Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(6):1403-1411. doi:10.7150/jca.31406 This issue Cite
Review
The Molecular Mechanism of Metabolic Remodeling in Lung Cancer
Ligong Chang*, Surong Fang*, Wei Gu ![]()
Department of Respiratory Medicine, Nanjing First Hospital, Nanjing Medical University. No. 68 Changle Road, Qinhuai District, Nanjing 210001,People's Republic of China.
*LC and SF contributed equally to the work.
Received 2018-11-11; Accepted 2019-10-23; Published 2020-1-13
Abstract
Metabolic remodeling is a key phenomenon in the occurrence and development of tumors. It not only offers materials and energy for the survival and proliferation of tumor cells, but also protects tumor cells so that they may survive, proliferate and transfer in the harsh microenvironment. This paper attempts to reveal the role of abnormal metabolism in the development of lung cancer by considering the processes of glycolysis and lipid metabolism, Identification of the molecules that are specifically used in the processes of glycolysis and lipid metabolism, and their underlying molecular mechanisms, is of great clinical and theoretical significance. We will focus on the recent progress in elucidating the molecular mechanism of metabolic remodeling in lung cancer.
Keywords: Lung cancer, Glycolysis, Lipid metabolism, Molecular mechanism
Introduction
Lung cancer (LC) is one of the most prevalent cancers and is the leading cause of cancer-related death worldwide. LC patients have low survival rates [1-2] with 56.2% of patients at stage III-IV at initial diagnosis; the five-year survival rate of LC is only 15% [3]. The poor prognosis of LC is due to the difficulty in early diagnosis and the current poor understanding of the mechanisms underlying LC. Metabolic remodeling has been widely accepted as the basis for novel tumor biomarkers [4]. Tumor cells, including LC cells, exhibit abnormal energy metabolism and significantly upregulated endogenous fatty acid metabolism. This upregulated metabolism, which is significantly different from the metabolism of normal cells, is called metabolic remodeling or reprograming. Metabolic remodeling takes place from the outset and throughout the development of LC, playing an important role in the evolution of LC [5].In this following sections we will explain metabolic remodeling and its importances from glycolysis metabolism and endogenous fatty acids metabolism in LC.
Active glucose metabolism in lung cancer
The significance of glycolysis
One century ago, Otto Warburg postulated that tumor cells still depend on glycolysis to produce adenosine triphosphate (ATP) when there is sufficient oxygen supply [6]. Warburg speculated that this apparent aerobic glycolysis (termed the Warburg effect) might be related to impairment in the mitochondrial function of tumor cells [7]. Generally, glycolysis is inferior to aerobic oxidation in terms of energy efficiency, the end product of aerobic glycolysisis lactic acid, which is fatal to cells. Therefore, some scholars regard aerobic glycolysis as a biological characteristic of tumor cells [8-9]. However, glycolysis not only offers energy quickly in the case of mitochondrial damage and anoxic conditions [10], but can also force tumor cells to absorb abundant glucose to provide materials for lipid metabolism, nucleic acid metabolism and amino acid metabolism [11]. The abundant intake of glucose by tumor cells, including LC cells, hinders nutrient supply to adjacent normal cells. Glycolysis can also induce deoxyribonucleic acid (DNA) mutation and the production of peroxide, both of which are beneficial for the proliferation and transfer of tumor cells [12].
There is still dispute over the mechanism underlying the Warburg effect. Warburg believed that the occurrence of a tumor is accompanied by mitochondrial damage; glycolysis plays an important role in energy metabolism [13]. However, other studies have demonstrated that aerobic oxidation is normal and even activated in some tumor cells [14], and most ATPs needed by tumor cells come from aerobic oxidation [15]. ATPs produced by glycolysis can account for 10-70% of ATP production in different tumors [16]. Even more intriguing is that most tumor cells can maintain growth by adjusting metabolism according to the microenvironment. For example, in hematological malignancy, primary and superficial tumor cells of solid tumors primarily utilize aerobic oxidation, while deep cells of solid tumors utilize glycolysis to gain energy due to the anoxic environment. The reliance of these cell subsets on different metabolisms can form a metabolic symbiont through the metabolic cooperation of the shuttle mechanism of lactic acids [17-18]. In a sense, the real significance of glycolysis is to provide intermediate products for other metabolisms [19].
Normal lungs contain the highest oxygen content, while nearly 50% of oxygen is used to make lactate; however, oxygen is rarely used to make proteins and fatty acids [20]. Relative to other tissues, lungs consume more glucose and are the highest producers of glutamine. LC tissue exhibits increased glucose contribution to tricarboxylic acid cycle (TCA) cycle relative to normal lung tissue, while LC cells have different glycolysis rates and mitochondrial capacities. The metabolic phenotypes of LC cells can self-regulate based on the tissue environment [21]. The proportion of hypoxic cells is consistently low in non small cell lung cancer (NSCLC), and there is no significant correlation between hypoxia and glucose metabolism in NSCLC [22-23]. Recently, metabolic remodeling has also been found in fresh LC surgical tissues using Stable Isotope Resolved Metabolomics (SIRM) technology. These LC tissues showed increased levels of glucose-derived TCA cycle intermediates (e.g., lactic acid, alanine, succinic acid, glutamic acid) relative to normal lung tissue [24]. Further, overexpression of pyruvate carboxylase (PC) and pyruvate was found in LC cells compared to normal lung tissues [25]. In short, an increasing evidence that metabolic remodeling is profoundly activated during carcinogenesis and malignant progression in LC [26].
Truncated TCA cycle and key molecules of glucose metabolism
Aside from ATP, TCA provides abundant intermediate products for the proliferation of tumor cells. Several studies have reported that TCA meets the needs of cell proliferation and invasion rather than providing ATP [27]. Warburg speculated that impairment of mitochondrial function was related to abnormal expression of key enzymes on the respiratory chain, dysfunction of the electron transmission chain and abnormal expression of mitochondrial genes [28-29]. In some tumors, damage of the respiratory chain induces a rise of reactive oxygen species (ROS) content. ROS can inhibit the key enzyme aconitase in TCA and thereby cause accumulation of citric acid in mitochondria. Citric acid can be decomposed into acetyl-CoA and acetoacetic acid. Acetyl-CoA is the raw material for synthesis of cholesterol and fat, which is called the truncated TCA cycle [30]. The synthesized macromolecular substances are carried to the cytoplasm to participate in synthesis of lipids and proteins. Active glycolysis offers sufficient energies to NSCLC cells. Different NSCLCcells have different glycolysis levels.The key enzyme hexokinase 2 (HK2), phosphofructokinase (PFK), pyruvate kinase (PKM) and lactate dehydrogenase (LDH) have been reported to be overexpressed LC [31]. Inhibiting expression of glycolysis metabolic enzymes obviously suppressed LC cells proliferation via by the AKT signaling pathway [32].
The glycolysis level is related to apoptosis signal transduction. Disturbing glycolysis can significantly inhibit the malignant biological behavior of NSCLC cells [33]. Metabolic remodeling of LC is related to drug resistance of epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) [34]. LC-driver genes (e.g. Kras and EGFR) can also lead to increased glucose metabolism in different modelling systems. Key drive gene mutant of LC cells exhibit increased glucose uptake [35]. The mutant Kras; p53fl/fl murine lung adenocarcinomas has similarities metabolic characteristics with human LC [36-37].KRAS mutations at codon-12 also had different metabolic remodeling and associated with different metabolomic profiles [38]. recent researches display increased glucose uptake and aerobic glycolysis of KRAS-induced LC, Enhanced aerobic glycolysis lead to LC cells extracellular matrix microenvironment changes, and the microenvironment can facilitate the occurrence and development of KRAS-induced LC [39-41].Similar results were found in EGFR-driven LC, and tissue environment is an important determinant of tumor metabolic phenotypes [42].
Active pentose phosphate pathway
To maintain their capacity for fast proliferation, tumor cells require not only ATP, but also genetic substances, a cytoskeleton, and functional proteins [43]. During culture of tumor cells without glucose, alternative pathways of phosphopentose still synthesize the necessary substances for proliferation. Intermediate products produced by glycolysis activate the pentose phosphate pathway (PPP) through glucose-6-phosphate dehydrogenase (G6PDH) or transketolase TKT [44-45]. The PPP that involves the catalyst, G6PDH, is the oxidative branch of PPP, while the PPP that involves the catalyst, TKT, is the non-oxidative branch of PPP. Products of glycolysis produce ribose 5-phosphate (R5P) through non-oxidative PPP, which is used in the synthesis of genetic substances. Meanwhile, PPP activation can induce abundant nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione. NADPH can provide metabolic substrates and reducing equivalents for lipid metabolism and nucleic acid metablishm [46]. An increasing number of studies have confirmed NADPH oxidase activity and expression related to malignant biological behavior of LC, and inhibition of NADPH oxidase function downregulates the proliferative and invasion of LC [47-48]. NADPH oxidase can also supports glycolysis and promotes glutamine metabolism of LC [49].The glutathione metabolic system is directly participated in the metabolism of platinum drugs [50].NADPH and glutathione can both enhance anti-apoptosis in tumor cells and the PPP pathways of tumor cells in activated state. The activation of alternative pathways of phosphopentose plays an important role in the evolution of tumors (Fig. 1).
Metabolic Remodeling in lung cancer. Lung cancer cells consume large amounts of glucose via GLUTs and convert glucose to G6P, which involves in PPP metabolism and produced genetic substances, cytoskeleton and functional proteins. Pyruvate, a production by glucose, offers lactate for microenvironment and citrate for glycolsis. Lactate is excreted and absorbed through MCTs, and this phenomenon is known as lactic acid shuttle system. Citrate cannot participate in metabolism smoothly due to ROS inhibits a key enzyme (aconitase) activity in TCA. However, citrate participates in endogenous fatty acids metabolism. Some citrate is converted to malate and continues to participate in TCA. As a result, endogenous fatty acids metabolism offers lung cancer cells the energy for proliferation and invasion, and is regulated by three key regulatory enzyme, which are negatively regulated by SREBPs. G6P Glucose-6-Phosphate), MCTs (Monocarboxylate transporters), ROS (Reactive oxygen species),
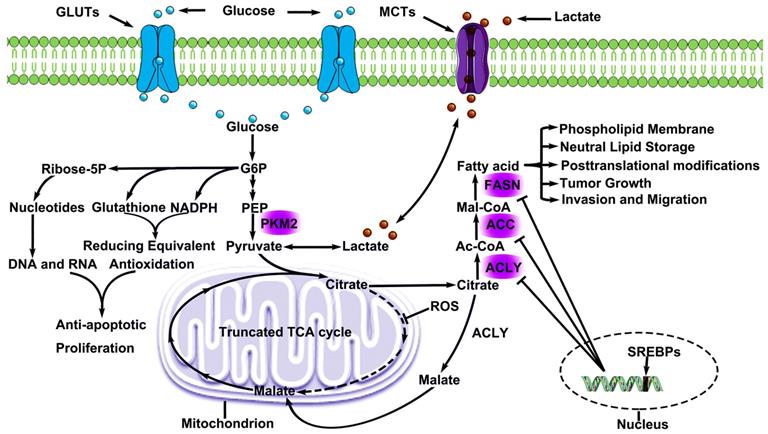
Accelerated endogenous fatty acid metabolism in LC
The regulation of endogenous fatty acids metabolism
Fatty acids gained by normal cells from blood circulation are called exogenous fatty acids. Generally, exogenous fatty acids can meet the metabolic demands of normal cells, so normal cells (except liver, fatty tissue and breast tissue during lactation) rarely make de novo synthesis of fatty acids [51]. Researchers discovered more than 50 years ago that the fatty acids required for the proliferation of tumor cells come mainly from de novo synthesis [52]. Such fatty acids are also called endogenous fatty acids. Due to demands of tumor cells for malignant proliferation, exogenous fatty acid cannot meet cell proliferation requirements and so tumor cells activate the metabolism of endogenous fatty acids. Therefore, most key enzymes involved in the metabolism of endogenous fatty acids in tumor cells, such as ATP citrate lyase (ACLY), fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), become highly activated [53]. Fatty acids metabolic abnormalities could result in lung cancer. A meta-analysis included 31 studies found that inverse correlation between excess body weight (BMI>25 kg/m2) and lung cancer incidence [54-55]. Another study has shown an association between total serum cholesterol (cut-off value; 5,3 mmol/L) and resectable NSCLC [56]. Cholesterol via releasing EGFR from lipid rafts increases EGFR signaling activity [57]. Endogenous fatty acids metabolisms were negatively correlation with EGFR expression and the fatty acids pathways may be valuable as a potential therapeutic target for lung adenocarcinoma [58].
Metabolic substrates (e.g. Ac-CoA) of endogenous fatty acids are acquired from the decomposition of citric acids by ACLY, while citric acid is produced by a truncated TCA cycle. Ac-CoA produces malonyl-CoA (Mal-CoA) under the catalysis of ACC while 9 Mal-CoAs aggregates into 16-C palmitic acids under the catalysis of FASN. Palmitic acids form essential lipids of cells under the catalysis effect of other specificity enzymes [59]. ACLY is the bridge between glucose metabolism and lipid metabolism. In vivo and in vitro studies all prove the key role of ACLY in the evolution of tumor. High ACLY expression patients exhibited shorter life span than negative ACLY expression patients [60]. Inhibiting ACLY disturbs NSCLC proliferation and ACLY can mediate occurrence of LC by participating in the metabolism of endogenous fatty acids. ACLY might be a new target for LC treatment [61-62]. FASN is a key enzyme that catalyzes lipid synthesis and it has high expression in LC tissues [63-64].
High expression of FASN is closely related to proliferation and anti-apoptosis capacity, invasion and metastatic capacity of LC cells, as well as prognosis [65]. ACC contains two subtypes (ACC1 and ACC2). ACC1 is the first key enzyme that catalyzes the denovo synthesis of fatty acids. Mal-CoA produced by ACC2 can repress the entrance of fatty acids into mitochondria for β oxidation of fatty acids, thus coordinating synthesis of liver fatty acids with β-oxidation and ketone synthesis. Inhibiting deficiency of ACC might cause complete blocking of the pathway for synthesis of fatty acids, which reflects that the pathways of fatty acid synthesis in LC cells are strictly regulated by ACC genes [66-68] (Fig. 1).
The rloe of endogenous fatty acids metabolism in LC
Synthesized fatty acids have extensive functions. β oxidation of fatty acids can produce Ac-CoA which offers raw materials for TCA. Moreover, fatty acids also participate in cell proliferation directly [69]. The metabolism of endogenous fatty acids can also contribute to epithelial-mesenchymal-transition (EMT) regulation, thus influencing the invasive and metastatic capacity of LC cells [70]. Lipid signals, such as prostaglandin E2 (PGE2), lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P), can collect macrophages and immune cells, and stimulate the production of tumor capillaries [71-72]. PGE2 can inhibit the activation of macrophages related to tumor, thus assisting tumor cells to escape from immunity monitoring [73]. Phospholipid is an important component of the cytomembrane and organelle membrane. Reduction of phospholipids can influence the bioelectricity transduction of organelles with membranes and cells. Besides, phospholipids participate in the acetylation of proteins and other protein modifications after translation. Phosphatidylinositol, phosphatidylserine and lecithin can form the lipid raft structure to promote the activation of growth factor and participate in the activation of important signal pathways, such as phosphoinositide 3-kinase (P13K)/protein kinase B (AKT), Ras and Wnt [74]. Multiple molecules can influence the activity of the metabolism of endogenous fatty acids. For example, sterol regulatory element binding proteins (SREBPs) are important transcription factors in sterol regulation and lipid synthesis and belong to one member of the basic helix-loop-helix (bHLH)-zip transcription factor family, with at least 3 spliceosomes (SREBP1a, SREBP1c and SREBP2). Under normal situations, SREBP and SREBP cleavage-activating protein (SCAP) form the composites in endoplasmic reticulum, and SREBP enters into the nucleus after cells are excited by stimulus signals. Meanwhile, the expressions of ACLY, ACC and FASN are regulated. Inhibiting pathways of lipid metabolism might be an alternative treatment for lung adenocarcinoma [75] (Fig. 1).
The significance of endogenous fatty acids metabolism
Tumor cells choose de novo synthesis of fatty acids at the cost of abundant valuable ATPs and metabolites. Such a metabolic approach gives LC cells traits of fast proliferation and invasion. Two different kinds of drive-gene mutant (KrasG12D;P53fl/fl and KrasG12D;LKB1fl/fl)mouse LC model have high rates of endogenous fatty acids metabolism [76], and the KrasG12D;LKB1fl/flLC model has higher rates of endogenous fatty acids metabolism than KrasG12D;P53fl/fl[77].This reveals that activated metabolism of endogenous fatty acids provides key substances for LC. Many anti-tumor drugs targeted at key enzymes of lipid metabolism have been developed based on the reported active lipid metabolism in LC cells. The discovery of high-efficiency anti-tumor targets based on lipid metabolism may become an edge in designing tools to defeat LC.
Molecular mechanism related to metabolic remodeling of LC
PI3K/AKT/mTOR signal pathway and LC
PI3K/AKT/mTOR is a typical signal pathway which is the focus of most current research. It mainly receives extracellular RTK (e.g. EGFR1/2/3/4, PDGFR, VEGFR, IGF-1R and HERs) signals to activate intra-cellular PI3K signals. PI3K signals activate the second receptor and the second receptor binds with the PH structure of AKT to activate AKT. AKT activates mTOR by inhibiting the formation of TSC1/TSC2 composite and PRAS40, an important negative regulatory factor in this pathway [78]. mTOR participates in the transcription and metabolism of cell proteins by regulating many downstream factors, and thereby influencing cell growth and proliferation. It is reported in studies on LC that 50-73% of NSCLC patients have high expression of AKT and suffer poor prognosis [79-80], while 2-5% of NSCLC patients have mutations of PI3K and AKT. Moreover, 70% of NSCLC patients have an absence of the negative regulatory factor PETN of the PI3K/AKT/mTOR signal pathways, which further results in a poor prognosis [81-82]. PI3K/AKT/mTOR pathway also participates in the regulation of EMT and glycolysis of LC [83-84].
Inhibiting the PI3K/AKT/mTOR pathway can inhibit NSCLC proliferation of TKI, indicating that interrupting the PI3K/AKT/mTOR pathway might be a treatment strategy for TKI drug resistance [85-86]. In summary, the PI3K/AKT/mTOR signal pathway participates in energy material metabolism, proliferation, autophagy, apoptosis and regulations of other biological functions of cells related to LC [87-90] (Fig. 2).
Related molecular mechanisms of metabolic remodeling in lung cancer. PI3K/AKT/mTOR and MEK/ERK/AMPK signaling pathways are both involved in metabolic remodeling of lung cancer cells. And, the two signaling pathways are regulated by extracellular signals (e.g. GFs, hormones, cytokines) to activate cascade response. Importantly, the interaction of the two signaling pathways affects glycolysis, TCA cycle, PPP, endogenous fatty acids metabolism. Consequently, the aggressive biological behaviors of lung cancer cells are activated by metabolic remodeling.
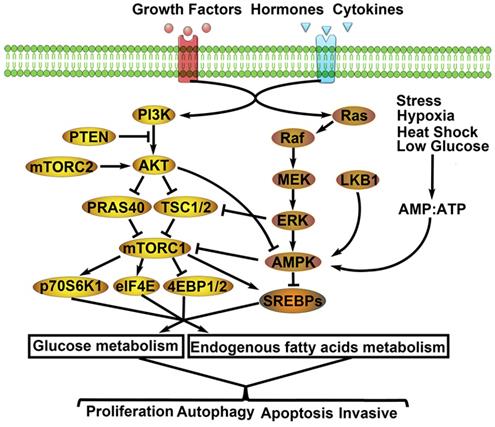
MEK/ERK/AMPK signal pathway and LC
Extracellular PTKs also can induce Raf activation when activating PI3K. The activated Raf then activates MEK, EPK and AMPK successively. Finally, the MEK/ERK/AMPK signal pathway is activated and participates in the occurrence and development of tumor cells. The MEK/ERK/AMPK signal pathway mainly perceives intracellular energy changes. Cells can activate AMPK automatically upon anoxia, ischemia, hunger and exercise. The activated AMPK can increase the supply of ATP and regulate the metabolic levels of glucose and lipids. Glycolysis inhibition sensitizes NSCLC with T790M Mutation to irreversible EGFR inhibitors via AMPK/mTOR/Mcl-1 pathway [91]. Therefore, AMPK is also regarded as the monitor of energy level changes [92-95]. According to studies on 3%-5% of NSCLC patients have mutation of BRAF [96] and some NSCLC patients have mutation of MEK, which could be used as the driving gene of NSCLC [97].
ERK can also participate in the formation of an inflammation microenvironment of LC cells [98]. The MEK/ERK/AMPK signal pathway can participate in the regulation of drug resistance to NSCLC [99], and its retardants have been applied to stage-II clinical studies [100]. MEK/ERK/AMPK pathway is important to the proliferation and apoptosis of LC cells [101-102] (Fig. 2).
Others molecular mechanisms for Metabolic Remodeling in LC
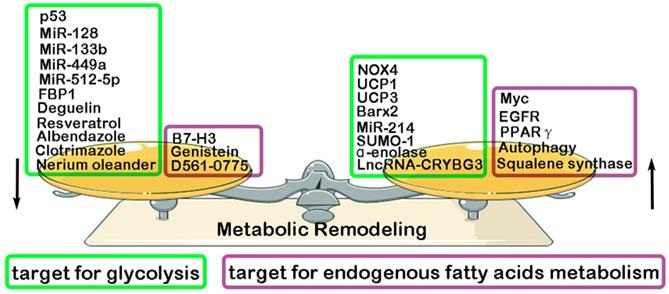
Previous studies have found that some key molecules exerts its inhibitory effect in LC progression through down-regulating glycolysis, such as MiR-128 (inhibiting AKT) [108], MiR-512-5p (inhibiting p21) [109], MiR-133b (targeting PKM2) [110], MiR-449a (targeting LDHA) [111], p53 (targeting RRAD) [112], Albendazole (inhibiting HIF-1α) [113], Resveratrol(inhibiting HK2) [114], Deguelin (inhibiting HK2) [115], FBP1 [116], Clotrimazole (targeting FDP) [117], Nerium oleander (targeting lactate) [118]. However, some other molecules have been reported to play a promotive effect in glycolysis and glutamine metabolism of LC, such as MiR-214 (targeting HK2) [119], NADPH oxidase 4 (NOX4) (targeting PI3K/AKT pathway) [120], α-enolase (targeting PI3K/AKT pathway) [121], LncRNA-CRYBG3 (targeting LDHA) [122], BarH-like homeobox 2 (Barx2) (targeting Wnt/β-catenin pathway) [123], Small ubiquitin-like modifier 1 (SUMO-1) (targeting PKM2) [124], Uncoupling protein1/3 (UCP1/3) (targeting HK2 and PFK) [125]. Compared with normal lung tissue, endogenous fatty acids metabolism is significantly enhanced in LC tissue. And, a high level of endogenous fatty acids metabolism has been reported to have a closely association with poor prognosis of LC patients [126-127]. Recent studies have proven some key molecules that exert its inhibitory effect in endogenous fatty acids metabolism of LC, resulting in the suppression of LC malignant biological behavior. They are Genistein (inhibiting SCD1) [128], B7-H3 (targeting SREBP1) [129], D561-0775(inhibiting AMPK) [130]. However, some other molecules were proven to exerts its promotive effect in endogenous fatty acids metabolism of LC, such as EGFR (targeting SCD1) [131], Autophagy [132], Myc (targeting COX and LOX pathway) [133], Squalene synthase (targeting TNFα) [134], PPARγ [135] (Fig. 3).
Regulatory factors of metabolic remodeling in lung cancer. Metabolic remodeling in lung cancer include glycolysis metabolism and endogenous fatty acids metabolism. Many negative/ positive regulatory factors involved in glycolysis and endogenous fatty acids metabolism.
Conclusions
Researchers have recognized many tumor characteristics through fighting tumors and formulating targeted treatments according to these characteristics. Tumor evolution is the consequence of both internal and external factors. The complexity of this evolution process is comparable with that of human evolution. For example, the first tumor of lung cancer may show significant damages. Although early screening, chemoradiotherapy, targeted treatment and immunotherapy have increased the diagnostic efficiency of lung cancer significantly, most patients with lung cancer develop unexpected progression of disease after multiple treatments. However, it is exciting that we have now recognized the metabolic difference between tumor cells and normal cells. These research conclusions reveal the relationship between abnormal metabolism and tumor evolution. The current study reviewed the molecular mechanisms of glycolysis metabolism and endogenous fatty acids metabolism of lung cancer, and offer a new opportunity for targeted tumor treatments.
Acknowledgements
This study is supported by Jiangsu Provincial Special Program of Medicine Science (No. BE2015611) and Jiangsu Natural Science Foundation (BK20171120).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. Ca A Cancer Journal for Clinicians. 2015;65:5-29
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. Ca A Cancer Journal for Clinicians. 2016;66:115-32
3. Zhang X, Xing P, Hao X, Li J. Analysis of the First Diagnosis Symptom and Its Influencing Factors in 500 Patients with Lung Cancer. Chinese Journal of Lung Cancer. 2018;21:408-12
4. Hanahan D, Weinberg RA. The hallmarks of cancer: the next generation. Cell. 2011;144:646-74
5. Weinhouse S, Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:267-72
6. Suvà M L, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567-70
7. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
8. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437-43
9. Swierczynski J, Hebanowska A, Sledzinski T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World Journal of Gastroenterology. 2014;20:2279-03
10. Gutschner T, Diederichs S. The hallmarks of cancer. Rna Biology. 2012;9:703-19
11. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer cell. 2008;13:472-82
12. Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Research. 2011;13:213-20
13. Gallardo-Pérez JC, Marín-Hernández A, Mandujano-Tinoco EA, Aguilar-Ponce JL, Mandujano-Tinoco EA, Meneses A. et al. Oxidative phosphorylation as a target to arrest malignant neoplasias. Current Medicinal Chemistry. 2011;18:3156-67
14. Koppenol WH, Bounds PL, Dang CV. Otto Warburg'S contributions to current concepts of cancer metabolism. Nature Reviews Cancer. 2011;11:325-37
15. Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A. et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013;12:1371-84
16. Kaldma A, Klepinin A, Chekulayev V, Mado K, Shevchuk I, Timohhina N. et al. An in situ study of bioenergetic properties of human colorectal cancer: The regulation of mitochondrial respiration and distribution of flux control among the components of ATP synthasome. The international journal of biochemistry & cell biology. 2014;55:171-86
17. Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochemical Journal. 2002;364:309-15
18. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation Science. 2009; 324: 1029-33.
19. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell. 2012;21:297-08
20. Fisher AB. Intermediary Metabolism of the Lung. Environmental Health Perspectives. 1984;55:149-58
21. Feinberg T, Herbig J, Kohl I, Las G, Cancilla JC, Torrecilla JS. et al. Cancer metabolism: the volatile signature of glycolysis—in vitro, model in lung cancer cells. Journal of Breath Research. 2017;11:1-11
22. Cherk MH, Foo SS, Poon MT, Knight SR, Murone C, Papenfuss AT. et al. Lack of Correlation of Hypoxic Cell Fraction and Angiogenesis with Glucose Metabolic Rate in Non-Small Cell Lung Cancer Assessed by 18F-Fluoromisonidazole and 18F-FDG PET. Journal of Nuclear Medicine Official Publication Society of Nuclear Medicine. 2006;47:1920-26
23. Hsieh IS, Cheng FJ, Tu CY, Chen TC, Hsia BW, Wang YL. et al. 12P Cigarette smoke promotes proliferation of non-small cell lung cancer (NSCLC) via enhancing glucose uptake and glycolysis. Journal of Thoracic Oncology. 2018;13:S6-S7
24. Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M. et al. Altered regulation of metabolic pathways in human lung cancer discerned by 13 C stable isotope-resolved metabolomics (SIRM). Molecular Cancer. 2009;8:839-50
25. Sellers K, Fox MP, Bousamra M, Slone SP, Higashi RM, Miller DM. et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. Journal of Clinical Investigation. 2015;125:687-98
26. Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. Febs Journal. 2017;285:28-41
27. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J. et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism. 2012;15:110-21
28. Vaitheesvaran B, Xu J, Yee J, QY L, Go VL, Xiao GG. et al. The Warburg effect: a balance of flux analysis. Metabolomics. 2015;11:787-96
29. Parlo RA, Coleman PS. Continuous pyruvate carbon flux to newly synthesized cholesterol and the suppressed evolution of pyruvate generated CO2 in tumors: further evidence for a persistent truncated Krebs cycle in hepatomas. Biochim Biophys Acta. 1986;886:169-76
30. Wu R, Galan-Acosta L, Norberg E. Glucose metabolism provide distinct prosurvival benefits to non-small cell lung carcinomas. Biochemical and biophysical research communications. 2015;460:572-77
31. Li XB, Gu JD, Zhou QH. Review of aerobic glycolysis and its key enzymes - new targets for lung cancer therapy. Thoracic Cancer. 2015;6:17-24
32. Scaglia N, Igal RA. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. International Journal of Oncology. 2008;33:839-50
33. Chiang CT, Demetriou AN, Ung N, Choudhury N, Ghaffarian K, Ruderman DL. et al. mTORC2 contributes to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor resistant cells in non-small cell lung cancer. Cancer letters. 2018;434:152-59
34. Takeda K. Overcoming resistance to EGFR-TKI in lung cancer. Gan to Kagaku Ryoho Cancer & Chemotherapy. 2009;36:552-56
35. Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. The FEBS Journal. 2018;285:28-41
36. Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. et al. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110-13
37. Hensley C, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J. et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164:681-94
38. Laura B, Elisa C, Mirko M, Broggini M, Pastorelli R. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget. 2014;5:4722-31
39. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364-73
40. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537-48
41. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70
42. Davidson S, Papagiannakopoulos T, Olenchock B, Heyman JE, Keibler MA, Luengo A. et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metabolism. 2016;23:517-28
43. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Oxidative stress and cancer: an overview. Ageing research reviews. 2013;12:376-90
44. Butler EB, Zhao Y, Muñoz-Pinedo C, Lu J, Tan M. Stalling the engine of resistance: targeting cancer metabolism to overcome therapeutic resistance. Cancer research. 2013;73:2709-17
45. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011;10:671-84
46. Benito A, Diaz-Moralli S, Coy JF, Josep J. CentellesMarta Cascante,et al. Role of the Pentose Phosphate Pathway in Tumour Metabolism[M]//Tumor Cell Metabolism. Springer Vienna. 2015:143-63
47. Han M, Zhang T, Yang L, Wang Z, Ruan J, Chang X. et al. Association between NADPH oxidase (NOX) and lung cancer: A systematic review and meta-analysis. Journal of Thoracic Disease. 2016;8:1704-11
48. Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F. et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes to Cells. 2010;10:1139-51
49. Zeng C, Wu Q, Wang J, Yao B, Ma L, Yang Z. et al. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free Radical Biology & Medicine. 2016;101:236-48
50. Yang P. Role of the Glutathione Metabolic Pathway in Lung Cancer Treatment and Prognosis: A Review. Journal of Clinical Oncology. 2006;24:1761-69
51. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews Cancer. 2007;7:763-77
52. Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue.IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13:27-29
53. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153-61
54. Yang Y, Dong J, Sun K, Zhao L, Zhao F, Wang L. et al. Obesity and incidence of lung cancer: a meta-analysis. International Journal of Cancer. 2013;132:1162-69
55. Duan P, Hu C, Quan C, Yi X, Zhou W, Yuan M. et al. Body mass index and risk of lung cancer: Systematic review and dose-response meta-analysis. Scientific Reports. 2015;5:16938
56. Sok M, Ravnik J, Ravnik M. Preoperative total serum cholesterol as a prognostic factor for survival in patients with resectable non-small-cell lung cancer. Wiener Klinische Wochenschrift. 2009;121:314-17
57. Ringerike T, Blystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. Journal of Cell Science. 2002;115:1331-40
58. Yano Kojiro. Lipid metabolic pathways as lung cancer therapeutic targets: A computational study. International Journal of Molecular Medicine. 2011;29:519-29
59. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer research. 2012;72:3709-14
60. Csanadi A, Kayser C, Donauer M, Gumpp V, Aumann K, Rawluk J. et al. Prognostic Value of Malic Enzyme and ATP-Citrate Lyase in Non-Small Cell Lung Cancer of the Young and the Elderly. Plos One. 2015;10:e0126357
61. Osugi J, Yamaura T, Muto S, Okabe N, Matsumura Y, Hoshino M. et al. Prognostic impact of the combination of glucose transporter 1 and ATP citrate lyase in node-negative patients with non-small lung cancer. Lung Cancer. 2015;88:310-18
62. Bauer DE, Hatzivassiliou G, Zhao F, Zhao FP, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314-22
63. Visca P, Sebastiani V, Botti C, Andreadis C, Thompson CB. Fatty Acid Synthase (FAS) is a Marker of Increased Risk of Recurrence in Lung Carcinoma. Anticancer Research. 2004;24:4169-73
64. Wang Y, Zhang X, Tan W, Fu J, Zhang W. Significance of fatty acid synthase expression in non-small cell lung cancer. Chinese journal of oncology. 2002;24:271-73
65. Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM. et al. Selective inhibition of fatty acid synthase for lung cancer treatment. Clinical Cancer Research. 2007;13:7139-45
66. Crunkhorn S. Anticancer agents: ACC inhibition suppresses lung cancer. Nature Reviews Drug Discovery. 2016;15:750
67. Mizojiri R, Asano M, Tomita D, Banno H, Nii N, Sasaki M. et al. Discovery of Novel Selective Acetyl-CoA Carboxylase (ACC) 1 Inhibitors. Journal of medicinal chemistry. 2018;61:1098-17
68. Svensson R, Harriman G, Greenwood J, Sathesh B, H.James H, Rosana K. et al. Abstract 2679: Acetyl-CoA carboxylase inhibition by ND646 reduces fatty acid synthesis and inhibits cell proliferation in human non-small cell lung cancer cells. Cancer Research. 2014;74:2679-79
69. Han J, Li E, Chen L, Zhang Y, Wei F, Liu J. et al. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature. 2015;524:243-56
70. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M. et al. Metabolic reprogramming during TGFβ1-induced pithelial-to-mesenchymal transition. Oncogene. 2015;34:3908-16
71. Kazlauskas A. Lysophosphatidic acid contributes to angiogenic homeostasis. Experimental cell research. 2015;333:166-70
72. Mendelson K, Evans T, Hla T. Sphingosine 1-phosphate signalling. Development. 2014;141:5-9
73. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proceedings of the National Academy of Sciences. 2015;112:15642-47
74. Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nature Reviews Cancer. 2016;16:732-49
75. Yano K. Lipid metabolic pathways as lung cancer therapeutic targets: A computational study. International Journal of Molecular Medicine. 2012;29:519-29
76. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN. et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small cell lung cancer in preclinical models. Nature Medicine. 2016;22:1108-19
77. Svensson RU, Shaw RJ. Lipid Synthesis Is a Metabolic Liability of Non-Small Cell Lung Cancer. Cold Spring Harbor Symposia on Quantitative Biology. 2016;81:93-03
78. Polivka J, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacology & therapeutics. 2014;142:164-75
79. Dennis P, Tsurutani J, Fukuoka J. et al. P-607 Evaluation of two phosphorylation sites establishes Akt activation as a poor prognostic factor for all stages of NSCLC tumors. Lung Cancer. 2005;49:S278-78
80. Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated AKT overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer. 2006;51:181-91
81. Lee S Y, Kim M J, Jin G, Jin G, Yoo SS, Park JY. et al. Somatic mutations in epidermal growth factor receptor signaling pathway genes in non-small cell lung cancers. Journal of Thoracic Oncology. 2010;5:1734-40
82. Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A. et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;517:576
83. Chen QY, Jiao DM, Wu YQ, Chen J, Wang J, Tang XL. et al. MiR-206 inhibits HGF-induced epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via c-Met /PI3k/Akt/mTOR pathway. Oncotarget. 2016;7:18247-61
84. Tianxiao G, Liuqing C, Haili W, Chen J, Wang J, Tang XL. et al. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. Journal of Translational Medicine. 2018;16:2-10
85. Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/AKT/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clinical Lung Cancer. 2013;14:322-32
86. Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K. et al. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clinical Cancer Research. 2011;17:2260-69
87. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR4. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochemical pharmacology. 2014;90:197-07
88. Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Translational lung cancer research. 2015;4:165-76
89. Papadimitrakopoulou V. Development of PI3K/AKT/mTOR Pathway Inhibitors and Their Application in Personalized Therapy for Non-Small-Cell Lung Cancer. Journal of Thoracic Oncology Official Publication of the International Association for the Study of Lung Cancer. 2012;7:1315-26
90. Li H, Jing H, Wu S, Wang L, Cao X, Zhang X. et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget. 2016;7:3548-58
91. Kim SM, Yun MR, Hong YK, Solca F, Kim JH, Kim HJ. et al. Glycolysis Inhibition Sensitizes Non-Small Cell Lung Cancer with T790M Mutation to Irreversible EGFR Inhibitors via Translational Suppression of Mcl-1 by AMPK Activation. Molecular Cancer Therapeutics. 2013;12:2145-56
92. Jeon S M, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Archives of pharmacal research. 2015;38:346-57
93. Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Current opinion in cell biology. 2015;33:1-7
94. Hardie DG, Ashford MLJ. AMPK: regulating energy balance at the cellular and whole body levels. Physiology. 2014;29:99-107
95. Hart P, Mao M, de Abreu A, Ansenberger-Fricano K, Ekoue DN, Ganini D. et al. MnSOD/SOD2 upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signaling in cancer. The FASEB Journal. 2015;29:884-62
96. Jetschke K, Viehweger H, Freesmeyer M, Warnke JP, Mawrin C. Primary pineal malignant melanoma with B-Raf V600E mutation: a case report and brief review of the literature. Acta neurochirurgica. 2015;157:1267-70
97. Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D. et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer research. 2008;68:9375-83
98. Zhang Y, Wang L, Zhang M, Jin M, Bai C, Wang X. Potential mechanism of interleukin-8 production from lung cancer cells: An involvement of EGF-EGFR-PI3K-Akt-Erk pathway. Journal of cellular physiology. 2012;227:35-43
99. Li H, Schmid-Bindert G, Wang D, Zhao Y, Yang X, Su B. et al. Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Advances in medical sciences. 2011;56:275-84
100. Rinehart J, Adjei AA, LoRusso PM, Waterhouse D, Hecht JR, Natale RB. et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. Journal of clinical oncology. 2004;22:4456-62
101. Nguyen TTT, Tran E, Nguyen TH, Do PT, Huynh TH. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis. 2004;25:647-59
102. Nguyen TTT, Tran E, Ong CK, Lee SK, Do PT, Huynh TT. et al. Kaempferol-induced growth inhibition and apoptosis in A549 lung cancer cells is mediated by activation of MEK-MAPK. Journal of cellular physiology. 2003;197:110-21
103. Idle J, Caporaso N, Dosemeci M, Hoover R, Ayesh R, Hetzel M. et al. Lung cancer risk, occupational exposure, and the debrisoquine metabolic phenotype. Cancer Research. 1989;49:3675-79
104. Caporaso NE, Tucker MA, Hoover RN, Hayes RB, Pickle LW, Issaq HJ. et al. Lung Cancer and the Debrisoquine Metabolic Phenotype. Journal of the National Cancer Institute (Bethesda). 1990;82:1264-72
105. Cote ML, Yoo W, Wenzlaff AS, Prysak GM, Santer SK, Claeys GB. et al. Tobacco and estrogen metabolic polymorphisms and risk of non-small cell lung cancer in women. Carcinogenesis. 2009;30:626-35
106. Suhua J, Zhengzhi Z, Peipei N, Wen R, Xiao Y, Tang J. Synergistic Effects between mTOR Complex 1/2 and Glycolysis Inhibitors in Non-Small-Cell Lung Carcinoma Cells. PLOS ONE. 2015;10:1-19
107. Ye M, Wang S, Wan T, Jiang R, Qiu Y, Pei L. et al. Combined Inhibitions of Glycolysis and AKT/autophagy Can Overcome Resistance to EGFR-targeted Therapy of Lung Cancer. J Cancer. 2017;8:3774-84
108. Yang J, Li J, Le Y, Zhou C, Zhang S, Gong Z. PFKL/miR-128 axis regulates glycolysis by inhibiting AKT phosphorylation and predicts poor survival in lung cancer. Am J Cancer Res. 2015;6:473-85
109. Chu K, Gao G, Yang X, Ren S, Li Y, Wu H. et al. MiR-512-5p induces apoptosis and inhibits glycolysis by targeting p21 in non-small cell lung cancer cells. International Journal of Oncology. 2015;48:577-86
110. Liu G, Li Y, Gao X. Overexpression of microRNA-133b sensitizes non-small cell lung cancer cells to irradiation through the inhibition of glycolysis. Oncology Letters. 2016;11:2903-08
111. Li L, Liu H, Du L, Xi P, Wang Q, Li Y. et al. MiR-449a Suppresses LDHA-Mediated Glycolysis to Enhance the Sensitivity of Non-Small Cell Lung Cancer Cells to Ionizing Radiation. Oncology Research. 2018;26(4):547-56
112. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J. et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5:5535-46
113. Zhou F, Du J, Wang J. Albendazole inhibits HIF-1α-dependent glycolysis and VEGF expression in non-small cell lung cancer cells. Molecular & Cellular Biochemistry. 2017;428:171-78
114. Li W, Ma X, Li N, Liu H, Dong Q, Zhang J. et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Experimental Cell Research. 2016;349:320-27
115. Li W, Gao F, Ma X, Wang R, Dong X, Wang W. Deguelin inhibits non-small cell lung cancer via down-regulating Hexokinases II-mediated glycolysis. Oncotarget. 2017;8:32586-99
116. Cong J, Wang X, Zheng X, Wang R, Dong X, Wang W. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metabolism. 2018;28:243-55
117. Penso J, Beitner R. Clotrimazole decreases glycolysis and the viability of lung carcinoma and colon adenocarcinoma cells. European Journal of Pharmacology. 2002;451:227-35
118. Calderón-Montaño JM, Burgos-Morón E, Orta ML, Mateos S, López-Lázaro M. A hydroalcoholic extract from the leaves of Nerium oleander inhibits glycolysis and induces selective killing of lung cancer cells. Planta Medica. 2013;79:1017-23
119. Zhang K, Zhang M, Jiang H, Liu F, Liu H, Li Y. Down-regulation of miR-214 inhibits proliferation and glycolysis in non-small-cell lung cancer cells via down-regulating the expression of hexokinase 2 and pyruvate kinase isozyme M2. Biomedicine & Pharmacotherapy. 2018;105:545-52
120. Zeng C, Wu Q, Wang J, Yao B, Ma L, Yang Z. et al. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free Radical Biology & Medicine. 2016;101:236-48
121. Fu Q F, Liu Y, Fan Y, Hua SN, Qu HY, Dong SW. et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. Journal of Hematology & Oncology. 2015;8:22
122. Hei TK. Long non-coding RNA CRYBG3 regulates glycolysis of lung cancer cells by interacting with lactate dehydrogenase A. Journal of Cancer. 2018;9:2580-88
123. Chen H, Zhang M, Zhang W, Li Y, Zhu J, Zhang X. et al. Downregulation of BarH-like homeobox 2 promotes cell proliferation, migration and aerobic glycolysis through Wnt/β-catenin signaling, and predicts a poor prognosis in non-small cell lung carcinoma. Thoracic Cancer. 2018;9:390-99
124. An S, Huang L, Miao P, Shi L, Shen M, Zhao X. et al. Small ubiquitin-like modifier 1 modification of pyruvate kinase M2 promotes aerobic glycolysis and cell proliferation in A549 human lung cancer cells. Onco Targets Ther. 2018;11:2097-09
125. Giatromanolaki A, Balaska K, Kalamida D, Kakouratos C, Sivridis E, Koukourakis MI. Thermogenic protein UCP1 and UCP3 expression in non-small cell lung cancer: relation with glycolysis and anaerobic metabolism. Cancer Biology & Medicine. 2017;14:396-04
126. Zieba M, Suwalski M, Kwiatkowska S, Piasecka G, Grzelewska-Rzymowska I, Stolarek R. et al. Comparison of hydrogen peroxide generation and the content of lipid peroxidation products in lung cancer tissue and pulmonary parenchyma. Respiratory Medicine. 2000;94:800-05
127. Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A. et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566:403-06
128. Hess D, Igal RA. Genistein downregulates de novo lipid synthesis and impairs cell proliferation in human lung cancer cells. Experimental Biology & Medicine. 2011;236:707-13
129. Luo D, Xiao H, Dong J, Li Y, Feng G, Cui M. et al. B7-H3 regulates lipid metabolism of lung cancer through SREBP1-Mediated expression of FASN. Biochemical & Biophysical Research Communications. 2016;482:1246-51
130. Chen X, Xie C, Fan XX, Jiang ZB, Wong VK. et al. Novel Direct AMPK activator suppresses non-small cell lung cancer through inhibition of lipid metabolism. Oncotarget. 2017;8:96089-02
131. Zhang J, Song F, Zhao X, Jiang H, Wu X, Wang B. et al. EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Molecular Cancer. 2017;16:27
132. Bhatt V, Khayati K, Hu ZS, Lee A1, Kamran W, Su X. et al. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019;1(33):150-65
133. Hall Z, Ament Z, Wilson CH, Burkhart DL, Ashmore T, Koulman A. et al. Myc expression drives aberrant lipid metabolism in lung cancer. Cancer Research. 2016;76:4608-18
134. Yang YF, Jan YH, Liu YP, Yang CJ, Su CY, Chang YC. et al. Squalene synthase induces tumor necrosis factor receptor 1 enrichment in lipid rafts to promote lung cancer metastasis. American Journal of Respiratory & Critical Care Medicine. 2014;190:675-87
135. Ai NHP, Vo VTA, Hua TNM, Kim MK, Jo SY, Choi JW. et al. PPARγ sumoylation-mediated lipid accumulation in lung cancer. Oncotarget. 2017;8:82491-05
Author contact
![]() Corresponding author: Wei Gu, No. 68 Changle Road, Qinhuai District, Nanjing 210001, People's Republic of China. Email: guwei123edu.cn; Fax: 86-025- 52271000.
Corresponding author: Wei Gu, No. 68 Changle Road, Qinhuai District, Nanjing 210001, People's Republic of China. Email: guwei123edu.cn; Fax: 86-025- 52271000.