl Cancer Inst Monogr. 1976;44:43-47
245. Thomas JA, Badini M. The role of innate immunity in spontaneous regression of cancer. Indian J Cancer. 2011;48:246-251
246. Bodey B, Bodey B Jr, Siegel SE, Kaiser HE. The spontaneous regression of neoplasms in mammals: possible mechanisms and their application in immunotherapy. In vivo. 1998;12:107-122
247. Kaiser HE, Bodey B Jr, Siegel SE, Groger AM, Bodey B. Spontaneous neoplastic regression: the significance of apoptosis. In vivo. 2000;14:773-788
248. Nagorsen D, Marincola FM, Kaiser HE. Bacteria-related spontaneous and therapeutic remission of human malignancies. In vivo. 2002;16:551-556
249. Jessy T. Immunity over inability: The spontaneous regression of cancer. J Nat Sci Biol Med. 2011;2:43-49
250. Ahmadi MP, Cornejo KM, Hutchinson L, Tomaszewicz K, Dresser K, Deng A. et al. Complete Spontaneous Regression of Merkel Cell Carcinoma After Biopsy: A Case Report and Review of the Literature. Am J Dermatopathol. 2016;38:e154-e158
251. Brodeur GM. Spontaneous regression of neuroblastoma. Cell Tissue Res. 2018 doi: 10.1007/s00441-017-2761-2
252. Buhler H, Pirovino M, Akobiantz A, Altorfer J, Weitzel M, Maranta E. et al. Regression of liver cell adenoma. A follow-up study of three consecutive patients after discontinuation of oral contraceptive use. Gastroenterology. 1982;82:775-782
253. Sakamaki A, Kamimura K, Abe S, Tsuchiya A, Takamura M, Kawai H. et al. Spontaneous regression of hepatocellular carcinoma: A mini-review. World J Gastroenterol. 2017;23:3797-3804
254. Bloom HJ, Richardson WW, Harries EJ. Natural history of untreated breast cancer (1805-1933). Comparison of untreated and treated cases according to histological grade of malignancy. Br Med J. 1962;2:213-221
255. Beller U, Beckman EM, Twombly GH. Spontaneous regression of advanced endometrial carcinoma. Gynecol Oncol. 1984;17:381-385
256. Hobohm U. Fever and cancer in perspective. Cancer Immunol Immunother. 2001;50:391-396
257. Jerry LM, Challis EB. Oncology. In Textbook of family practice. 3rd edn. edition. Edited by Rakel, RE. 1952:1061-1081
258. Chang WY. Complete spontaneous regression of cancer: four case reports, review of literature, and discussion of possible mechanisms involved. Hawaii Med J. 2000;59:379-387
259. Smith JL Jr, Stehlin JS Jr. Spontaneous regression of primary malignant melanomas with regional metastases. Cancer. 1965;18:1399-1415
260. Ribero S, Gualano MR, Osella-Abate S, Scaioli G, Bert F, Sanlorenzo M. et al. Association of Histologic Regression in Primary Melanoma With Sentinel Lymph Node Status: A Systematic Review and Meta-analysis. JAMA Dermatol. 2015;151:1301-1307
261. Pique-Duran E, Palacios-Llopis S, Martinez-Martin M, Perez-Cejudo JA. Complete regression of melanoma associated with vitiligo. Dermatol Online J. 2011;17:4-https //escholarship.org/uc/item/7sn7h2j7#main
262. Ong SF, Harden M, Irandoust S, Lee RW. Spontaneous regression of pulmonary metastatic melanoma. Respirol Case Rep. 2016;4:7-9
263. Khosravi H, Akabane AL, Alloo A, Nazarian RM, Boland GM. Metastatic melanoma with spontaneous complete regression of a thick primary lesion. JAAD Case Rep. 2016;2:439-441
264. Kang S, Barnhill RL, Mihm MC Jr, Sober AJ. Histologic regression in malignant melanoma: an interobserver concordance study. J Cutan Pathol. 1993;20:126-129
265. Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181
266. Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res. 2009;19:275-282
267. Cervinkova M, Kucerova P, Cizkova J. Spontaneous regression of malignant melanoma - is it based on the interplay between host immune system and melanoma antigens? Anticancer Drugs. 2017;28:819-830
268. Ribero S, Moscarella E, Ferrara G, Piana S, Argenziano G, Longo C. Regression in cutaneous melanoma: a comprehensive review from diagnosis to prognosis. J Eur Acad Dermatol Venereol. 2016;30:2030-2037
269. Drobyski WR, Qazi R. Spontaneous regression in non-Hodgkin's lymphoma: clinical and pathogenetic considerations. Am J Hematol. 1989;31:138-141
270. Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol. 2014;11:704-713
271. Fisher JPH, Tweddle DA. Neonatal neuroblastoma. Semin Fetal Neonatal Med. 2012;17:207-215
272. Cole WH, Everson TC. Spontaneous regression of cancer: preliminary report. Ann Surg. 1956;144:366-383
273. Cole WH. Spontaneous regression of cancer and the importance of finding its cause. Natl Cancer Inst Monogr. 1976;44:5-9
274. Cole WH. Efforts to explain spontaneous regression of cancer. J Surg Oncol. 1981;17:201-209
275. Everson TC, Cole WH. Spontaneous regression of malignant disease. J Am Med Assoc. 1959;169:1758-1759
276. Strong LC. Studies of the regression of spontaneous adenocarcinomata in mice. Z Krebsforsch. 1969;72:32-35
277. Asashima M, Komazaki S, Satou C, Oinuma T. Seasonal and geographical changes of spontaneous skin papillomas in the Japanese newt Cynops pyrrhogaster. Cancer Res. 1982;42:3741-3746
278. Asashima M, Oinuma T, Matsuyama H, Nagano M. Effects of temperature on papilloma growth in the newt, Cynops pyrrhogaster. Cancer Res. 1985;45:1198-1205
279. Asashima M, Koyama H, Shimada K, Pfeiffer CJ. Temperature-induced alterations in protein composition of newt papilloma cells. Cell Mol Biol. 1989;35:669-677
280. Getchell RG, Gasey JW, Bowser PR. Seasonal occurence of virally induced skin tumors in wild fish. J Aquat Anim Health. 1998;10:191-201
281. Holzschu D, Lapierre LA, Lairmore MD. Comparative pathogenesis of epsilonretroviruses. J Virol. 2003;77:12385-12391
282. Peters G, Peters N. Temperature-dependent growth and regression of epidermal tumors in the european eel (Anguilla anguilla L.). Ann N Y Acad Sci. 1978;298:245-260
283. Pfeiffer CJ, Nagai T, Fujimura M, Tobe T. Spontaneous regressive epitheliomas in the Japanese newt, Cynops pyrrhogaster. Cancer Res. 1979;39:1904-1910
284. Wojciechowska S, Zeng Z, Lister JA, Ceol CJ, Patton EE. Melanoma Regression and Recurrence in Zebrafish. Methods Mol Biol. 2016;1451:143-153
285. Seppanen ED, McKinnell RG, Tarin D, Rollins-Smith LA, Hanson W. Temperature-dependent dissociation of Lucke renal adenocarcinoma cells. Differentiation. 1984;26:227-230
286. Riker AJ. Studies on the influence of some environmental factors on the development of crown gall. J Agric Res. 1926;32:83-96
287. Braun AC. Thermal studies on the factors responsible for tumor initiation in crown gall. Am J Bot. 1947;34:234-240
288. Riker AJ. Plant tumors: Introduction. Proc Natl Acad Sci U S A. 1958;44:338-339
289. Riker AJ, Hildebrandt AC. Pathological plant growth. Annu Rev Microbiol. 1951;5:223-240
290. Riker AJ, Lyneis MM, Locke SB. Comparative physiology of crown gall, attenuated crown gall, radiobacter, and hairy root bacteria. Phytopathology. 1941;31:964-977
291. Adami JG. An Address on the Causation of Cancerous and other New Growths: Delivered before the Yale University Medical Alumni Association, New Haven, Conn. Br Med J. 1901;1:621-628
292. Brand RA. Biographical sketch: James Stephen Ewing, MD (1844-1943). Clin Orthop Relat Res. 2012;470:639-641
293. Knauss S, Klein A. From aneuploidy to cancer: the evolution of a new species? J Biosci. 2012;37:211-220
294. Huxley JS. Cancer biology: Comparative and genetic. Biol Rev. 1956;31:474-513
295. Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283-350
296. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1:407-422
297. Duesberg P, Mandrioli D, McCormack A, Nicholson JM. Is carcinogenesis a form of speciation? Cell Cycle. 2011;10:2100-2114
298. Salmina K, Huna A, Kalejs M, Pjanova D, Scherthan H, Cragg MS. et al. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes (Basel). 2019:10
299. Davies PC, Lineweaver CH. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys Biol. 2011;8:015001-doi 10.1088/1478-3975/8/1/015001
300. Erenpreisa J, Salmina K, Huna A, Jackson TR, Vazquez-Martin A, Cragg MS. The "virgin birth", polyploidy, and the origin of cancer. Oncoscience. 2015;2:3-14
301. Green H, Todaro GJ. The mammalian cell as differentiated microorganism. Annu Rev Microbiol. 1967;21:573-600
302. Liu X, Yang W, Guan Z, Yu W, Fan B, Xu N. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell Biosci. 2018;8:6-doi 10.1186/s13578-018-0206-6
303. Shi M, Zhou H, Lei M, Chen L, Zellmer L, He Y. et al. Spontaneous Cancers, But Not Many Induced Ones in Animals, Resemble Semi-New Organisms that Possess a Unique Programmed Cell Death Mode Different from Apoptosis, Senescent Death, Necrosis and Stress-Induced Cell Death. J Cancer. 2018;9:4726-4735
304. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101
305. Greene HS. A conception of tumor autonomy based on transplantation studies: a review. Cancer Res. 1951;11:899-903
306. Farber E. Origins of human cancers. Toxicol Pathol. 1985;13:86-89
307. Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006;28:515-524
308. Tomlinson IP, Bodmer WF. Modelling the consequences of interactions between tumour cells. Br J Cancer. 1997;75:157-160
309. Weber RJ, Desai TA, Gartner ZJ. Non-autonomous cell proliferation in the mammary gland and cancer. Curr Opin Cell Biol. 2017;45:55-61
310. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M. et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016;8:327ra26
311. Subramanian S. Quantifying harmful mutations in human populations. Eur J Hum Genet. 2012;20:1320-1322
312. Subramanian S. Europeans have a higher proportion of highfrequency deleterious variants than Africans. Hum Genet. 2016;135:1-7
313. Rubin H. Cell damage, aging and transformation: a multilevel analysis of carcinogenesis. Anticancer Res. 1999;19:4877-4886
314. Chow M, Rubin H. Relation of the slow growth phenotype to neoplastic transformation: possible significance for human cancer. In vitro Cell Dev Biol Anim. 1999;35:449-458
315. Teotonio H, Rose MR. Perspective: reverse evolution. Evolution. 2001;55:653-660
316. Teotonio H, Chelo IM, Bradic M, Rose MR, Long AD. Experimental evolution reveals natural selection on standing genetic variation. Nat Genet. 2009;41:251-257
317. Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40:721-728
318. Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140-148
319. Pasmooij AM, Jonkman MF, Uitto J. Revertant mosaicism in heritable skin diseases: mechanisms of natural gene therapy. Discov Med. 2012;14:167-179
320. van DE, Pretorius PJ. Point mutation instability (PIN) mutator phenotype as model for true back mutations seen in hereditary tyrosinemia type 1 - a hypothesis. J Inherit Metab Dis. 2012;35:407-411
321. Ashworth A. Drug resistance caused by reversion mutation. Cancer Res. 2008;68:10021-10023
322. Bouwman P, Jonkers J. Molecular Pathways: How Can BRCA-Mutated Tumors Become Resistant to PARP Inhibitors? Clin Cancer Res. 2014;20:540-547
323. Dhillon KK, Swisher EM, Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011;102:663-669
324. Baum JK, Bookstein JJ, Holtz F, Klein EW. Possible association between benign hepatomas and oral contraceptives. Lancet. 1973;2:926-929
325. Horvath E, Kovacs K, Ross RC. Letter: Benign hepatoma in a young woman on contraceptive steroids. Lancet. 1974;1:357-358
326. Knapp WA, Ruebner BH. Letter: Hepatomas and oral contraceptives. Lancet. 1974;1:270-271
327. Lingeman CH. Letter: Liver-cell neoplasms and oral contraceptives. Lancet. 1974;1:64
328. No Authors Available. Oral contraceptives and cancer. Lancet. 1972;2:911-DOI https://doi.org/10.1016/S0140-6736(72)92541-X
329. Thalassinos NC, Lymberatos C, Hadjioannou J, Gardikas C. Letter: Liver-cell carcinoma after long-term oestrogen-like drugs. Lancet. 1974;1:270
330. Wendel HA. Oral contraceptives and cancer. Lancet. 1972;2:1139
331. AGNEW LR, GARDNER WU. The incidence of spontaneous hepatomas in C3H, C3H (low milk factor), and CBA mice and the effect of estrogen and androgen on the occurrence of these tumors in C3H mice. Cancer Res. 1952;12:757-761
332. Pullinger BD, Head MA. Hepatoma in intact c3hf male and virgin female mice and after gonadectomy alone or sebsequent treatment with oestrogen. Br J Cancer. 1964;13:521-527
333. Andervont HB, Dunn TB. Transplantation of spontaneous and induced hepatomas in inbred mice. J Natl Cancer Inst. 1952;13:455-503
334. Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de BM. et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342:575-577
335. Wundisch T, Thiede C, Morgner A, Dempfle A, Gunther A, Liu H. et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018-8024
336. Morgner A, Thiede C, Bayerdorffer E, Alpen B, Wundisch T, Neubauer A. et al. Long-term follow-up of gastric MALT lymphoma after H. pylori eradication. Curr Gastroenterol Rep. 2001;3:516-522
337. Morgner A, Miehlke S, Fischbach W, Schmitt W, Muller-Hermelink H, Greiner A. et al. Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol. 2001;19:2041-2048
338. Wundisch T, Dieckhoff P, Greene B, Thiede C, Wilhelm C, Stolte M. et al. Second cancers and residual disease in patients treated for gastric mucosa-associated lymphoid tissue lymphoma by Helicobacter pylori eradication and followed for 10 years. Gastroenterology. 2012;143:936-942
339. Bertoni F, Conconi A, Capella C, Motta T, Giardini R, Ponzoni M. et al. Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: early analysis of the LY03 cooperative trial. Blood. 2002;99:2541-2544
340. Park JB, Koo JS. Helicobacter pylori infection in gastric mucosa-associated lymphoid tissue lymphoma. World J Gastroenterol. 2014;20:2751-2759
341. El HH, El-Sabban M, Hasegawa H, Zaatari G, Ablain J, Saab ST. et al. Therapy-induced selective loss of leukemia-initiating activity in murine adult T cell leukemia. J Exp Med. 2010;207:2785-2792
342. Gill PS, Harrington W Jr, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA. et al. Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med. 1995;332:1744-1748
343. Hermine O, Bouscary D, Gessain A, Turlure P, Leblond V, Franck N. et al. Brief report: treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332:1749-1751
344. Axelrod R. Launching "the evolution of cooperation". J Theor Biol. 2012;299:21-24
345. Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A. 2006;103:13474-13479
346. Bruns P. Die Heilwirkung des Erysipels auf Geschwulste. Beiträge zur Klinischen Chirurgie. 1887;3:443-446
347. Busch W. Uber den Einfluss welche heftigere Erysipeln zuweilig auf organisierte Neubildungenausuben. Verhandlungen des Naturhistorischen Vereines der Preussischen Rheinlande und Westphalens. 1866;23:28-30
348. Busch W. Aus der sitzung der medicinischen section. Berliner Klinische Wochenschrift. 1868;5:137-138
349. Fehleisen F. Ueber die Züchtung der Erysipelkokken auf künstlichem Nährboden und ihre Übertragbarkeit auf den Menschen. Dtsch Med Wochenschr. 1882;8:553-554
350. Hobohm U. Fever therapy revisited. Br J Cancer. 2005;92:421-425
351. Hobohm U. Healing heat: Harnessing infection to flight cancer. Am Sci. 2009;97:34-41
352. Hobohm U. Toward general prophylactic cancer vaccination. Bioessays. 2009;31:1071-1079
353. Hoption Cann SA, van Netten JP, van NC. Dr William Coley and tumour regression: a place in history or in the future. Postgrad Med J. 2003;79:672-680
354. Hoption Cann SA, van Netten JP, van NC. Acute infections as a means of cancer prevention: opposing effects to chronic infections? Cancer Detect Prev. 2006;30:83-93
355. Hoption Cann SA. Peak fever: helpful or harmful? Heart Lung. 2011;40:585-586
356. Kienle GS. Fever in Cancer Treatment: Coley's Therapy and Epidemiologic Observations. Glob Adv Health Med. 2012;1:92-100
357. Nauts HC, FOWLER GA, BOGATKO FH. A review of the influence of bacterial infection and of bacterial products (Coley's toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley's mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med Scand Suppl. 1953;276:1-103
358. Nauts HC. The beneficial effects of bacterial infections on host resistance to cancer. End results in 449 cases. A study and abstracts of reports in the world medical literature (1775-1980) and personal communication. Cancer Research Institute Monograph. 1980;8:1-225
359. Nauts HC. Bacterial pyrogens: beneficial effects on cancer patients. Prog Clin Biol Res. 1982;107:687-696
360. Nauts HC. Bacteria and cancer-antagonisms and benefits. Cancer Surv. 1989;8:713-723
361. Nauts HC, McLaren JR. Coley toxins-the first century. Adv Exp Med Biol. 1990;267:483-500
362. Repasky EA, Evans SS, Dewhirst MW. Temperature Matters! And Why it Should Matter to Tumor Immunologists. Cancer Immunol Res. 2013;1:210-216
363. Tang ZY, Zhou HY, Zhao G, Chai LM, Zhou M, Lu JZ. et al. Preliminary result of mixed bacterial vaccine as adjuvant treatment of hepatocellular carcinoma. Med Oncol Tumor Pharmacother. 1991;8:23-28
364. Wiemann B, Starnes CO. Coley's toxins, tumor necrosis factor and cancer research: a historical perspective. Pharmacol Ther. 1994;64:529-564
365. Orange M, Reuter U, Hobohm U. Coley's Lessons Remembered: Augmenting Mistletoe Therapy. Integr Cancer Ther. 2016;15:502-511
366. Kucerova P, Cervinkova M. Spontaneous regression of tumour and the role of microbial infection - possibilities for cancer treatment. Anticancer Drugs. 2016
367. Bodey B, Bodey B Jr, Siegel SE, Kaiser HE. The spontaneous regression of neoplasms in mammals: possible mechanisms and their application in immunotherapy. In vivo. 1998;12:107-122
368. Liu B, Ezeogu L, Zellmer L, Yu B, Xu N, Liao DJ. Protecting the normal in order to better kill the cancer. Cancer Med. 2015;4:1394-1403
369. Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83:155-158
370. Prasad KN, Cole WC, Yan XD, Nahreini P, Kumar B, Hanson A. et al. Defects in cAMP-pathway may initiate carcinogenesis in dividing nerve cells: a review. Apoptosis. 2003;8:579-586
371. Prasad KN, Hovland AR, Nahreini P, Cole WC, Hovland P, Kumar B. et al. Differentiation genes: are they primary targets for human carcinogenesis? Exp Biol Med (Maywood ). 2001;226:805-813
372. Elston DM. Mechanisms of regression. Clin Med Res. 2004;2:85-88
373. Bayer-Garner IB, Ivan D, Schwartz MR, Tschen JA. The immunopathology of regression in benign lichenoid keratosis, keratoacanthoma and halo nevus. Clin Med Res. 2004;2:89-97
374. Loeb L. On Transplantation of tumors. J Med Res. 1901;6:28-38
375. Loeb L. On some conditions of tissue growth, especially in culture media. Science. 1911;34:414-415
376. Twombly GH, Meisel D. The growth of mammalian tumors in fertile eggs; is a filterable cancer virus produced? Cancer Res. 1946;6:82-91
377. Heilman FR, Bittner JJ. Observations on mouse tumors cultivated in the yolk sac of the embryonic chick. Cancer Res. 1912;4:578-582
378. Mcduffie NG Jr, Gibson BS, Taylor A. Study of toxic factors associated with mouse mammary carcinomas in egg cultures. Cancer Res. 1960;20:1631-1635
379. Taylor A, Carmichael N. Toxic factor associated with egg-cultivated tumors. Cancer Res. 1960;20:1636-1639
380. Dalal UC, Taylor A, McKenna GF. The effect of plan extracts on egg cultivated tumor tissue. Tex Rep Biol Med. 1958;16:439-442
381. Murphy JB, Rous P. The behavior of chicken sarcoma implanted in the developing embryo. J Exp Med. 1912;15:119-132
382. Taylor A, Carmichael N. Egg cultivated tumor protects embryo against vaccinia virus. Proc Soc Exp Biol Med. 1953;83:676-678
383. Taylor A, Carmichael N, McKenna GF, Burlage HM. Inhibition of the growth of egg cultivated tumor tissue by extracts of Cooperia pedunculata Herb. Proc Soc Exp Biol Med. 1951;77:841-843
384. Taylor A, Carmichael N, Norris T. Temperature level and the growth of embryo and tumor of tumor-bearing eggs. Proc Soc Exp Biol Med. 1947;66:165-171
385. Taylor A, Carmichael N. Stromal malignancy in mouse-grown transplants of egg-cultivated mouse mammary carcinoma. Cancer Res. 1947;7:78-87
386. Murphy JB. Transplantability of malignant tumors to the embryos of a foreign species. J A M A. 1912;59:874-875
387. Kalirai H, Shahidipour H, Coupland SE, Luyten G. Use of the Chick Embryo Model in Uveal Melanoma. Ocul Oncol Pathol. 2015;1:133-140
388. Ribatti D. The chick embryo chorioallantoic membrane as a model for tumor biology. Exp Cell Res. 2014;328:314-324
389. Kain KH, Miller JW, Jones-Paris CR, Thomason RT, Lewis JD, Bader DM. et al. The chick embryo as an expanding experimental model for cancer and cardiovascular research. Dev Dyn. 2014;243:216-228
390. Jefferies B, Lenze F, Sathe A, Truong N, Anton M, von Eisenhart-Rothe R. et al. Non-invasive imaging of engineered human tumors in the living chicken embryo. Sci Rep. 2017;7:4991-doi 10.1038/s41598-017-04572-1
391. Ribatti D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod Toxicol. 2017;70:97-101
392. Rovithi M, Avan A, Funel N, Leon LG, Gomez VE, Wurdinger T. et al. Development of bioluminescent chick chorioallantoic membrane (CAM) models for primary pancreatic cancer cells: a platform for drug testing. Sci Rep. 2017;7:44686-doi 10.1038/srep44686
393. Beedie SL, Rore HM, Barnett S, Chau CH, Luo W, Greig NH. et al. In vivo screening and discovery of novel candidate thalidomide analogs in the zebrafish embryo and chicken embryo model systems. Oncotarget. 2016;7:33237-33245
394. Kim Y, Williams KC, Gavin CT, Jardine E, Chambers AF, Leong HS. Quantification of cancer cell extravasation in vivo. Nat Protoc. 2016;11:937-948
395. Haraguchi S, Matsubara Y, Hosoe M. Chick embryos can form teratomas from microinjected mouse embryonic stem cells. Dev Growth Differ. 2016;58:194-204
396. Herrmann A, Moss D, See V. The Chorioallantoic Membrane of the Chick Embryo to Assess Tumor Formation and Metastasis. Methods Mol Biol. 2016;1464:97-105
397. Deryugina EI. Chorioallantoic Membrane Microtumor Model to Study the Mechanisms of Tumor Angiogenesis, Vascular Permeability, and Tumor Cell Intravasation. Methods Mol Biol. 2016;1430:283-298
398. Ames JJ, Henderson T, Liaw L, Brooks PC. Methods for Analyzing Tumor Angiogenesis in the Chick Chorioallantoic Membrane Model. Methods Mol Biol. 2016;1406:255-269
399. Leighton J. Invasion and metastasis of heterologous tumors in the chick embryo. Prog Exp Tumor Res. 1964;4:98-125
400. Bracke ME, Roman BI, Stevens CV, Mus LM, Parmar VS, De WO. et al. Chick Heart Invasion Assay for Testing the Invasiveness of Cancer Cells and the Activity of Potentially Anti-invasive Compounds. J Vis Exp. 2015 e52792-doi: 10.3791/52792
401. Bracke ME, Parmar VS, Depass AL, Stevens CV, Vanhoecke BW, Mareel MM. Chick heart invasion assay. Methods Mol Biol. 2014;1070:93-106
402. Bracke ME, Boterberg T, Mareel MM. Chick heart invasion assay. Methods Mol Med. 2001;58:91-102
403. Engers R, Gerharz CD, Donner A, Mrzyk S, Krause-Paulus R, Petek O. et al. In vitro invasiveness of human epithelioid-sarcoma cell lines: association with cell motility and inverse correlation with the expression of tissue inhibitor of metalloproteinases. Int J Cancer. 1999;80:406-412
404. Easty GC, Easty DM. An organ culture system for the examination of tumor invasion. Nature. 1963;199:1104-1105
405. Mareel M, Kint J, Meyvisch C. Methods of study of the invasion of malignant C3H-mouse fibroblasts into embryonic chick heart in vitro. Virchows Arch B Cell Pathol Incl Mol Pathol. 1979;30:95-111
406. Scherer WF, Syverton JT, Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695-710
407. Billingham RE. Transplantation: past, present and future. J Invest Dermatol. 1963;41:165-180
408. Klein G. The usefulness and limitations of tumor transplantation in cancer research: a review. Cancer Res. 1959;19:343-358
409. KALISS N. Immunological enhancement of tumor homografts in mice: a review. Cancer Res. 1958;18:992-1003
410. HAY LJ. A review of mammary carcinoma in mice and of transplantation of carcinoma. Surg Staff Semin U S Veterans Adm Hosp Minneap. 1947;3:126-134
411. Barrett MK. Some immunogenetic influences upon transplanted tumors. Cancer Res. 1952;12:535-542
412. Barrett MK, Deringer MK, Hansen WH. Induced adaptation in a tumor: specificity of the change. J Natl Cancer Inst. 1953;14:381-394
413. Barrett MK, Deringer MK. An induced adaptation in a transplantable tumor of mice. J Natl Cancer Inst. 1950;11:51-59
414. Dunham LJ, Stewart HL. A survey of transplantable and transmissible animal tumors. J Natl Cancer Inst. 1953;13:1299-1377
415. Gorer PA. Some recent work on tumor immunity. Adv Cancer Res. 1956;4:149-186
416. Hauschka TS. Tissue genetics of neoplastic cell populations. Proc Can Cancer Conf. 1957;2:305-345
417. Hauschka TS. Methods of conditioning the graft in tumor transplantation. J Natl Cancer Inst. 1953;14:723-739
418. Hirsch HM. Some aspects of the problem of immunity against transplanted and spontaneous tumors. Bacteriol Rev. 1962;26:336-353
419. Hirsch HM. Tumor immunity and tissue transplantation. J Lancet. 1959;79:340-347
420. Hirsch HM. Tumor isoimmunity. Experientia. 1958;14:269-271
421. Kaliss N. The transplanted tumor as a research tool in cancer immunology. Cancer Res. 1961;21:1203-1208
422. Law LW. Genetic studies in experimental cancer. Adv Cancer Res. 1954;2:281-352
423. Cornil V. Sur les greffes et inoculations de cancer. Bull Acad Med. 1891;25:906-909
424. Hahn E. Ueber transplantation von carcinomatoser. Haut Berlin Klin Wchschr. 1888;25:413-415
425. Senn N. The present status of the carcinoma question. J A M A. 1901;37:811
426. Cazin M, Duplay S. Experimentelle tumoren bei thieren. Centralbl Allgem Pathol Pathol Anat. 1894;5:399-400
427. Doutrelepont J. Versuche über die uebertragung der carcinoma von their auf thier. Arch Pathol Anat Physiol. 1869;45:507
428. Duplay S, Cazin M. Des greffes cancereuses. Semaine Med (Paris). 1892;12:61
429. Duplay S, Cazin M. Contagion et inoculabilite du cancer. Semaine Med (Paris). 1893;13:329-332
430. Fischer E. Ueber transplantationen von organischen material. Deut Chir. 1882;17:61-92
431. Greene HS. On the development of cancer. Sci Am. 1948;179:40-43
432. Greene HS, Newton BL. Evolution of cancer of the uterine fundus in the rabbit. Cancer. 1948;1:82-99
433. Greene HS. Heterologous transplantation of the Brown-Pearce tumors. Cancer Res. 1949;9:728-35
434. Greene HS. Attributes of embryonic tissues after growth and development in heterologous hosts. Cancer Res. 1955;15:170-172
435. Greene HS. Pathology in fields collateral to tissue culture. J Natl Cancer Inst. 1957;19:711-721
436. Greene HS. The significance of transplantability. Trans Stud Coll Physicians Phila. 1957;24:101-104
437. Greene HS. Heterotransplantation of tumors. Ann N Y Acad Sci. 1957;69:818-829
438. Greene HS, Harvey EK. Metastasis of heterologously transplanted tumors. Cancer Res. 1964;24:1678-1687
439. Vivarelli S, Wagstaff L, Piddini E. Cell wars: regulation of cell survival and proliferation by cell competition. Essays Biochem. 2012;53:69-82
440. Wagstaff L, Kolahgar G, Piddini E. Competitive cell interactions in cancer: a cellular tug of war. Trends Cell Biol. 2013;23:160-167
441. Allard D, Stoker M, Gherardi E. A G2/M cell cycle block in transformed cells by contact with normal neighbors. Cell Cycle. 2003;2:484-487
442. Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N. et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. 2011;128:2793-2802
443. Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:9176-9181
444. Furuta S, Jeng YM, Zhou L, Huang L, Kuhn I, Bissell MJ. et al. IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells. Sci Transl Med. 2011;3:78ra31-doi 10.1126/scitranslmed.3001374
445. Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem. 2012;287:1397-1405
446. Stoker M. Regulation of growth and orientation in hamster cells transformed by polyoma virus. Virology. 1964;24:165-174
447. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239-242
448. Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C. et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231-245
449. Wang G, Chen L, Yu B, Zellmer L, Xu N, Liao DJ. Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors. J Cancer. 2016;7:436-445
450. Rubin H. Rethinking "cancer as a dynamic developmental disorder" a quarter century later. Cancer Res. 2009;69:2171-2175
451. Rubin H. Microenvironmental regulation of the initiated cell. Adv Cancer Res. 2003;90:1-62
452. Rubin H. Contact interactions between cells that suppress neoplastic development: can they also explain metastatic dormancy? Adv Cancer Res. 2008;100:159-202
453. Rubin H. Ordered heterogeneity and its decline in cancer and aging. Adv Cancer Res. 2007;98:117-147
454. Aktipis CA, Nesse RM. Evolutionary foundations for cancer biology. Evol Appl. 2013;6:144-159
455. Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC. et al. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond B Biol Sci 2015, 370: -pii. 2014 0219. doi: 10.1098/rstb.2014.0219
456. Thomas F, Kareva I, Raven N, Hamede R, Pujol P, Roche B. et al. Evolved Dependence in Response to Cancer. Trends Ecol Evol. 2018;33:269-276
457. Thomas F, Vavre F, Tissot T, Vittecoq M, Giraudeau M, Bernex F. et al. Cancer Is Not (Only) a Senescence Problem. Trends Cancer. 2018;4:169-172
458. Thomas F, Jacqueline C, Tissot T, Henard M, Blanchet S, Loot G. et al. The importance of cancer cells for animal evolutionary ecology. Nat Ecol Evol. 2017;1:1592-1595
459. Hennings H, Robinson VA, Michael DM, Pettit GR, Jung R, Yuspa SH. Development of an in vitro analogue of initiated mouse epidermis to study tumor promoters and antipromoters. Cancer Res. 1990;50:4794-4800
460. Strickland JE, Ueda M, Hennings H, Yuspa SH. A model for initiated mouse skin: suppression of papilloma but not carcinoma formation by normal epidermal cells in grafts on athymic nude mice. Cancer Res. 1992;52:1439-1444
461. Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10:232-233
462. Perez-Caro M, Cobaleda C, Gonzalez-Herrero I, Vicente-Duenas C, Bermejo-Rodriguez C, Sanchez-Beato M. et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009;28:8-20
463. Tsujiuchi T, Nakae D, Konishi Y. Multi-step lung carcinogenesis model induced by oral administration of N-nitrosobis(2-hydroxypropyl)amine in rats. Exp Toxicol Pathol. 2014;66:81-88
464. Holmquist GP. Cell-selfish modes of evolution and mutations directed after transcriptional bypass. Mutat Res. 2002;510:141-152
465. Jia QW, Chen XH, Jia YP, Dou XX, Ezeogu L, Xu NZ. et al. Is type 2 diabetes one of such aging phenomena that lack an irreversible structural change? J Diabetes Metab 2105, 6: 543-doi: 10.4172/2155-6156.1000543.
466. Dou X, Chen L, Lei M, Zellmer L, Jia Q, Ling P. et al. Evaluating the Remote Control of Programmed Cell Death, with or without a Compensatory Cell Proliferation. Int J Biol Sci. 2018;14:1800-1812
467. Liao DJ. The scavenger cell hypothesis of apoptosis: apoptosis redefined as a process by which a cell in living tissue is destroyed by phagocytosis. Med Hypotheses. 2005;65:23-28
468. Liu B, Xu N, Man Y, Shen H, Avital I, Stojadinovic A. et al. Apoptosis in Living Animals Is Assisted by Scavenger Cells and Thus May Not Mainly Go through the Cytochrome C-Caspase Pathway. J Cancer. 2013;4:716-723
469. Zellmer L, Han YP, Chen LC, Xu NZ, Liao DJ. Does the cytochrome c-caspase pathway of cell death occur physiologically in animals? Journal Tumor Med Prev. 2017;1:JTMP.MS.ID.555557.pdf
470. Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8-doi 10.1126/scisignal.3145re8
471. Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell!. Trends Cell Biol. 2008;18:467-473
472. Sun G, Irvine KD. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev Biol. 2011;350:139-151
473. Mollereau B, Perez-Garijo A, Bergmann A, Miura M, Gerlitz O, Ryoo HD. et al. Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ. 2013;20:181
474. Liu XD, Yang WX, Guan ZZ, Yu WF, Fan B, Xu NZ. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell & Bioscience. 2018;8:doi.org /10.1186/s13578-018-0206-6
475. Ewing J. The General Pathological Conception of Cancer. Can Med Assoc J. 1935;33:125-135
476. No Authors Available. Reviews. Br Med J. 1903;1:376-378
477. Newbold RF, Overell RW, Connell JR. Induction of immortality is an early event in malignant transformation of mammalian cells by carcinogens. Nature. 1982;299:633-635
478. Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. 1983;304:648-651
479. Newbold RF. Multistep malignant transformation of mammalian cells by carcinogens: induction of immortality as a key event. Carcinog Compr Surv. 1985;9:17-28
480. Newbold RF. Malignant transformation of mammalian cells in culture: delineation of stages and role of cellular oncogene activation. IARC Sci Publ. 1985;67:31-53
481. Newbold RF, Cuthbert AP, Themis M, Trott DA, Blair AL, Li W. Cell immortalization as a key, rate-limiting event in malignant transformation: approaches toward a molecular genetic analysis. Toxicol Lett. 1993;67:211-230
482. Thomas F, Nesse RM, Gatenby R, Gidoin C, Renaud F, Roche B. et al. Evolutionary Ecology of Organs: A Missing Link in Cancer Development? Trends Cancer. 2016;2:409-415
483. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614-636
484. Polymenis M, Kennedy BK. Unbalanced Growth, Senescence and Aging. Adv Exp Med Biol. 2017;1002:189-208
485. Passaro F, Testa G. Implications of Cellular Aging in Cardiac Reprogramming. Front Cardiovasc Med. 2018;5:43-doi 10.3389/fcvm.2018.00043
486. Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, Segers V. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018 -doi: 10.1152/ajpheart.00287.2018
487. Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139-1153
488. Falandry C, Bonnefoy M, Freyer G, Gilson E. Biology of cancer and aging: a complex association with cellular senescence. J Clin Oncol. 2014;32:2604-2610
489. Sikora E, Bielak-Zmijewska A, Mosieniak G. Cellular senescence in ageing, age-related disease and longevity. Curr Vasc Pharmacol. 2014;12:698-706
490. Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15:643-660
491. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439-446
492. Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E. et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path-a mini-review. Gerontology. 2008;54:92-99
493. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-1217
494. de Magalhaes JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018;170:2-9
495. Faragher RG, McArdle A, Willows A, Ostler EL. Senescence in the aging process. F1000Res. 2017;6:1219-doi 10.12688/f1000research.10903.1
496. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129-5143
497. Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB. Cellular Senescence as the Causal Nexus of Aging. Front Genet. 2016;7:13-doi 10.3389/fgene.2016.00013
498. Sapieha P, Mallette FA. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018 -pii: S0962-8924(18)30059-X. doi: 10.1016/j.tcb.2018.03.003
499. Markert CL. Neoplasia: a disease of cell differentiation. Cancer Res. 1968;28:1908-1914
500. Vassilopoulou-Sellin R, Ajani J. Islet cell tumors of the pancreas. Endocrinol Metab Clin North Am. 1994;23:53-65
501. Isaacs M, Lee P. Preoperative alpha-blockade in phaeochromocytoma and paraganglioma: is it always necessary? Clin Endocrinol (Oxf). 2017;86:309-314
502. Tan S, Khumalo N, Bayat A. Understanding Keloid Pathobiology From a Quasi-Neoplastic Perspective: Less of a Scar and More of a Chronic Inflammatory Disease With Cancer-Like Tendencies. Front Immunol. 2019;10:1810
503. Lim KH, Itinteang T, Davis PF, Tan ST. Stem Cells in Keloid Lesions: A Review. Plast Reconstr Surg Glob Open. 2019;7:e2228
504. Tsai CH, Ogawa R. Keloid research: current status and future directions. Scars Burn Heal. 2019;5:2059513119868659
505. Pitot HC. Some aspects of the developmental biology of neoplasia. Cancer Res. 1968;28:1880-1887
506. Yamasaki H, Mesnil M, Nakazawa H. Interaction and distinction of genotoxic and non-genotoxic events in carcinogenesis. Toxicol Lett. 1992:64-65 Spec No: 597-604
507. Solt DB, Farber E. A new principle for the analysis of chemical carcinogenesis. Nature. 1976;263:702-703
508. Solt DB, Medline A, Farber E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am J Pathol. 1977;88:595-618
509. Liao D, Porsch-Hallstrom I, Gustafsson JA, Blanck A. Sex differences at the initiation stage of rat liver carcinogenesis-influence of growth hormone. Carcinogenesis. 1993;14:2045-2049
510. Liao DZ, Blanck A, Gustafsson JA, Hallstrom IP. Expression of the c-jun, jun-B, ets-2 and liver regeneration factor-1 (LRF-1) genes during promotion and progression of rat liver carcinogenesis in the resistant hepatocyte model. Cancer Lett. 1996;100:215-221
511. Blanck A, Liao D, Gustafsson JA, Hallstrom IP. Hormonal regulation of sex differentiated parameters in liver nodules from rats treated in the resistant hepatocyte model. Carcinogenesis. 1995;16:231-235
512. Flodby P, Liao DZ, Blanck A, Xanthopoulos KG, Hallstrom IP. Expression of the liver-enriched transcription factors C/EBP alpha, C/EBP beta, HNF-1, and HNF-4 in preneoplastic nodules and hepatocellular carcinoma in rat liver. Mol Carcinog. 1995;12:103-109
513. Farber E. The Biology of Carcinogen-Induced Hepatocyte Nodules and Related Liver Lesions in the Rats. Toxicol Pathol. 1982;10:197-201
514. Farber E. The step-by-step development of epithelial cancer: from phenotype to genotype. Adv Cancer Res. 1996;70:21-48
515. Herceg Z, Lambert MP, van VK, Demetriou C, Vineis P, Smith MT. et al. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis. 2013;34:1955-1967
516. Herceg Z, Vaissiere T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics. 2011;6:804-819
517. Herceg Z. Epigenetic Mechanisms as an Interface Between the Environment and Genome. Adv Exp Med Biol. 2016;903:3-15
518. Patel S, Shah K, Mirza S, Daga A, Rawal R. Epigenetic regulators governing cancer stem cells and epithelial-mesenchymal transition in oral squamous cell carcinoma. Curr Stem Cell Res Ther. 2015;10:140-152
519. Salemi R, Marconi A, Di S, V, Franco S, Rapisarda V, Libra M. Epigenetic alterations and occupational exposure to benzene, fibers, and heavy metals associated with tumor development (Review). Mol Med Rep. 2017;15:3366-3371
520. Thomson JP, Moggs JG, Wolf CR, Meehan RR. Epigenetic profiles as defined signatures of xenobiotic exposure. Mutat Res Genet Toxicol Environ Mutagen. 2014;764-765:3-9
521. Berenblum I. A re-evaluation of the concept of cocarciongenesis. Prog Exp Tumor Res. 1969;11:21-30
522. Berenblum I. Challenging problems in cocarcinogenesis. Cancer Res. 1985;45:1917-1921
523. Cairns J. Cocarcinogenesis and biological effects of tumor promoters. Conclusions and perspectives. Carcinog Compr Surv. 1982;7:647-651
524. Brash D, Cairns J. The mysterious steps in carcinogenesis: addendum. Br J Cancer. 2009;101:1490
525. Cairns J. Mutation and cancer: the antecedents to our studies of adaptive mutation. Genetics. 1998;148:1433-1440
526. Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142-145
527. Kennedy AR. Is a mutagenic event involved in radiation induced malignant transformation? Mutat Res. 1996;350:81-91
528. Kennedy AR. Is there a critical target gene for the first step in carcinogenesis? Environ Health Perspect. 1991;93:199-203
529. Kennedy AR, Cairns J, Little JB. Timing of the steps in transformation of C3H 10T 1/2 cells by X-irradiation. Nature. 1984;307:85-86
530. Kennedy AR, Fox M, Murphy G, Little JB. Relationship between x-ray exposure and malignant transformation in C3H 10T1/2 cells. Proc Natl Acad Sci U S A. 1980;77:7262-7266
531. Rous P. Recent Advances in Cancer Research. Bull N Y Acad Med. 1947;23:65-78
532. Blagosklonny MV. Cell immortality and hallmarks of cancer. Cell Cycle. 2003;2:296-299
533. Alsabti EA. Tumor dormancy (a review). Neoplasma. 1979;26:351-361
534. Gordon-Taylor G. The incomputable factor in cancer prognosis. Br Med J. 1959;1:455-462
535. Burrow GN, Wortzman G, Rewcastle NB, Holgate RC, Kovacs K. Microadenomas of the pituitary and abnormal sellar tomograms in an unselected autopsy series. N Engl J Med. 1981;304:156-158
536. Chambers EF, Turski PA, LaMasters D, Newton TH. Regions of low density in the contrast-enhanced pituitary gland: normal and pathologic processes. Radiology. 1982;144:109-113
537. Costello RT. Subclinical Adenoma of the Pituitary Gland. Am J Pathol. 1936;12:205-216
538. Muhr C, Bergstrom K, Grimelius L, Larsson SG. A parallel study of the roentgen anatomy of the sella turcica and the histopathology of the pituitary gland in 205 autopsy specimens. Neuroradiology. 1981;21:55-65
539. Parent AD, Bebin J, Smith RR. Incidental pituitary adenomas. J Neurosurg. 1981;54:228-231
540. Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120:817-820
541. Rich AR. On the frequency of occurrence of occult carcinoma of the prostate. J Urol. 1935;33:3-7
542. Rich AR. Classics in oncology. On the frequency of occurrence of occult carcinoma of the prostate: Arnold Rice Rich, M.D, Journal of Urology 33:3, 1935. CA Cancer J Clin. 1979;29:115-119
543. Rich AR. On the frequency of occurrence of occult carcinoma of the prostrate. 1934. Int J Epidemiol. 2007;36:274-277
544. Ashley DJ. On the incidence of carcinoma of the prostate. J Pathol Bacteriol. 1965;90:217-224
545. Montgomery TR, Whitlock GF, Nohlgren JE, Lewis AM. What becomes of the patient with latent or occult carcinoma of the prostate. J Urol. 1961;86:655-658
546. Munsie WJ, Foster EA. Unsuspected very small foci of carcinoma of the prostate in transurethral resection specimens. Cancer. 1968;21:692-698
547. Mortensen JD, Bennett WA, Woolner LB. Incidence of carcinoma in thyroid glands removed at 1000 consecutive routine necropsies. Surg Forum. 1955;5:659-663
548. Mortensen JD, Woolner LB, Bennett WA. Gross and microscopic findings in clinically normal thyroid glands. J Clin Endocrinol Metab. 1955;15:1270-1280
549. Beckwith JB, Perrin EV. In situ neuroblastomas: a contribution to the natural history of neural crest tumors. Am J Pathol. 1963;43:1089-1104
550. Kolquist KA, Ellisen LW, Counter CM, Meyerson M, Tan LK, Weinberg RA. et al. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat Genet. 1998;19:182-186
551. Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197-200
552. Cairns J. Cancer and the immortal strand hypothesis. Genetics. 2006;174:1069-1072
553. FISHER JC. Multiple-mutation theory of carcinogenesis. Nature. 1958;181:651-652
554. Beckman RA, Loeb LA. Evolutionary dynamics and significance of multiple subclonal mutations in cancer. DNA Repair (Amst). 2017;56:7-15
555. Fox EJ, Loeb LA. Lethal mutagenesis: targeting the mutator phenotype in cancer. Semin Cancer Biol. 2010;20:353-359
556. Prindle MJ, Fox EJ, Loeb LA. The mutator phenotype in cancer: molecular mechanisms and targeting strategies. Curr Drug Targets. 2010;11:1296-1303
557. Venkatesan RN, Loeb LA. The multiplicity of mutations in human cancers. Adv Exp Med Biol. 2005;570:3-17
558. Venkatesan RN, Bielas JH, Loeb LA. Generation of mutator mutants during carcinogenesis. DNA Repair (Amst). 2006;5:294-302
559. Bodmer WF, Cottrell S, Frischauf AM, Kerr IB, Murday VA, Rowan AJ. et al. Genetic analysis of colorectal cancer. Princess Takamatsu Symp. 1989;20:49-59
560. Koorey DJ, McCaughan GW. Tumour suppressor genes and colorectal neoplasia. J Gastroenterol Hepatol. 1993;8:174-184
561. Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993;2:425-434
562. Nakamura Y, Nishisho I, Kinzler KW, Vogelstein B, Miyoshi Y, Miki Y. et al. Mutations of the adenomatous polyposis coli gene in familial polyposis coli patients and sporadic colorectal tumors. Princess Takamatsu Symp. 1991;22:285-292
563. Williams AC, Browne SJ, Manning AM, Hague A, van der Stappen JW, Paraskeva C. Biological consequences of the genetic changes which occur during human colorectal carcinogenesis. Semin Cancer Biol. 1993;4:153-159
564. Risio M. Reprint of: the natural history of adenomas. Best Pract Res Clin Gastroenterol. 2010;24:397-406
565. Risio M. The natural history of colorectal adenomas and early cancer. Pathologe. 2012;33(Suppl 2):206-210
566. Williams RD, Fish JC. Multiple polyposis, polyp regression, and carcinoma of the colon. Am J Surg. 1966;112:846-849
567. Rubin H. Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture. Bioessays. 2011;33:224-231
568. Farber E. Putative precursor lesions: summary and some analytical considerations. Cancer Res. 1976;36:2703-2705
569. Williams D. Thyroid Growth and Cancer. Eur Thyroid J. 2015;4:164-173
570. Berger AH, Pandolfi PP. Haplo-insufficiency: a driving force in cancer. J Pathol. 2011;223:137-146
571. Kopelovich L, Shea-Herbert B. Heritable one-hit events defining cancer prevention? Cell Cycle. 2013;12:2553-2557
572. Yeung AT, Patel BB, Li XM, Seeholzer SH, Coudry RA, Cooper HS. et al. One-hit effects in cancer: altered proteome of morphologically normal colon crypts in familial adenomatous polyposis. Cancer Res. 2008;68:7579-7586
573. Tyzzer EE. A Series of spontaneous tumors in Mice with Observations on the Influence of Heredity on the Frequency of their Occurrence. J Med Res. 1909;21:479-518
574. Tyzzer EE. A Study of Heredity in Relation to the Development of tumors in Mice. J Med Res. 1907;17:199-211
575. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759-767
576. Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1-84
577. Farber E, Solt D, Cameron R, Laishes B, Ogawa K, Medline A. Newer insights into the pathogenesis of liver cancer. Am J Pathol. 1977;89:477-482
578. Farber E. Pre-cancerous steps in carcinogenesis. Their physiological adaptive nature. Biochim Biophys Acta. 1984;738:171-180
579. Farber E, Rubin H. Cellular adaptation in the origin and development of cancer. Cancer Res. 1991;51:2751-2761
580. Farber E. Cell proliferation as a major risk factor for cancer: a concept of doubtful validity. Cancer Res. 1995;55:3759-3762
581. Farber E. Cell proliferation is not a major risk factor for cancer. Mod Pathol. 1996;9:606
582. Farber E. Risk assessment for possible carcinogens: a critical look. Drug Metab Rev. 2000;32:143-151
583. Chao MP, Majeti R, Weissman IL. Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer. 2012;12:58-67
584. McCullough KD, Coleman WB, Smith GJ, Grishan JW. Age-dependent regulation of the tumorigenic potential of neoplastically transformed rat liver epithelial cells by the liver microenvironment. Cancer Res. 1994;54:3668-3671
585. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853-2868
586. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072-12077
587. Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117-3126
588. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424-1435
589. Canino C, Mori F, Cambria A, Diamantini A, Germoni S, Alessandrini G. et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene. 2012;31:3148-3163
590. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118
591. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273-283
592. Ghosh K, Capell BC. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J Invest Dermatol. 2016;136:2133-2139
593. Greten TF, Eggert T. Cellular senescence associated immune responses in liver cancer. Hepat Oncol. 2017;4:123-127
594. Lecot P, Alimirah F, Desprez PY, Campisi J, Wiley C. Context-dependent effects of cellular senescence in cancer development. Br J Cancer. 2016;114:1180-1184
595. Pare R, Yang T, Shin JS, Lee CS. The significance of the senescence pathway in breast cancer progression. J Clin Pathol. 2013;66:491-495
596. Valenzuela CA, Quintanilla R, Moore-Carrasco R, Brown NE. The Potential Role of Senescence As a Modulator of Platelets and Tumorigenesis. Front Oncol. 2017;7:188-doi 10.3389/fonc.2017.00188
597. Eriksson LC, Blanck A, Bock KW, Mannervik B. Metabolism of xenobiotics in hepatocyte nodules. Toxicol Pathol. 1987;15:27-42
598. Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, Brothman AR. et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle. 2014;13:3423-3435
599. Lorens JB. The immortality two-step. Cell Cycle. 2015;14:798
600. Zimonjic D, Brooks MW, Popescu N, Weinberg RA, Hahn WC. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001;61:8838-8844
601. Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9:M57-M60
602. Carnero A, Blanco-Aparicio C, Kondoh H, LLeonart ME, Martinez-Leal JF, Mondello C. et al. Disruptive chemicals, senescence and immortality. Carcinogenesis. 2015;36(Suppl 1):S19-S37
603. Maqsood MI, Matin MM, Bahrami AR, Ghasroldasht MM. Immortality of cell lines: challenges and advantages of establishment. Cell Biol Int. 2013;37:1038-1045
604. Rhim JS. Development of human cell lines from multiple organs. Ann N Y Acad Sci. 2000;919:16-25
605. Rhim JS. Neoplastic transformation of human cells in vitro. Crit Rev Oncog. 1993;4:313-335
606. Chen QY, Costa M. A comprehensive review of metal-induced cellular transformation studies. Toxicol Appl Pharmacol. 2017;331:33-40
607. Steinberg P. In vitro-In vivo Carcinogenicity. Adv Biochem Eng Biotechnol. 2017;157:81-96
608. Heeg S, Doebele M, von WA, Opitz OG. In vitro transformation models: modeling human cancer. Cell Cycle. 2006;5:630-634
609. Klocke R, Gomez-Lechon MJ, Ehrhardt A, Mendoza-Figueroa T, Donato MT, Lopez-Revilla R. et al. Establishment and characterization of immortal hepatocytes derived from various transgenic mouse lines. Biochem Biophys Res Commun. 2002;294:864-871
610. Powell AJ, Gates PB, Wylie D, Velloso CP, Brockes JP, Jat PS. Immortalization of rat embryo fibroblasts by a 3'-untranslated region. Exp Cell Res. 1998;240:252-262
611. Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299-313
612. Todaro GJ, Green H, Goldberg BD. Transformation of properties of an established cell line by sv40 and polyoma virus. Proc Natl Acad Sci U S A. 1964;51:66-73
613. Nilausen K, Green H. Reversible arrest of growth in G1 of an established fibroblast line (3T3). Exp Cell Res. 1965;40:166-168
614. Gunes C, Avila AI, Rudolph KL. Telomeres in cancer. Differentiation. 2017;99:41-50
615. Russo I, Silver AR, Cuthbert AP, Griffin DK, Trott DA, Newbold RF. A telomere-independent senescence mechanism is the sole barrier to Syrian hamster cell immortalization. Oncogene. 1998;17:3417-3426
616. Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593-1603
617. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138-141
618. Brash D, Cairns J. The mysterious steps in carcinogenesis. Br J Cancer. 2009;101:379-380
619. Brash D, Cairns J. The mysterious steps in carcinogenesis: addendum. Br J Cancer. 2009;101:1490
620. Rubin H, Rubin AL. Phenotypic selection as the biological mode of epigenetic conversion and reversion in cell transformation. Proc Natl Acad Sci U S A. 2018;115:E725-E732
621. Shvemberger IN. Conversion of malignant cells into normal ones. Int Rev Cytol. 1986;103:341-386
622. Gonzalez-Garcia I, Sole RV, Costa J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci U S A. 2002;99:13085-13089
623. Stevens LC. Origin of testicular teratomas from primordial germ cells in mice. J Natl Cancer Inst. 1967;38:549-552
624. Stevens LC. Experimental production of testicular teratomas in mice. Proc Natl Acad Sci U S A. 1964;52:654-661
625. Stevens LC. The development of teratomas from intratesticular grafts of tubal mouse eggs. J Embryol Exp Morphol. 1968;20:329-341
626. Buta C, David R, Dressel R, Emgard M, Fuchs C, Gross U. et al. Reconsidering pluripotency tests: do we still need teratoma assays? Stem Cell Res. 2013;11:552-562
627. Damjanov I, Andrews PW. Teratomas produced from human pluripotent stem cells xenografted into immunodeficient mice - a histopathology atlas. Int J Dev Biol. 2016;60:337-419
628. Solter D, Dominis M, Damjanov I. Embryo-derived teratocarcinoma: I. The role of strain and gender in the control of teratocarcinogenesis. Int J Cancer. 1979;24:770-772
629. Damjanov I. Teratocarcinoma: neoplastic lessons about normal embryogenesis. Int J Dev Biol. 1993;37:39-46
630. Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res. 2008;100:133-158
631. Bustamante-Marin X, Garness JA, Capel B. Testicular teratomas: an intersection of pluripotency, differentiation and cancer biology. Int J Dev Biol. 2013;57:201-210
632. Pierce GB. Neoplasms, differentiations and mutations. Am J Pathol. 1974;77:103-118
633. Pierce GB. Neoplastic stem cells. Adv Pathobiol. 1977:141-152
634. Sell S. Cellular origin of cancer: dedifferentiation or stem cell maturation arrest? Environ Health Perspect. 1993;101(Suppl 5):15-26
635. Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6-22
636. Sell S, Nicolini A, Ferrari P, Biava PM. Cancer: A Problem of Developmental Biology; Scientific Evidence for Reprogramming and Differentiation Therapy. Curr Drug Targets. 2016;17:1103-1110
637. Martin GR. Teratocarcinomas and mammalian embryogenesis. Science. 1980;209:768-776
638. Martin GR. Teratocarcinomas as a model system for the study of embryogenesis and neoplasia. Cell. 1975;5:229-243
639. Arechaga J, Damjanov I. Above the borderland between normal and neoplastic development. Int J Dev Biol. 2012;56:939-948
640. Solter D, Skreb N, Damjanov I. Extrauterine growth of mouse egg-cylinders results in malignant teratoma. Nature. 1970;227:503-504
641. Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21:364-382
642. Skreb N, Svajger A, Levak-Svajger B. Growth and differentiation of rat egg-cylinders under the kidney capsule. J Embryol Exp Morphol. 1971;25:47-56
643. Svajger A, Levak-Svajger B, Skreb N. Rat embryonic ectoderm as renal isograft. J Embryol Exp Morphol. 1986;94:1-27
644. Damjanov I. Development of teratomas from embryos transplanted into outbred and inbred adult hamsters. J Natl Cancer Inst. 1978;61:911-915
645. Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I. et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340-345
646. Villodre ES, Felipe KB, Oyama MZ, Oliveira FH, Lopez PLDC, Solari C. et al. Silencing of the transcription factors Oct4, Sox2, Klf4, c-Myc or Nanog has different effect on teratoma growth. Biochem Biophys Res Commun. 2019;517:324-329
647. Zakrzewski W, Dobrzynski M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10:68
648. Bulic-Jakus F, Katusic BA, Juric-Lekic G, Vlahovic M, Sincic N. Teratoma: from spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip Rev Dev Biol. 2016;5:186-209
649. Hultman I, Bjork L, Blomberg E, Sandstedt B, Ahrlund-Richter L. Experimental teratoma: at the crossroad of fetal- and onco-development. Semin Cancer Biol. 2014;29:75-79
650. Cunningham JJ, Ulbright TM, Pera MF, Looijenga LH. Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotechnol. 2012;30:849-857
651. Peterson SE, Garitaonandia I, Loring JF. The tumorigenic potential of pluripotent stem cells: What can we do to minimize it? Bioessays. 2016;38(Suppl 1):S86-S95
652. Solter D. From teratocarcinomas to embryonic stem cells and beyond: a history of embryonic stem cell research. Nat Rev Genet. 2006;7:319-327
653. Papaioannou VE, McBurney MW, Gardner RL, Evans MJ. Fate of teratocarcinoma cells injected into early mouse embryos. Nature. 1975;258:70-73
654. Papaioannou VE. Ontogeny, pathology, oncology. Int J Dev Biol. 1993;37:33-37
655. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72:3585-3589
656. Illmensee K, Stevens LC. Teratomas and chimeras. Sci Am. 1979;240:120-132
657. Illmensee K. Reversion of malignancy and normalized differentiation of teratocarcinoma cells in chimeric mice. Basic Life Sci. 1978;12:3-25
658. Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A. 1976;73:549-553
659. Brinster RL. The effect of cells transferred into the mouse blastocyst on subsequent development. J Exp Med. 1974;140:1049-1056
660. Brinster RL. Participation of teratocarcinoma cells in mouse embryo development. Cancer Res. 1976;36:3412-3414
661. Brinster RL. Stem cells and transgenic mice in the study of development. Int J Dev Biol. 1993;37:89-99
662. Lust JM, Carlson DL, Kowles R, Rollins-Smith L, Williams JW III, McKinnell RG. Allografts of tumor nuclear transplantation embryos: differentiation competence. Proc Natl Acad Sci U S A. 1991;88:6883-6887
663. McKinnell RG, Deggins BA, Labat DD. Transplantation of pluripotential nuclei from triploid frog tumors. Science. 1969;165:394-396
664. McKinnell RG. Frog tumor controversy. J Natl Cancer Inst. 1972;49:1471-1474
665. McKinnell RG. Neoplastic cells. Modulation of the differentiated state. Dev Biol (N Y 1985 ). 1989;6:199-236
666. McKinnell RG, Lust JM, Sauerbier W, Rollins-Smith LA, Williams JW III, Williams CS. et al. Genomic plasticity of the Lucke renal carcinoma: a review. Int J Dev Biol. 1993;37:213-219
667. McKinnell RG. Reduced oncogenic potential associated with differentiation of the Lucke renal adenocarcinoma. In vivo. 1994;8:65-69
668. McKinnell RG, Carlson DL. Lucke renal adenocarcinoma, an anuran neoplasm: studies at the interface of pathology, virology, and differentiation competence. J Cell Physiol. 1997;173:115-118
669. Coleman WB, Wennerberg AE, Smith GJ, Grisham JW. Regulation of the differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenvironment. Am J Pathol. 1993;142:1373-1382
670. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7:246-255
671. Pierce GB, Pantazis CG, Caldwell JE, Wells RS. Specificity of the control of tumor formation by the blastocyst. Cancer Res. 1982;42:1082-1087
672. Podesta AH, Mullins J, Pierce GB, Wells RS. The neurula stage mouse embryo in control of neuroblastoma. Proc Natl Acad Sci U S A. 1984;81:7608-7611
673. Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW. et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci U S A. 2008;105:4329-4334
674. Webb CG, Gootwine E, Sachs L. Developmental potential of myeloid leukemia cells injected into midgestation embryos. Dev Biol. 1984;101:221-224
675. Kulesa PM, Kasemeier-Kulesa JC, Teddy JM, Margaryan NV, Seftor EA, Seftor RE. et al. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc Natl Acad Sci U S A. 2006;103:3752-3757
676. Diez-Torre A, Andrade R, Eguizabal C, Lopez E, Arluzea J, Silio M. et al. Reprogramming of melanoma cells by embryonic microenvironments. Int J Dev Biol. 2009;53:1563-1568
677. Gerschenson M, Graves K, Carson SD, Wells RS, Pierce GB. Regulation of melanoma by the embryonic skin. Proc Natl Acad Sci U S A. 1986;83:7307-7310
678. Kasemeier-Kulesa JC, Teddy JM, Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. et al. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev Dyn. 2008;237:2657-2666
679. Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer Res. 1971;31:127-134
680. Braun AC. An epigenetic model for the origin of cancer. Q Rev Biol. 1981;56:33-60
681. Binns A, Meins F. Habituation of tobacco pith cells for factors promoting cell division is heritable and potentially reversible. Proc Natl Acad Sci U S A. 1973;70:2660-2662
682. Carlson PS, Smith HH, Dearing RD. Parasexual interspecific plant hybridization. Proc Natl Acad Sci U S A. 1972;69:2292-2294
683. Meins F Jr, Binns A. Epigenetic variation of cultured somatic cells: evidence for gradual changes in the requirement for factors promoting cell division. Proc Natl Acad Sci U S A. 1977;74:2928-2932
684. Sacristan MD, Melchers G. The caryological analysis of plants regenerated from tumorous and other callus cultures of tobacco. Mol Gen Genet. 1969;105:317-333
685. Smith HH, Kao KN, Combatti NC. Interspecific hybridization by protoplast fusion in nicotiana. J Heredity. 1976;67:123-128
686. Braun AC, Wood HN. Suppression of the neoplastic state with the acquisition of specialized functions in cells, tissues, and organs of crown gall teratomas of tobacco. Proc Natl Acad Sci U S A. 1976;73:496-500
687. Wood HN, Binns AN, Braun AC. Differential expression of oncogenicity and nopaline synthesis in intact leaves derived from crown gall teratomas of tobacco. Differentiation. 1978;11:175-180
688. Rubin H. The suppression of morphological alterations in cells infected with Rous sarcoma virus. Virology. 1960;12:14-31
689. Stoker MG, Shearer M, O'Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci. 1966;1:297-310
690. Deng CT, Boettiger D, Macpherson I, Varmus HE. The persistence and expression of virus-specific DNA in revertants of Rous sarcoma virus-transformed BHK-21 cells. Virology. 1974;62:512-521
691. Macpherson I. Reversion in Hamster Cells Transformed by Rous Sarcoma Virus. Science. 1965;148:1731-1733
692. Visfeldt J. Transformation of sympathicoblastoma into ganglioneuroma, with a case report. Acta Pathol Microbiol Scand. 1963;58:414-428
693. Kimhi Y, Palfrey C, Spector I, Barak Y, Littauer UZ. Maturation of neuroblastoma cells in the presence of dimethylsulfoxide. Proc Natl Acad Sci U S A. 1976;73:462-466
694. Enane FO, Saunthararajah Y, Korc M. Differentiation therapy and the mechanisms that terminate cancer cell proliferation without harming normal cells. Cell Death Dis. 2018;9:912
695. Kenny PA, Bissell MJ. Tumor reversion: correction of malignant behavior by microenvironmental cues. Int J Cancer. 2003;107:688-695
696. Friend C, Scher W, Holland JG, Sato T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci U S A. 1971;68:378-382
697. Andersson LC, Jokinen M, Gahmberg CG. Induction of erythroid differentiation in the human leukaemia cell line K562. Nature. 1979;278:364-365
698. Lozzio CB, Lozzio BB, Machado EA, Fuhr JE, Lair SV, Bamberger EG. Effects of sodium butyrate on human chronic myelogenous leukaemia cell line K562. Nature. 1979;281:709-710
699. Andersson LC, Nilsson K, Gahmberg CG. K562-a human erythroleukemic cell line. Int J Cancer. 1979;23:143-147
700. Rutherford TR, Clegg JB, Weatherall DJ. K562 human leukaemic cells synthesise embryonic haemoglobin in response to haemin. Nature. 1979;280:164-165
701. Metcalf D. The granulocyte-macrophage colony-stimulating factors. Science. 1985;229:16-22
702. Sachs L. Cell differentiation and malignancy. Cell Biophys. 1986;9:225-242
703. Lotem J, Sachs L. Control of in vivo differentiation of myeloid leukemic cells. Leukemia. 1988;2:24S-37S
704. Reitsma PH, Rothberg PG, Astrin SM, Trial J, Bar-Shavit Z, Hall A. et al. Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature. 1983;306:492-494
705. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8:pii a019505. doi: 10.1101/cshperspect.a019505
706. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596-602
707. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3:355-359
708. Krelin Y, Zhang L, Kang TB, Appel E, Kovalenko A, Wallach D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008;15:1350-1355
709. Knight RA, Vaux DL. A tumour suppressor function of caspase-8? Cell Death Differ. 2008;15:1337-1338
710. Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q. et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8:1105-1114
711. Gordon K, Clouaire T, Bao XX, Kemp SE, Xenophontos M, de Las Heras JI. et al. Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression. Nucleic Acids Res. 2014;42:3529-3541
712. Chang S, DePinho RA. Telomerase extracurricular activities. Proc Natl Acad Sci U S A. 2002;99:12520-12522
713. Belair CD, Yeager TR, Lopez PM, Reznikoff CA. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci U S A. 1997;94:13677-13682
714. Reddel RR. The role of senescence and immortalization in carcinogenesis. Carcinogenesis. 2000;21:477-484
715. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-361
716. Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321:1841-1844
717. Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14:283-284
718. Creton S, Aardema MJ, Carmichael PL, Harvey JS, Martin FL, Newbold RF. et al. Cell transformation assays for prediction of carcinogenic potential: state of the science and future research needs. Mutagenesis. 2012;27:93-101
719. Pickles JC, Pant K, Mcginty LA, Yasaei H, Roberts T, Scott AD. et al. A mechanistic evaluation of the Syrian hamster embryo cell transformation assay (pH 6.7) and molecular events leading to senescence bypass in SHE cells. Mutat Res Genet Toxicol Environ Mutagen. 2016;802:50-58
720. Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1:580-590
721. Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38:627-637
722. Stewart TA, Bellve AR, Leder P. Transcription and promoter usage of the myc gene in normal somatic and spermatogenic cells. Science. 1984;226:707-710
723. Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115-122
724. Guy CT, Cardoso G. Transgenic animal models. Methods Mol Med. 2001;58:231-249
725. Fantozzi A, Christofori G. Mouse models of breast cancer metastasis. Breast Cancer Res. 2006;8:212
726. Menezes ME, Das SK, Emdad L, Windle JJ, Wang XY, Sarkar D. et al. Genetically engineered mice as experimental tools to dissect the critical events in breast cancer. Adv Cancer Res. 2014;121:331-382
727. Anisimov VN, Ukraintseva SV, Yashin AI. Cancer in rodents: does it tell us about cancer in humans? Nat Rev Cancer. 2005;5:807-819
728. Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331-341
729. Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer. 2003;3:952-959
730. Braun AC. Studies on tumor inception in the crown-gall disease. Am J Botany. 1943;30:674-677
731. Braun AC. Recovery of crown-gall tumor cells. Cancer Res. 1951;11:839-844
732. Braun AC, Mandle RJ. Studies on the inactivation of the tumor-inducing principle in crown gall. Growth. 1948;12:255-269
733. Braun AC. Recovery of tumor cells from effects of the tumor-inducing principle in crown gall. Science. 1951;113:651-653
734. Braun AC. Cellular autonomy in crown gall. Phytopathology. 1951;41:963-966
735. Braun AC. Tissue culture as a tool for studying the development of autonomy in neoplastic plant cells. J Natl Cancer Inst. 1957;19:753-759
736. Braun AC. A Physiological Basis for Autonomous Growth of the Crown-Gall Tumor Cell. Proc Natl Acad Sci U S A. 1958;44:344-349
737. Braun AC. On the origin of the cancer cells. Am Sci. 1970;58:307-320
738. Braun AC. Plant tumors. Biochim Biophys Acta. 1978;516:167-191
739. Braun AC. Genetic and biochemical studies on the suppression of and a recovery from the tumorous state in higher plants. In vitro. 1980;16:38-48
740. Marin G, Macpherson I. Reversion in polyoma-transformed cells: retransformation, induced antigens and tumorigenicity. J Virol. 1969;3:146-149
741. Macpherson I. Reversion in virus-transformed cells. Biochem Pharmacol. 1971;20:1005-1008
742. Wiblin CN, Macpherson I. Reversion in hybrids between SV40-transformed hamster and mouse cells. Int J Cancer. 1973;12:148-161
743. Macera-Bloch L, Houghton J, Lenahan M, Jha KK, Ozer HL. Termination of lifespan of SV40-transformed human fibroblasts in crisis is due to apoptosis. J Cell Physiol. 2002;190:332-344
744. Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A. et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12:439-451
745. Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest. 2010;120:2619-2626
746. Sacco A, Kawano Y, Moschetta M, Zavidij O, Huynh D, Reagan M. et al. A novel in vivo model for studying conditional dual loss of BLIMP-1 and p53 in B-cells, leading to tumor transformation. Am J Hematol. 2017;92:E138-E145
747. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B. et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599-607
748. Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X. et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A. 2012;109:20035-20040
749. Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681-686
750. Sakairi T, Abe Y, Kajiyama H, Bartlett LD, Howard LV, Jat PS. et al. Conditionally immortalized human podocyte cell lines established from urine. Am J Physiol Renal Physiol. 2010;298:F557-F567
751. Kawata S, Suzuki J, Maruoka M, Mizutamari M, Ishida-Kitagawa N, Yogo K. et al. Retrovirus-mediated conditional immortalization and analysis of established cell lines of osteoclast precursor cells. Biochem Biophys Res Commun. 2006;350:97-104
752. Lidington EA, Rao RM, Marelli-Berg FM, Jat PS, Haskard DO, Mason JC. Conditional immortalization of growth factor-responsive cardiac endothelial cells from H-2K(b)-tsA58 mice. Am J Physiol Cell Physiol. 2002;282:C67-C74
753. O'Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C. et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci U S A. 2001;98:646-651
754. Noble M, Groves AK, Ataliotis P, Ikram Z, Jat PS. The H-2KbtsA58 transgenic mouse: a new tool for the rapid generation of novel cell lines. Transgenic Res. 1995;4:215-225
755. Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L. et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88:5096-5100
756. Tegtmeyer P. Function of simian virus 40 gene A in transforming infection. J Virol. 1975;15:613-618
757. Petit CA, Gardes M, Feunteun J. Immortalization of rodent embryo fibroblasts by SV40 is maintained by the A gene. Virology. 1983;127:74-82
758. Zaret KS, DiPersio CM, Jackson DA, Montigny WJ, Weinstat DL. Conditional enhancement of liver-specific gene transcription. Proc Natl Acad Sci U S A. 1988;85:9076-9080
759. Radna RL, Caton Y, Jha KK, Kaplan P, Li G, Traganos F. et al. Growth of immortal simian virus 40 tsA-transformed human fibroblasts is temperature dependent. Mol Cell Biol. 1989;9:3093-3096
760. Jat PS, Sharp PA. Cell lines established by a temperature-sensitive simian virus 40 large-T-antigen gene are growth restricted at the nonpermissive temperature. Mol Cell Biol. 1989;9:1672-1681
761. Wojcik BE, Dermody JJ, Ozer HL, Mun B, Mathews CK. Temperature-sensitive DNA mutant of Chinese hamster ovary cells with a thermolabile ribonucleotide reductase activity. Mol Cell Biol. 1990;10:5688-5699
762. Jia Y, Chen L, Ma Y, Zhang J, Xu N, Liao DJ. To Know How a Gene Works, We Need to Redefine It First but then, More Importantly, to Let the Cell Itself Decide How to Transcribe and Process Its RNAs. Int J Biol Sci. 2015;11:1413-1423
763. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789-799
764. Cobaleda C, Sanchez-Garcia I. Stem cell aging and cancer: immortal but vulnerable. Cell Cycle. 2011;10:2823-2824
765. Alspach E, Fu Y, Stewart SA. Senescence and the pro-tumorigenic stroma. Crit Rev Oncog. 2013;18:549-558
766. Pazolli E, Stewart SA. Senescence: the good the bad and the dysfunctional. Curr Opin Genet Dev. 2008;18:42-47
767. Ruhland MK, Coussens LM, Stewart SA. Senescence and cancer: An evolving inflammatory paradox. Biochim Biophys Acta. 2016;1865:14-22
768. Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531-557
769. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51-57
770. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223-233
771. Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472-476
772. Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle. 2005;4:1722-1724
773. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M. et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642-doi 10.1038/436642a
774. Soo JK, Mackenzie Ross AD, Kallenberg DM, Milagre C, Heung CW, Chow J. et al. Malignancy without immortality? Cellular immortalization as a possible late event in melanoma progression. Pigment Cell Melanoma Res. 2011;24:490-503
775. Wells S. The Morton Lecture on Cancer and Cancerous Diseases. Br Med J. 1888;2:1201-1205
776. Pierce GB. On the boundary between development and neoplasia. An interview with Professor G. Barry Pierce. Interview by Juan Arechaga. Int J Dev Biol. 1993;37:5-16
777. Potter VR. Phenotypic diversity in experimental hepatomas: the concept of partially blocked ontogeny. The 10th Walter Hubert Lecture. Br J Cancer. 1978;38:1-23
778. Trosko JE. Review paper: cancer stem cells and cancer nonstem cells: from adult stem cells or from reprogramming of differentiated somatic cells. Vet Pathol. 2009;46:176-193
779. Trosko JE. Human adult stem cells as the target cells for the initiation of carcinogenesis and for the generation of "cancer stem cells". Int J Stem Cells. 2008;1:8-26
780. Trosko JE. From adult stem cells to cancer stem cells: Oct-4 Gene, cell-cell communication, and hormones during tumor promotion. Ann N Y Acad Sci. 2006;1089:36-58
781. Trosko JE, Carruba G. "Bad Luck Mutations": DNA Mutations Are not the Whole Answer to Understanding Cancer Risk. Dose Response. 2017;15:1559325817716585
782. Trosko JE, Lenz HJ. What roles do colon stem cells and gap junctions play in the left and right location of origin of colorectal cancers? J Cell Commun Signal. 2017;11:79-87
783. Trosko JE. Induction of iPS cells and of cancer stem cells: the stem cell or reprogramming hypothesis of cancer? Anat Rec (Hoboken ). 2014;297:161-173
784. Markert CL. Neoplasia: a disease of cell differentiation. Cancer Res. 1968;28:1908-1914
785. O'Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T. et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180-187
786. Smalley M, Ashworth A. Stem cells and breast cancer: A field in transit. Nat Rev Cancer. 2003;3:832-844
787. van Staveren WC, Solis DY, Hebrant A, Detours V, Dumont JE, Maenhaut C. Human cancer cell lines: Experimental models for cancer cells in situ? For cancer stem cells? Biochim Biophys Acta. 2009;1795:92-103
788. Brinckerhoff CE. Cancer Stem Cells (CSCs) in melanoma: There's smoke, but is there fire? J Cell Physiol. 2017;232:2674-2678
789. Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36(Suppl 1):59-72
790. Carruba G, Trosko JE. The Long Evolutionary Journey of Cancer from Ancestor to Modern Humans. Crit Rev Oncog. 2017;22:323-352
791. Waring RH, Harris RM, Mitchell SC. In utero exposure to carcinogens: Epigenetics, developmental disruption and consequences in later life. Maturitas. 2016;86:59-63
792. Lemercier C, To RQ, Swanson BJ, Lyons GE, Konieczny SF. Mist1: a novel basic helix-loop-helix transcription factor exhibits a developmentally regulated expression pattern. Dev Biol. 1997;182:101-113
793. Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R. et al. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006;66:242-247
794. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954-961
795. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737
796. Sutherland HJ, Blair A, Zapf RW. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood. 1996;87:4754-4761
797. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-648
798. Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111-1115
799. Chiodi I, Belgiovine C, Dona F, Scovassi AI, Mondello C. Drug treatment of cancer cell lines: a way to select for cancer stem cells? Cancers (Basel). 2011;3:1111-1128
800. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497-512
801. Chaffer CL, Weinberg RA. How does multistep tumorigenesis really proceed? Cancer Discov. 2015;5:22-24
802. Bakhshinyan D, Adile AA, Qazi MA, Singh M, Kameda-Smith MM, Yelle N. et al. Introduction to Cancer Stem Cells: Past, Present, and Future. Methods Mol Biol. 2018;1692:1-16
803. Mertins SD. Cancer stem cells: a systems biology view of their role in prognosis and therapy. Anticancer Drugs. 2014;25:353-367
804. Muinao T, Deka Boruah HP, Pal M. Diagnostic and Prognostic Biomarkers in ovarian cancer and the potential roles of cancer stem cells - An updated review. Exp Cell Res. 2018;362:1-10
805. Schwitalla S. Tumor cell plasticity: the challenge to catch a moving target. J Gastroenterol. 2014;49:618-627
806. Valle S, Martin-Hijano L, Alcala S, Alonso-Nocelo M, Sainz B Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers (Basel). 2018;10:pii E33. doi: 10.3390/cancers10020033
807. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267-284
808. FURTH J, Kahn MC. The transmission of leukemia of mice with a single cell. Am J Cancer. 1937;31:276-282
809. Kleinsmith LJ, Pierce GB Jr. multipotentiality of single embryonal carcinoma cells. Cancer res. 1964;24:1544-1551
810. van den Brenk HA. Effect of immunological attenuation on cell dosage required to establish single or double tumour homografts. Br J Cancer. 1961;15:798-803
811. Ishibashi K. Studies on the number of cells necessary for the transplantation of Yoshida sarcoma; transmission of the tumor with a single cell. Gan. 1950;41:1-14
812. Hewitt HB. Transplantation of mouse sarcoma with small numbers of single cells. Nature. 1952;170:622-623
813. Hewitt HB. Studies of the quantitative transplantation of mouse sarcoma. Br J Cancer. 1953;7:367-383
814. Visvader JE. Cells of origin in cancer. Nature. 2011;469:314-322
815. Antoniou A, Hebrant A, Dom G, Dumont JE, Maenhaut C. Cancer stem cells, a fuzzy evolving concept: a cell population or a cell property? Cell Cycle. 2013;12:3743-3748
816. Maenhaut C, Dumont JE, Roger PP, van Staveren WC. Cancer stem cells: a reality, a myth, a fuzzy concept or a misnomer? An analysis. Carcinogenesis. 2010;31:149-158
817. Floor S, van Staveren WC, Larsimont D, Dumont JE, Maenhaut C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: distinct, overlapping or same populations. Oncogene. 2011;30:4609-4621
818. Ksiazek K. Let's stop overlooking bacterial aging. Biogerontology. 2010;11:717-723
819. Gomez JM. Aging in bacteria, immortality or not-a critical review. Curr Aging Sci. 2010;3:198-218
820. Florea M. Aging and immortality in unicellular species. Mech Ageing Dev. 2017;167:5-15
821. Hallsworth JE. Stress-free microbes lack vitality. Fungal Biol. 2018;122:379-385
822. da Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, Munoz P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol. 2018;53:48-58
823. Groisman GM, Benkov KJ, Adsay V, Dische MR. Osseous metaplasia in benign colorectal polyps. Arch Pathol Lab Med. 1994;118:64-65
824. Zhang Y, Toy KA, Kleer CG. Metaplastic breast carcinomas are enriched in markers of tumor-initiating cells and epithelial to mesenchymal transition. Mod Pathol. 2012;25:178-184
825. Barnes PJ, Boutilier R, Chiasson D, Rayson D. Metaplastic breast carcinoma: clinical-pathologic characteristics and HER2/neu expression. Breast Cancer Res Treat. 2005;91:173-178
826. Catroppo JF, Lara JF. Metastatic metaplastic carcinoma of the breast (MCB): an uncharacteristic pattern of presentation with clinicopathologic correlation. Diagn Cytopathol. 2001;25:285-291
827. Chhieng C, Cranor M, Lesser ME, Rosen PP. Metaplastic carcinoma of the breast with osteocartilaginous heterologous elements. Am J Surg Pathol. 1998;22:188-194
828. Sasano H, Shizawa S, Nagura H, Yamaki T. Mucinous adenocarcinoma arising in a giant urachal cyst associated with pseudomyxoma peritonei and stromal osseous metaplasia. Pathol Int. 1997;47:502-505
829. Eble JN, Young RH. Carcinoma of the urinary bladder: a review of its diverse morphology. Semin Diagn Pathol. 1997;14:98-108
830. Haque S, Eisen RN, West AB. Heterotopic bone formation in the gastrointestinal tract. Arch Pathol Lab Med. 1996;120:666-670
831. Kirchhof N, Steinhauer D, Fey K. Equine adenocarcinomas of the large intestine with osseous metaplasia. J Comp Pathol. 1996;114:451-456
832. Bennett JH, Jones J, Speight PM. Odontogenic squamous cell carcinoma with osseous metaplasia. J Oral Pathol Med. 1993;22:286-288
833. Hanada M, Nakano K, Ii Y, Yamashita H. Carcinosarcoma of the esophagus with osseous and cartilagenous production. A combined study of keratin immunohistochemistry and electron microscopy. Acta Pathol Jpn. 1984;34:669-678
834. Caluori D, Gallo P. Case report of heterotopic bone formation in metastatic carcinoma of the colon. Tumori. 1979;65:345-351
835. Fox EJ, Prindle MJ, Loeb LA. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev. 2013;32:353-361
836. Miyahira AK, Den RB, Carlo MI, de LR, Hope TA, Karzai F. et al. Tumor cell heterogeneity and resistance; report from the 2018 Coffey-Holden Prostate Cancer Academy Meeting. Prostate. 2019;79:244-258
837. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018;20:1349-1360
838. Heppner GH, Shekhar M. Tumor heterogeneity is fundamental to the tumor ecosystem. Oncology (Williston Park). 2014;28:780-781
839. Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259-2265
840. Allison KH, Sledge GW. Heterogeneity and cancer. Oncology (Williston Park). 2014;28:772-778
841. Kusoglu A, Biray AC. Cancer stem cells: A brief review of the current status. Gene. 2019;681:80-85
842. Ben-David U, Beroukhim R, Golub TR. Genomic evolution of cancer models: perils and opportunities. Nat Rev Cancer. 2019;19:97-109
843. O'Conor CJ, Chen T, Gonzalez I, Cao D, Peng Y. Cancer stem cells in triple-negative breast cancer: a potential target and prognostic marker. Biomark Med. 2018;17:813-820
844. Condiotti R, Guo W, Ben-Porath I. Evolving views of breast cancer stem cells and their differentiation States. Crit Rev Oncog. 2014;19:337-348
845. Nakshatri H, Srour EF, Badve S. Breast cancer stem cells and intrinsic subtypes: controversies rage on. Curr Stem Cell Res Ther. 2009;4:50-60
846. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-3988
847. Lapinska K, Faria G, McGonagle S, Macumber KM, Heerboth S, Sarkar S. Cancer Progenitor Cells: The Result of an Epigenetic Event? Anticancer Res. 2018;38:1-6
848. Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S. et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. 2013;14:21087-21113
849. Sarkar S, Goldgar S, Byler S, Rosenthal S, Heerboth S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: sensitization of cancer cells by combination therapy. Epigenomics. 2013;5:87-94
850. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111
851. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18:669-680
852. Liao DZ, Porsch-Hallstrom I, Gustafsson JA, Blanck A. Persistent sex differences in growth control of early rat liver lesions are programmed during promotion in the resistant hepatocyte model. Hepatology. 1996;23:835-839
Author contact
![]() Corresponding authors: Xixi Dou, Shandong Freda Pharmaceutical Group Co., Ltd., Jinan 250101, Shandong Province, P.R. China; Email: doxxcom. Pingzhen Tong, Department of Pathology, The Second Hospital, Guizhou University of Traditional Chinese Medicine, Guiyang 550001, Guizhou Province, P.R. China; Email: 2376554com. Hai Huang, Center for Clinical Laboratories, The Affiliated Hospital of Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China; Email: huanghai828edu.cn. Joshua Liao, Department of Pathology, the Second Hospital, Guizhou University of Traditional Chinese Medicine, Guiyang 550001, Guizhou Province, P.R. China; Email: djliaoedu.cn.
Corresponding authors: Xixi Dou, Shandong Freda Pharmaceutical Group Co., Ltd., Jinan 250101, Shandong Province, P.R. China; Email: doxxcom. Pingzhen Tong, Department of Pathology, The Second Hospital, Guizhou University of Traditional Chinese Medicine, Guiyang 550001, Guizhou Province, P.R. China; Email: 2376554com. Hai Huang, Center for Clinical Laboratories, The Affiliated Hospital of Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China; Email: huanghai828edu.cn. Joshua Liao, Department of Pathology, the Second Hospital, Guizhou University of Traditional Chinese Medicine, Guiyang 550001, Guizhou Province, P.R. China; Email: djliaoedu.cn.