Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(9):2645-2655. doi:10.7150/jca.40726 This issue Cite
Research Paper
Transcriptomic analysis reveals the oncogenic role of S6K1 in hepatocellular carcinoma
Keng Po Lai1,2 ![]() , Angela Cheung2, Cheuk Hin Ho2, Nathan Yi-Kan Tam2, Jing Woei Li2, Xiao Lin3, Ting Fung Chan3,4, Nikki Pui-Yue Lee5, Rong Li1
, Angela Cheung2, Cheuk Hin Ho2, Nathan Yi-Kan Tam2, Jing Woei Li2, Xiao Lin3, Ting Fung Chan3,4, Nikki Pui-Yue Lee5, Rong Li1
1. Guanxi Key Laboratory of Tumor Immunology and Microenvironmental Regulation, Guilin Medical University, Guilin, PR China;
2. Department of Chemistry, City University of Hong Kong, Hong Kong SAR, China;
3. School of Life Sciences, The Chinese University of Hong Kong, Hong Kong SAR, China;
4. State Key Laboratory of Agrobiotechnology, Chinese University of Hong Kong, Hong Kong SAR, China;
5. Department of Surgery, University of Hong Kong, Hong Kong SAR, China.
Received 2019-11-22; Accepted 2020-1-19; Published 2020-2-19
Abstract
The p70 ribosomal protein S6 kinase 1 (S6K1), a serine/threonine kinase, is commonly overexpressed in a variety of cancers. However, its expression level and functional roles in hepatocellular carcinoma (HCC), which ranks as the third leading cause of cancer-related death worldwide, is still largely unknown. In the current report, we show the in vivo and in vitro overexpression of S6K1 in HCC. In the functional analysis, we demonstrate that S6K1 is required for the proliferation and colony formation abilities in HCC. By using comparative transcriptomic analysis followed by gene ontology enrichment analysis and Ingenuity Pathway Analysis, we find that the depletion of S6K1 can elevate the expression of a cluster of apoptotic genes, tumor suppressor genes and immune responsive genes. Moreover, the knockdown of S6K1 is predicted to reduce the tumorigenicity of HCC through the regulation of hubs of genes including STAT1, HDAC4, CEBPA and ONECUT1. In conclusion, we demonstrate the oncogenic role of S6K1 in HCC, suggesting the possible use of S6K1 as a therapeutic target for HCC treatment.
Keywords: S6K1, hepatocellular carcinoma, proliferation, tumorigenicity, transcriptome
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies, ranking as the third leading cause of cancer-related death worldwide. The main reason for the high mortality rate is due to its poor prognosis and the lack of satisfactory curative pharmacological treatment [1]. Sorafenib, a small multi-kinase inhibitor, is the only FDA approved drug-based therapy for advanced HCC [2]. Therefore, it is necessary to identify an alternative approach for HCC treatment. It has been reported that a number of cell signaling pathways such as NF-κB, TAK, ZFX, PI3K, PKB and GSK-3β play important roles in the tumorigenicity of HCC [3-7]. A better understanding on the mechanism underlying the cell signaling which regulates the development and growth of HCC may help us to develop a novel molecular therapeutic target for HCC treatment.
PI3K-Akt-mTOR pathway is a major tumor-initiating pathway in hepatocellular carcinoma, and is elevated in up to 50% of tumors [8]. The p70 ribosomal protein S6 kinase 1 (S6K1), a serine/threonine kinase, is a downstream effector of the mTOR pathway. S6K1 has been reported to be commonly overexpressed in a variety of cancers such as ovarian cancer, prostate cancer and leukemia [9-11]. Additionally, we have previously demonstrated that S6K1 acts as a multifaceted regulator of the Mdm2-p53 pathway [12]. Also, p53 is important key mediator which controls cell proliferation, apoptosis and cell cycle in HCC [13-14], suggesting that S6K1 might also play an important functional role in the development of HCC. In this report, we showed that S6K1 is commonly overexpressed in HCC both in vivo and in vitro. In addition, we depleted the expression of S6K1 in three HCC cell lines including Hep3B, PLC5 and HepG2, followed by functional and transcriptomic analyses, to understand the role of S6K1 in HCC and the mechanisms underlying the hepatocarcinogenicity of S6K1, respectively. We showed that the knockdown of S6K1 could reduce the cell proliferation and colony formation abilities of HCC cells. In the comparative transcriptomic analysis, we demonstrated that the depletion of S6K1 recused the expression of tumor suppressor genes and cell apoptotic genes. The Ingenuity Pathways Analysis (IPA) further highlighted that the depletion of S6K1 could reduce the tumorigenicity of HCC through the regulation of gene cluster including STAT1, HDAC4, CEBPA and ONECUT1. In this study, the identification and functional characterization of S6K1 in HCC provides new insights into the tumorigenic role of S6K1 in HCC and the possible use of S6K1 as a therapeutic target for HCC treatment.
Materials and Methods
Patient sample collection
Retrospectively collected clinical specimens were used in this study. Patient consents have been obtained. IRB has approved on the use of clinical specimens for research. Tissue specimens were preserved immediately in RNAlater (for RNA extraction) or fixed in formalin for preparation of paraffin blocks. The collection and processing of clinical specimens were performed by Surgical Tissue Bank staff in Department of Surgery, The University of Hong Kong. Patients treated with tumor resection in Queen Mary Hospital were included. Patients treated with neoadjuvant or adjuvant treatment were excluded. Clinico-pathological parameters required for this study are available from Surgical Tissue Bank.
Cell culture
The human HCC cell lines Hep3B, PLC5 and HepG2 were cultured in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum. The cells were supplied with 95% of air and 5% of carbon dioxide (CO2), and were kept at 37°C.
Lentiviral packaging and transduction
HEK293FT cells were transfected with packaging plasmids pCMV-VSV-G, pRSV-Rev and pMDLg/pRRE along with MISSION® non-target shRNA plasmid or MISSION® shS6K1 plasmid (Sigma) using the Lipofectamine 2000 transfection reagent (Intvitrogen). After 48 hours post-transfection, the viral supernatant was precipitated with PEG-it Virus Precipitation Solution at a 1:4 ratio to produce a concentrated viral stock. Hep3B, PLC5 and HepG2 were plated 1 day before transduction. After 48 hr post-transduction, cells were selected in medium containing 2 µg/ml puromycin for 10 days. The knockdown efficiency of S6K1 was examined using qPCR and Western Blotting.
cDNA synthesis and quantitative RT-PCR (qRT-PCR)
RNA was isolated from the cell lines using Trizol reagent (Invitrogen) according to the manufacturer's instruction. For each sample, 1 µg of total RNA was subjected to first strand cDNA synthesis using SuperScript® VILO™ cDNA Synthesis Kit (Thermo Fisher). The expression levels of the target genes were determined using the KAPA SYBR FAST qPCR kit. The primer sequences were listed in Supplementary Table S1. To adjust for variations in starting template, gene expression was normalized against GAPDH.
Western Blot Analysis
50 µg of protein was resolved on 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrophoretically onto a polyvinylidene fluoride membrane. The blots were incubated with specified antibodies against S6K1 (1:1000; Cell Signaling), or beta-actin (1:10000; Chemicon). After incubation with secondary antibody conjugated to horseradish peroxidase, the results were visualized using enhanced chemiluminescence detection (GE Healthcare, Piscataway, NJ).
Cell proliferation assays
Stable transfected HCC cells were seeded in a 96-well plate at a cell density of 3 x 103 cells per well, with 8 replicate wells. The cells were allowed to incubate for 1, 3 and 7 days. After the incubation, cell proliferation was measured by the WST-1 assay (Sigma). The colorimetric product formed was measured at an absorbance of 570nm.
Colony formation assay
In order to determine the role of S6K1 in HCC tumorigenicity, colony formation was conducted as previously described [3]. Briefly, stable transfected HCC cells were seeded in a 6-well plate (200 cells per well). After an incubation period of 14 days, the cells were stained with 1% crystal violet (in 100% methanol) for 15 min, followed by detaining with water. Colonies (more than 20 cells/colony) were counted.
Comparative transcriptome analysis
Total RNA was isolated from the stable transfected cells (shCtrl and shS6K1) from the Hep3B and PLC5 cell lines using the mirVanaTM RNA isolation kit (Applied Biosystems). The samples with a RNA Integrity Number (RIN) greater than 8 were used for RNA library construction. The RNA library was sequenced by the Beijing Genomics Institute (Wuhan, China). Single-end reads with a 50 bp read-length were sequenced on the BGISEQ-500RS sequencer. Sequence-reads were in turn dynamically trimmed according to BWA's - q algorithm. All downstream analyses were based on quality-trimmed reads [15,16]. Quality-trimmed sequence reads were mapped to the Human genome reference hg19 using STAR v2.3.0e. Read-counts of genes were quantified against Gencode v19 using HTSeq-count (v0.6.0). Read-count data were then subjected to differential expression analysis using the edgeR package [17]. Genes with a |log2 (fold change: shS6K1/shCtrl)| > 0.6 were considered as differentially expressed genes (DEGs).
Statistical Analysis
Quantitative data was described as the means with standard deviation (SD). Wilcoxon matched-pairs signed rank test was used to compare expression level difference of S6K1 between studied groups. In the functional study, the effect of S6K1 was compared using the paired Student t-test. A p value < 0.05 was considered as statistically significant. All statistical analyses were performed using GraphPad Prism 3.02 (GraphPad Software Inc., San Diego, CA).
Results
S6K1 is commonly overexpressed in human HCC
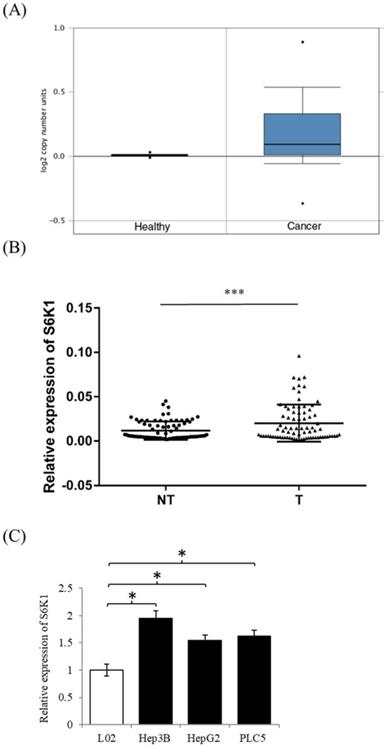
In order to examine the expression level of S6K1 in HCC, we firstly used the publicly available datasets from Oncomine(available at: https://www.oncomine.org) to survey DNA copy-number variation of S6K1 in HCC. There was a significant amplification in the copy-number-gain of S6K1 in HCC as compared to normal liver tissue (Figure 1A). We then conducted qPCR analysis to determine the expression of S6K1 in a cohort of 80 HCC tumors (T) and paired adjacent non-tumoral livers (NT). Our results demonstrated a significant induction of S6K1 in HCC tumors (42.5%), as compared with their adjacent nonmalignant livers (log10 T/NT > 0.2, p = 0.0001) (Figure 1B). We also attempted to correlate the expression of S6K1 in HCC tumors with the clinicopathological features of patients. But no significant correlation was observed (Supplementary Table S2). Additionally, we demonstrated that HCC cell lines (Hep3B, HepG2 and PLC5) had elevated S6K1 expression as compared to the normal liver cell line L02 (Figure 1C). Taken together, our results showed that S6K1 is commonly overexpressed in HCC both in vivo and in vitro.
In vivo and in vitro overexpression of S6K1 in HCC. (A) copy-number-gain of S6K1 in human hepatocellular carcinoma. DNA copy-number variation of TCGA data is obtained from Oncomine (https://www.oncomine.org). (B) Overexpression of S6K1 transcript in a cohort of 80 HCC tumors (T) and paired adjacent non-tumoral livers (NT). (C) Upregulation of S6K1 in HCC cell lines, Hep3B, PLC5 and HepG2 as compared to the normal liver cell line L02. The mRNA expression level of S6K1 was determined by qRT-PCR. Data are presented as the mean ± SD values (***p = 0.0001, *p < 0.05).
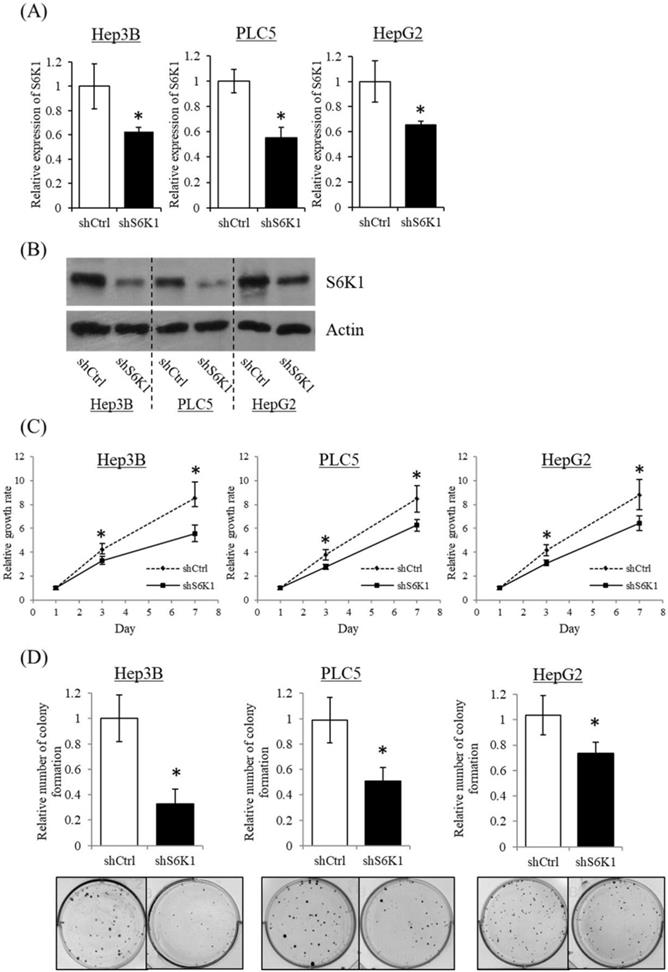
Depletion of S6K1 suppresses HCC cell proliferation
To gain insight into the functional role of increased S6K1 expression in HCC, three HCC cell lines (Hep3B, HepG2 and PLC5) showing elevated S6K1 expression were used to establish S6K1-deficient stable clones (shS6K1) by lentiviral transfection. The knockdown efficiency of S6K1 was determined by qRT-PCR (Figure 2A) and Western blot (Figure 2B). Our results demonstrated the successful depletion of S6K1 in these 3 HCC cell lines. In the functional analysis, the deficiency of S6K1 significantly reduced the in vitro proliferation of HCC in the WST-1 assay (Figure 2C). In addition, smaller-sized and smaller numbers of colonies were formed in S6K1-depleted cells in the colony formation assay (Figure 2D). Collectively, our results suggested that S6K1 is required for the proliferation and growth of HCC.
Depletion of S6K1 suppresses in vitro proliferation of HCC cells. (A) Knockdown of S6K1 in HCC cell lines. qRT-PCR and western blotting demonstrated the knockdown efficiency of S6K1 in S6K1-depleted Hep3B, PLC5 and HepG2. (B-C) Depletion of S6K1 suppressed in vitro cell proliferation of HCC. The cell proliferation of S6K1-depleted cell line was determined by (B) WST-1 assay and (C) colony formation assay. Data are presented as the mean ± s.e.m. values (*p < 0.05).
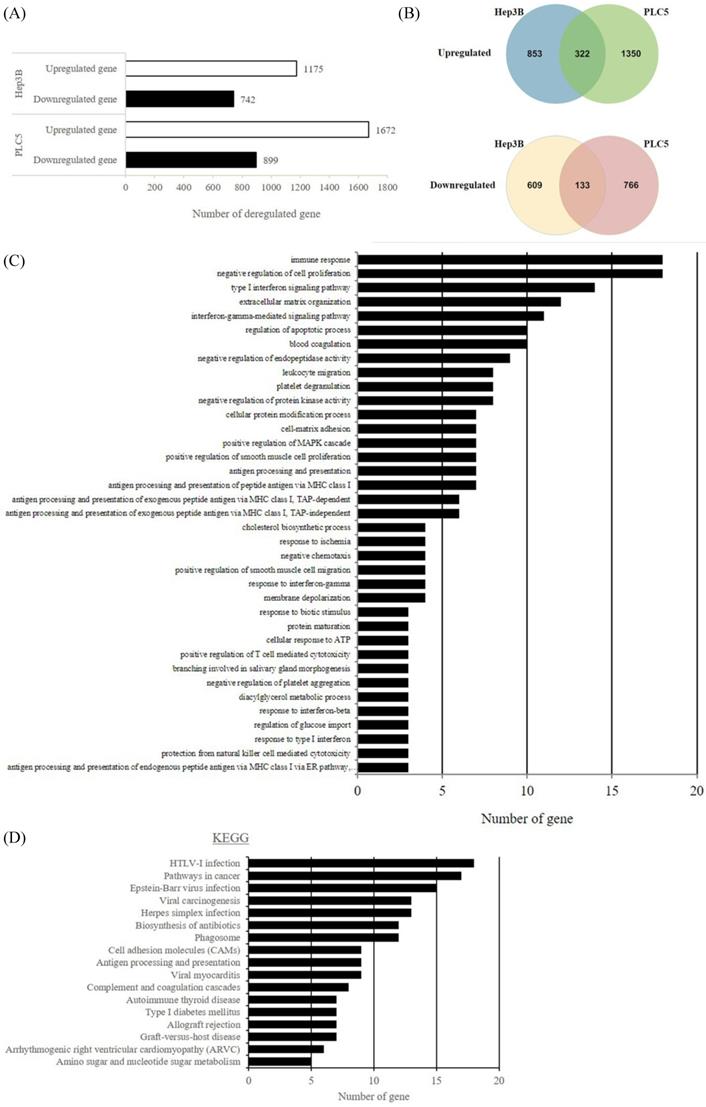
S6K1 is required for the expression of apoptotic and tumor suppressor genes in HCC
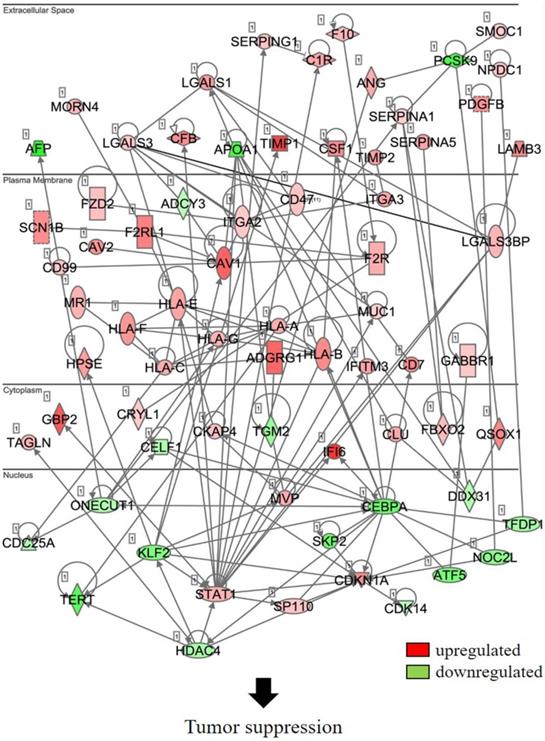
In an attempt to understand the mechanism underlying the hepatocarcinogenicity of S6K1, comparative transcriptomic analysis (shCtrl vs shS6K1) was conducted on the Hep3B and PLC5 cells. 1917 and 2571 differentially expressed genes (DEGs) were found in Hep3B (Figure 3A, Supplementary Table S3) and PLC5 (Figure 3A, Supplementary Table S4), respectively after the depletion of S6K1. Of these genes, 322 upregulated and 133 downregulated genes were shared in both Hep3B and PLC5 with S6K1 knockdown (Figure 3B, Supplementary Table S5). The gene set enrichment analysis on these 455 DEGs using the DAVID tool highlighted 37 biological processes that were controlled by S6K1 (p < 0.05) (Figure 3C, Supplementary Table S6). It included immune response, antigen processing, interferon-related cell signaling pathways, negative regulation of cell proliferation, and regulation of apoptotic process. We also found that S6K1 is required for the regulation of the apoptotic process through a cluster of genes such as TP53I3, DAPK2, SKP2 and SERBP1 in HCC cells (Table 1). In addition, the depletion of S6K1 would stimulate the immune response by overexpressing the major histocompatibility complex (MHC) class I cell surface receptor family such as human leukocyte antigen HLA-A, HLA-B and HLA-C. KEGG pathway analysis further highlighted the role of S6K1 in cancer progression (Figure 3D, Supplementary Table S7), in which the depletion of S6K1 could re-express a group of tumor suppressor genes including RASSF5, RASSF6, STAT1, CEACAM1, BNIP3L and SOCS2 (Table 1). More importantly, the results of the Ingenuity Pathway Analysis (IPA) in diseases or functions annotation demonstrated that the depletion of SK61 could reduce the tumorigenicity of HCC through the control of hubs of gene including STAT1, HDAC4, CEBPA and ONECUT1 (Figure 4), which is reflected by the inactivation of several cancers including liver tumor (Figure 5 and Table 2, Supplementary Table S8). Taken together, our results suggested the anti-apoptotic and oncogenic role of S6K1 in the HCC.
Depletion of S6K1 reduces the proliferation and regains the apoptosis in HCC. (A) Gene-annotation enrichment analysis using The Database for Annotation, Visualization and Integrated Discovery (DAVID). Biological processes with p < 0.05 were considered statistically significant. (B) KEGG pathway analysis using DAVID.
S6K1 controlled apoptotic and tumor suppressor genes in HCC.
| Gene symbol | Gene name | Reference |
|---|---|---|
| RASSF5 | Ras Association Domain Family Member 5 | 41,42 |
| RASSF6 | Ras Association Domain Family Member 6 | 43-45 |
| TP53I3 | Tumor Protein P53 Inducible Protein 3 | 38,46-48 |
| LGALS1 | Galectin 1 | 49-51 |
| BNIP3L | BCL2 Interacting Protein 3 Like | 52-55 |
| NDRG1 | N-Myc Downstream Regulated 1 | 56-59 |
| DAPK2 | Death Associated Protein Kinase 2 | 60-63 |
| STAT1 | Signal Transducer And Activator Of Transcription 1 | 64-66 |
| SERBP1 | SERPINE1 MRNA Binding Protein 1 | 34,67 |
| SKP2 | S-Phase Kinase-Associated Protein 2 | 32,68-70 |
| CEACAM1 | carcinoembryonic antigen related cell adhesion molecule 1 | 68-73 |
| SOCS2 | suppressor of cytokine signaling 2 | 74-76 |
Depletion of S6K1 regulates the gene network to suppress the tumor development.
Schematic diagram to summarize the oncogenic role of S6K1 in HCC.
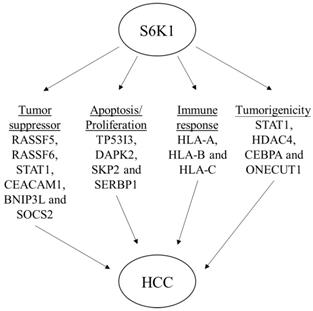
Depletion of S6K1 reduced the tumorigenicity of HCC.
| Diseases or Functions Annotation | p-value | Predicted Activation State | Activation z-score |
|---|---|---|---|
| lymphocytic cancer | 0.00000862 | Decreased | -2.183 |
| breast or colorectal cancer | 0.0000549 | Decreased | -2.18 |
| endocrine gland tumor | 0.000176 | Decreased | -2.219 |
| neuroendocrine tumor | 0.000196 | Decreased | -2.219 |
| lymphoid cancer | 0.000291 | Decreased | -2.183 |
| breast cancer | 0.000916 | Decreased | -2.415 |
| breast or ovarian cancer | 0.00104 | Decreased | -2.415 |
| lymphoproliferative malignancy | 0.00126 | Decreased | -2.371 |
| liver tumor | 0.00264 | Decreased | -2.109 |
Validation of transcriptome sequencing using qPCR analysis
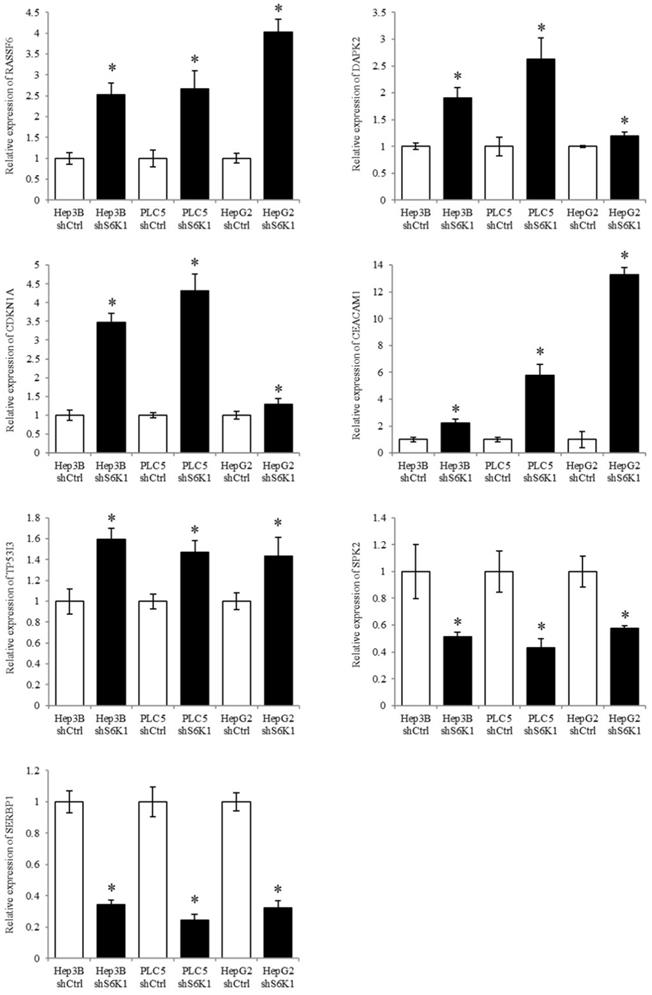
S6K1 controlled tumor suppressor, cell proliferative and cell apoptotic genes (RASSF6, DAPK2, CDKN1A, CEACAM1, TP53I3, SPK2, and SERBP1) were selected to be validated in the Hep3B, PLC5 and HepG2 cell lines using qPCR analysis. The result of qPCR analysis agreed with the transcriptome sequencing data that RASSF6, DAPK2, CDKN1A, CEACAM1 and TP53I3 were induced in the S6K1-depleted HCC cell lines, and SPK2 and SERBP1 were suppressed under the S6K1 knockdown (Figure 6).
Quantitative PCR analysis validates the result of transcriptome sequencing. The expression of tumor suppressor genes, apoptotic genes and cell proliferative genes were determined in S6K1-depleted HCC cell lines, as compared to shCtrl cells using qPCR. Data are presented as the mean ± s.e.m. values (*p < 0.05) (n = 8).
Discussion
p70 ribosomal protein S6 kinase 1 (S6K1), an important downstream kinase of Akt-mTOR pathway, has been reported to play roles including tumor growth, chemoresistance and self-renewal ability in numerous of cancers such as leukemia, bladder cancer, colorectal carcinomas, neuroblastoma and breast cancer [9, 18-20]. But its expression level and functional role in hepatocellular carcinoma (HCC) is still largely unknown. Here, we retrieved the data from Oncomine [21], a cancer microarray database and web-based data-mining platform of genome-wide expression, to show the copy number gain of S6K1 in HCC. Further clinic analysis demonstrated the overexpression of S6K1 transcript in patients with HCC. These results suggested that the upregulation of S6K1 expression in HCC may be due to the chromosomal aberration in genomic level. DNA copy number changes are commonly found in HCC. For instance, microarray-based comparative genomic hybridization (array CGH) analysis has demonstrated the amplification of chromosome 1q21 - q22 and 8q24 regions in HCC which is associated with the promotion of cell motility and epithelial-to-mesenchymal transition in HCC, respectively [22, 23].
Then, we conducted functional and mechanistic characterization of S6K1 in 3 HCC cell lines including Hep3B, HepG2 and PLC5, which covered HCC with different cell properties and ethnic origins [24]. HepG2 is hepatitis B virus negative and non-tumorigenic, but Hep3B is hepatitis B virus positive and tumorigenic [25, 26]. PLC5 is also hepatitis virus B [27]. In the loss-of-functional analysis, we showed the depletion of S6K1 could reduce the proliferation of HCC cells, suggesting that S6K1 is necessary for the growth of HCC. In the mechanistic analysis, we performed comparative transcriptome sequencing to dissect gene networks and pathways related to the tumorigenicity of HCC which are controlled by S6K1. Interestingly, the depletion of S6K1 could alter a number of immune responses such as the activation of MHC class I cell surface receptor members, and the alteration of antigen processing and interferon signaling. The result of this finding suggested a possible use of S6K1 inhibitor together with bevacizumab (anti-VEGF antibody) for regulating immune response against HCC and immunotherapeutic approaches for HCC [28, 29].
Due to the importance of S6K1 in the growth of HCC, we then further looked at the pathways and gene clusters related to cell apoptosis and proliferation in the DVAID analysis. In which, we discovered 2 clusters of gene were controlled by S6K1; one is related to cell apoptosis or cell proliferation including TP53I3, DAPK2, SKP2 and SERBP1. The other cluster including RASSF5, RASSF6, STAT1, CEACAM1, BNIP3L and SOCS2 is related to tumor suppression. S-phase kinase-associated protein 2 (SKP2), forms a feedback loop with p21 and p27 that control cell cycle entry and G1/S transition [26]. It has been reported that the inhibition of SKP2 resulted in cell cycle arrest in HCC [27, 28]. Serpine mRNA binding protein 1 (SERBP1) is a protein that binds to the plasminogen activator inhibitor type 1 (PAI-1) mRNA, leading to the regulation of plasminogen activation [29]. Previous report showed that the overexpression of SERBP1 in human breast cancer is correlated with favorable prognosis [30]. In addition, the activation of SERBP1 promotes cell proliferation, migration, and invasion in prostate cancer [31]. So the suppression of SERBP1 in S6K1 depletion cells may result in the decrease in tumorigenicity of HCC. Besides, the S6K1 knockdown could re-express a group of apoptotic genes. For instance, tumor protein p53 inducible protein 3 (TP53I3), a downstream molecule of the tumor suppressor p53, is reported to activate apoptosis in many cancers such as glioblastoma, papillary thyroid cancer and lung adenoma [32-34]. In addition, the re-expression of DAPK2 may be able to reduce the drug resistance of HCC [35, 36].
By using both functional characterization and comparative transcriptomic analysis, we have demonstrated the importance of S6K1 in the proliferation and tumorigenicity of HCC. The discovered regulatory role of S6K1 on the apoptotic and tumor suppressive gene clusters further support that S6K1 is a potential functional target for the application in HCC therapeutics.
Supplementary Material
Supplementary tables.
Acknowledgements
Keng Po Lai is supported by Hong Kong SAR, Macao SAR and Taiwan Province Talent Young Scientist Program of Guangxi.
Funding
This work is partly supported by the Start-up Grant, City University of Hong Kong (7200499) for KPL and National Natural Science Foundation of China for RL (81560134).
Authors' contributions
AC and CHH carried out sample preparation and functional analysis. NPYL and RL participated in the clinic data analysis. JWL carried out the sequencing data analysis and drafted the manuscript. TFC participated in the design of the study and performed the statistical analysis. KPL participated in experimental design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Retrospectively collected clinical specimens were used in this study. Patient consents have been obtained. IRB has approved on the use of clinical specimens for research. The collection and processing of clinical specimens were performed by Surgical Tissue Bank staff in Department of Surgery, The University of Hong Kong.
Availability of data and material
The sequence data from this study have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under the accession number SRP096313.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liao H, Huang Y, Guo B. et al. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res. 2014;5:125-139
2. Giannelli G, Rani B, Dituri F. et al. Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut. 2014;63:1668-1676
3. Lai KP, Chen J, He M. et al. Overexpression of ZFX confers self-renewal and chemoresistance properties in hepatocellular carcinoma. Int J Cancer. 2014;135:1790-1799
4. Liu L, Dai Y, Chen J. et al. Maelstrom promotes hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition by way of Akt/GSK-3beta/Snail signaling. Hepatology. 2014;59:531-543
5. Ma S, Lee TK, Zheng BJ. et al. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749-1758
6. Zhao N, Wang R, Zhou L. et al. MicroRNA-26b suppresses the NF-kappaB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol Cancer. 2014;13:35
7. Zhou Q, Lui VW, Yeo W. Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol. 2011;7:1149-1167
8. Bhat M, Sonenberg N, Gores G. The mTOR Pathway in Hepatic Malignancies. Hepatology. 2013;58:810-818
9. Ghosh J, Kobayashi M, Ramdas B. et al. S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J Clin Invest. 2016;126:2621-2625
10. Hsiao YH, Yu TH, Chia YH. et al. PYK2 via S6K1 Regulates the Function of Androgen Receptors and the Growth of Prostate Cancer Cells. Endocr Relat Cancer. 2016;23:651-663
11. Lam SSN, Ip CKM, Mak ASC. et al. A novel p70 S6 kinase-microRNA biogenesis axis mediates multicellular spheroid formation in ovarian cancer progression. Oncotarget. 2016;7:38064-38077
12. Lai KP, Leong WF, Chau JFL. et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010;29:2994-3006
13. Hussain SP, Schwank J, Staib F. et al. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166-2176
14. Meng X, Franklin DA, Dong J. et al. MDM2-p53 Pathway in Hepatocellular Carcinoma. Cancer Res. 2014;74:7161-7167
15. Lai KP, Li JW, Wang SY. et al. Tissue-specific transcriptome assemblies of the marine medaka Oryzias melastigma and comparative analysis with the freshwater medaka Oryzias latipes. BMC Genomics. 2015;16:135
16. Li JW, Lai KP, Ching AK. et al. Transcriptome sequencing of Chinese and Caucasian population identifies ethnic-associated differential transcript abundance of heterogeneous nuclear ribonucleoprotein K (hnRNPK). Genomics. 2013;103:56-64
17. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-140
18. Chen Y, Wang S, Zhang L. et al. Identification of ULK1 as a novel biomarker involved in miR-4487 and miR-595 regulation in neuroblastoma SH-SY5Y cell autophagy. Sci Rep. 2015;5:11035
19. Karlsson E, Magić I, Bostner J. et al. Revealing different roles of the mTOR-targets S6K1 and S6K2 in breast cancer by expression profiling and structural analysis. PloS One. 2015;10:e0145013
20. Yuge K, Kikuchi E, Hagiwara M. et al. Nicotine induces tumor growth and chemoresistance through activation of the PI3K/Akt/mTOR pathway in bladder cancer. Mol Cancer Ther. 2015;14:2112-2120
21. Nakayama S, Matsushita A, Ichiba S. et al. [Clinical evaluation of traditional Chinese medicine for chronic idiopathic thrombocytopenic purpura]. Rinsho ketsueki. 1992;33:157-161
22. Cheng IK, Tsang BC, Lai KP. et al. GEF-H1 over-expression in hepatocellular carcinoma promotes cell motility via activation of RhoA signalling. J Pathol. 2012;228:575-585
23. Chung KY, Cheng IKC, Ching AKK. et al. Block of proliferation 1 (BOP1) plays an oncogenic role in hepatocellular carcinoma by promoting epithelial-to-mesenchymal transition. Hepatology. 2011;54:307-318
24. Qiu GH, Xie X, Xu F. et al. Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology. 2015;67:1-12
25. Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209:497-499
26. Knasmüller S, Parzefall W, Sanyal R. et al. Use of metabolically competent human hepatoma cells for the detection of mutagens and antimutagens. Mutat Res. 1998;402:185-202
27. MacNab GM, Alexander JJ, Lecatsas G. et al. Hepatitis B surface antigen produced by a human hepatoma cell line. Br J Cancer. 1976;34:509-515
28. Tsuchiya N, Sawada Y, Endo I. et al. Potentiality of immunotherapy against hepatocellular carcinoma. World J Gastroenterol. 2015;21:10314-10326
29. Hato T, Zhu AX, Duda DG. Rationally combining anti-VEGF therapy with checkpoint inhibitors in hepatocellular carcinoma. Immunotherapy. 2016;8:299-313
30. Barr AR, Cooper S, Heldt FS. et al. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8:14728
31. Liao YJ, Bai HY, Li ZH. et al. Longikaurin A, a natural ent-kaurane, induces G2/M phase arrest via downregulation of Skp2 and apoptosis induction through ROS/JNK/c-Jun pathway in hepatocellular carcinoma cells. Cell Death Dis. 2014;5:e1137
32. Wang ST, Ho HJ, Lin JT. et al. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8:e2626
33. Koensgen D, Mustea A, Klaman I. et al. Expression analysis and RNA localization of PAI-RBP1 (SERBP1) in epithelial ovarian cancer: association with tumor progression. Gynecol Oncol. 2007;107:266-273
34. Serce NB, Boesl A, Klaman I. et al. Overexpression of SERBP1 (Plasminogen activator inhibitor 1 RNA binding protein) in human breast cancer is correlated with favourable prognosis. BMC Cancer. 2012;12:597
35. Guo K, Zheng S, Xu Y. et al. Loss of miR-26a-5p promotes proliferation, migration, and invasion in prostate cancer through negatively regulating SERBP1. Tumor Biol. 2016;37:12843-12854
36. Jiang Y, Shan S, Chi L. et al. Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumor Biol. 2016;37:3785-3795
37. Quan J, Li Y, Jin M. et al. Suppression of p53-inducible gene 3 is significant for glioblastoma progression and predicts poor patient prognosis. Tumour Biol. 2017;39:1010428317694572
38. Xu J, Cai J, Jin X. et al. PIG3 plays an oncogenic role in papillary thyroid cancer by activating the PI3K/AKT/PTEN pathway. Oncol Rep. 2015;34:1424-1430
39. Isshiki K, Hirase T, Matsuda S. et al. Death-associated protein kinase 2 mediates nocodazole-induced apoptosis through interaction with tubulin. Biochem Biophys Res Commun. 2015;468:113-118
40. Su CM, Wang MY, Hong CC. et al. miR-520h is crucial for DAPK2 regulation and breast cancer progression. Oncogene. 2016;35:1134-1142
41. Park J, Kang SI, Lee SY. et al. Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J Biol Chem. 2010;285:35029-35038
42. Zhou XH, Yang CQ, Zhang CL. et al. RASSF5 inhibits growth and invasion and induces apoptosis in osteosarcoma cells through activation of MST1/LATS1 signaling. Oncol Rep. 2014;32:1505-1512
43. Chen E, Yang F, He H. et al. Decreased level of RASSF6 in sporadic colorectal cancer and its anti-tumor effects both in vitro and in vivo. Oncotarget. 2016;7:19813-19823
44. Ikeda M, Hirabayashi S, Fujiwara N. et al. Ras-association domain family protein 6 induces apoptosis via both caspase-dependent and caspase-independent pathways. Exp Cell Res. 2007;313:1484-1495
45. Liang YY, Zheng LS, Wu YZ. et al. RASSF6 promotes p21 (Cip1/Waf1)-dependent cell cycle arrest and apoptosis through activation of the JNK/SAPK pathway in clear cell renal cell carcinoma. Cell Cycle. 2014;13:1440-1449
46. Ilienko IN, Bazyka DA. Overexpression of TP53, TP53I3 and BIRC5, alterations of gene regulation of apoptosis and aging of human immune cells in a remote period after radiation exposure. Probl Radiac Med Radiobiol. 2016;21:238-246
47. Wang H, Luo K, Tang LZ. et al. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. J Biol Chem. 2012;287:16890-16902
48. Zhang W, Luo J, Chen F. et al. BRCA1 regulates PIG3-mediated apoptosis in a p53-dependent manner. Oncotarget. 2015;6:7608-7618
49. Geiger P, Mayer B, Wiest I. et al. Binding of galectin-1 to breast cancer cells MCF7 induces apoptosis and inhibition of proliferation in vitro in a 2D- and 3D- cell culture model. BMC Cancer. 2016;16:870
50. Yan B, Zhao D, Yao Y. et al. Deguelin Induces the Apoptosis of Lung Squamous Cell Carcinoma Cells through Regulating the Expression of Galectin-1. Int J Biol Sci. 2016;12:850-860
51. You Y, Tan JX, Dai HS. et al. MiRNA-22 inhibits oncogene galectin-1 in hepatocellular carcinoma. Oncotarget. 2016;7:57099-57116
52. Chen JL, David J, Cook-Spaeth D. et al. Autophagy Induction Results in Enhanced Anoikis Resistance in Models of Peritoneal Disease. Mol Cancer Res. 2017;15:26-34
53. Pedanou VE, Gobeil S, Tabariès S. et al. The histone H3K9 demethylase KDM3A promotes anoikis by transcriptionally activating pro-apoptotic genes BNIP3 and BNIP3L. Elife. 2016;5:e16844
54. Roca-Alonso L, Castellano L, Mills A. et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015;6:e1754
55. Wilfinger N, Austin S, Scheiber-Mojdehkar B. et al. Novel p53-dependent anticancer strategy by targeting iron signaling and BNIP3L-induced mitophagy. Oncotarget. 2016;7:1242-1261
56. Liu W, Zhang B, Hu Q. et al. A new facet of NDRG1 in pancreatic ductal adenocarcinoma: Suppression of glycolytic metabolism. Int J Oncol. 2017;50:1792-1800
57. Mi L, Zhu F, Yang X. et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin-1 ubiquitylation in human colorectal cancer cells. Oncogene. 2017;36:4323-4335
58. Sharma A, Mendonca J, Ying J. et al. The prostate metastasis suppressor gene NDRG1 differentially regulates cell motility and invasion. Mol Oncol. 2017;11:655-669
59. Sun J, Zhang D, Bae DH. et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis. 2013;34:1943-1954
60. Tsai YT, Chuang MJ, Tang SH. et al. Novel Cancer Therapeutics with Allosteric Modulation of the Mitochondrial C-Raf-DAPK Complex by Raf Inhibitor Combination Therapy. Cancer Res. 2015;75:3568-3582
61. Yan LH, Chen ZN, Li L. et al. E2F-1 promotes DAPK2-induced anti-tumor immunity of gastric cancer cells by targeting miR-34a. Tumour Biol. 2016;37:15925-15936
62. Zhang J, Liu L, Sun Y. et al. MicroRNA-520g promotes epithelial ovarian cancer progression and chemoresistance via DAPK2 repression. Oncotarget. 2016;7:26516-26534
63. Zhao J, Zhao D, Poage GM. et al. Death-associated protein kinase 1 promotes growth of p53-mutant cancers. J Clin Invest. 2015;135:2707-2720
64. Ahn R, Sabourin V, Bolt AM. et al. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat Commun. 2017;8:14638
65. Wang H, Zhang Y, Yun H. et al. ERK expression and its correlation with STAT1 in esophageal squamous cell carcinoma. Oncotarget. 2017;8:45249-45258
66. Zhang X, Li X, Tan F. et al. STAT1 Inhibits miR-181a Expression to Suppress Colorectal Cancer Cell Proliferation through PTEN/Akt. J Cell Biochem. 2017;118:3435-3443
67. Kuo IY, Wu CC, Chang JM. et al. Low SOX17 expression is a prognostic factor and drives transcriptional dysregulation and esophageal cancer progression. Int J Cancer. 2014;35:563-573
68. Ohshima K, Hatakeyama K, Nagashima T. et al. Integrated analysis of gene expression and copy number identified potential cancer driver genes with amplification-dependent overexpression in 1,454 solid tumors. Sci Rep. 2017;7:641
69. Ruan D, He J, Li CF. et al. Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene. 2017;36:4299-4310
70. Watanabe A, Araki K, Yokobori T. et al. Stathmin 1 promotes the proliferation and malignant transformation of pancreatic intraductal papillary mucinous neoplasms. Oncol Lett. 2017;13:1783-1788
71. Ortenberg R, Galore-Haskel G, Greenberg I. et al. CEACAM1 promotes melanoma cell growth through Sox-2. Neoplasia. 2014;16:451-460
72. Shiokawa D, Sato A, Ohata H. et al. The Induction of Selected Wnt Target Genes by Tcf1 Mediates Generation of Tumorigenic Colon Stem Cells. Cell Rep. 2017;19:981-994
73. Wegwitz F, Lenfert E, Gerstel D. et al. CEACAM1 controls the EMT switch in murine mammary carcinoma in vitro and in vivo. Oncotarget. 2016;7:63730-63746
74. Das R, Gregory PA, Fernandes RC. et al. MicroRNA-194 Promotes Prostate Cancer Metastasis by Inhibiting SOCS2. Cancer Res. 2017;77:1021-1034
75. Vitali C, Bassani C, Chiodoni C. et al. SOCS2 Controls Proliferation and Stemness of Hematopoietic Cells under Stress Conditions and Its Deregulation Marks Unfavorable Acute Leukemias. Cancer Res. 2015;75:2387-2399
76. Wang X, Liu S, Zhou Z. et al. A herpes simplex virus type 2-encoded microRNA promotes tumor cell metastasis by targeting suppressor of cytokine signaling 2 in lung cancer. Tumour Biol. 2017;39:1010428317701633
Author contact
![]() Corresponding author: Prof. Keng Po Lai, Guanxi Key Laboratory of Tumor Immunology and Microenvironmental Regulation, Guilin Medical University, Guilin, PR China. Phone (+86)-773-5895881; email: kengplaiedu.hk
Corresponding author: Prof. Keng Po Lai, Guanxi Key Laboratory of Tumor Immunology and Microenvironmental Regulation, Guilin Medical University, Guilin, PR China. Phone (+86)-773-5895881; email: kengplaiedu.hk