Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(25):6457-6465. doi:10.7150/jca.33005 This issue Cite
Research Paper
Association between gene methylation and HBV infection in hepatocellular carcinoma: A meta-analysis
Cheng Zhang1 ![]() , Changxin Huang1, Xinbing Sui1, Xueqing Zhong2, Wenjun Yang3, Xiangrong Hu3, Yongqiang Li1,4
, Changxin Huang1, Xinbing Sui1, Xueqing Zhong2, Wenjun Yang3, Xiangrong Hu3, Yongqiang Li1,4 ![]()
1. Department of Medical Oncology, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, Zhejiang, China
2. Department of Gastroenterology, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, Zhejiang, China
3. Department of Pathology, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, Zhejiang, China
4. Key Laboratory of Elemene Class Anti-cancer Chinese Medicine of Zhejiang Province, Hangzhou, Zhejiang, China
Received 2019-1-10; Accepted 2019-8-28; Published 2019-10-20
Abstract
Gene methylation is an epigenetic alteration in hepatocellular carcinoma (HCC), and hepatitis B virus (HBV) plays a crucial role in carcinogenesis of HCC. However, the association between gene methylation and HBV infection in HCC remains unclear. In our study, we conducted a comprehensive meta-analysis to evaluate the association. A total of 1,148 studies were initially retrieved from some literature database. After a four-step filtration, we obtained 69 case-control studies in this meta-analysis. Our results showed six genes (p16, RASSF1A, GSTP1, APC, p15 and SFRP1) in HBV-positive carcinoma tissues, one gene (GSTP1) in HBV-positive adjacent tissues and two gene (p16 and APC) in HBV-positive carcinoma serums, which were significantly hypermethylated. Subgroup meta-analysis by geographical populations revealed that GSTP1 methylation was significantly higher in HBV-positive carcinoma tissues in China and Japan. In addition, p16 and RASSF1A methylation was significantly higher in China but not in Japan. Our study indicated that HBV infection could induce DNA methylation in HCC and DNA methylation could lead to the development of HBV-related HCC.
Keywords: meta-analysis, gene methylation, hepatitis B virus, hepatocellular carcinoma
Introduction
Primary liver cancer (PLC) is one of the most common malignant diseases, ranked sixth in incidence and fourth in mortality worldwide [1, 2]. Hepatocellular carcinoma (HCC) is the most common type of PLC and it accounts for approximate 90% of PLC. HCC develops from normal liver tissues and cirrhosis through a multistep process. Hepatocarcinogenesis results from the interaction of environmental and hereditary factors, including aflatoxin exposure, alcohol abuse, genetic hemochromatosis and hepatitis virus infection [3-7]. Hepatitis B virus (HBV) infection is the most prominent risk factor for the carcinogenesis of HCC, and HBV-related HCC accounts for over 80% of all HCC patients globally [8]. Pathogenesis of HBV-induced HCC involves genetic and epigenetic mechanisms [9-11], especially DNA methylation [12, 13].
DNA methylation, an essential epigenetic modification, plays a crucial role in transcriptional regulation and gene expression [14-16]. Methylation of the gene promoter may induce chromatin alteration to inhibit the access of the transcriptional machinery and cause gene silencing and expression decrease, thus affecting cellular biological processes [17, 18]. Hypermethylation of tumor suppressor genes (TSGs) can lead to carcinogenesis of many types of carcinomas, including hepatocellular carcinoma [19, 20]. In HCC, gene methylation may be enhanced by HBV infection to induce hepatocarcinogenesis [21-23]. HBV-induced DNA methylation may provide a useful target for the treatment of HBV-related HCC.
Several studies also showed that geographic variation existed in DNA methylation of HBV-related HCC [19, 24]. However, these studies had a small number of samples which might lead to inaccurate results. In this study, we conducted a comprehensive meta-analysis based on HBV-related HCC association studies on DNA methylation. In addition, we performed subgroup meta-analysis to evaluate DNA methylation in HBV-related HCC in geographical populations. This meta-analysis provided new clues that HBV infection could induce DNA methylation in HCC and also showed that DNA methylation could contribute to development of HBV-related HCC in some geographical populations.
Materials and methods
Study identification
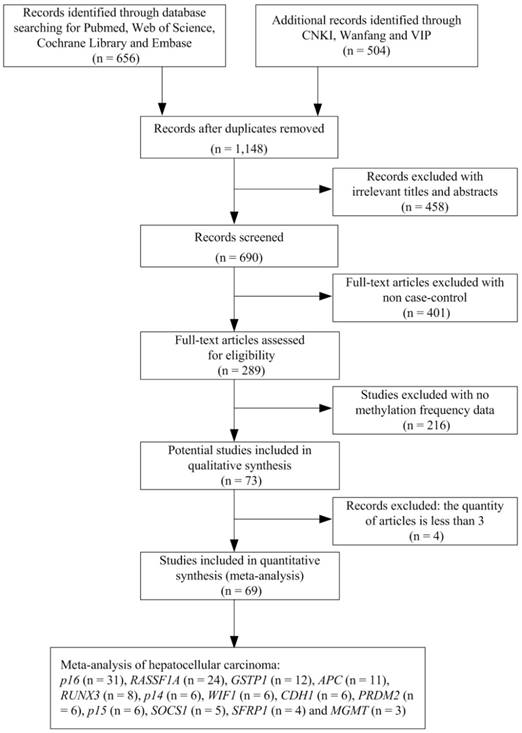
We undertook a systematic review of the literature via PubMed, Web of Science, Embase, CNKI, Wanfang and VIP using the following search terms: “hepatocellular carcinoma or primary liver cancer or hepatic carcinoma or liver tumor or HCC” and “Hepatitis B virus or HBV” and “DNA methylation or gene methylation” as keywords in titles and abstracts. The search was updated until July 1, 2018. The selection of studies in this meta-analysis had to meet the following criteria: (1) they were the case-control studies with gene methylation and HBV infection in human HCC; (2) they contained the sufficient gene methylation data in order to calculate the odds ratios (ORs) and 95 % confidence intervals (CIs). We excluded the studies such as letters, books, reviews, case reports, abstracts, editorials and conference articles. Articles without case-control or methylation data were also excluded. In addition, we removed the genes with the quantity of articles less than three. The stepwise procedure of the selected studies was shown in the flow diagram of Figure 1. The meta-analysis was based on the Preferred Reporting Items for Systematic Reviews and Meta- Analysis (PRISMA) statement (Supplementary Document 1).
Flow diagram of the stepwise selection from the relevant studies.
Data extraction and quality assessment
Four authors (CZ, CH, XS and WY) independently applied the inclusion criteria, retrieved and extracted the data, and resolved any inconsistency through discussion with the fifth author (YL). For each included study, we extracted the following information: first author's name, publication year, sample types, HBV infection status, geographical populations and the numbers of cases and controls (Supplementary Table 1).
Four authors (CZ, CH, XS and XH) independently conducted the quality assessment, and resolved any inconsistency through discussion with the fifth author (YL). The Newcastle-Ottawa Scale was used for the methodological quality assessment of the included studies.
Meta-analysis
The Review Manager software (version 5.2, Cochrane Collaboration, Oxford, UK) was used in the meta-analysis. The ORs and the corresponding 95% CIs were calculated and computed in the forest plots for each gene to evaluate the contribution of gene methylation and HBV infection to development of HCC. A fixed-effect model was applied for the meta-analysis with the moderate heterogeneity (I2 < 50%), otherwise a random-effect model was used. STATA software (version 12.0 Stata Corporation College Station, TX) was used for the meta-regression analysis to assess the potential sources of heterogeneity. Funnel plots were used to check the publication bias among the included studies. P values (p < 0.05) were considered to be significant.
Results
A total of 1,148 studies were initially retrieved by our literature search, using the keywords “hepatocellular carcinoma or primary liver cancer or hepatic carcinoma or liver tumor or HCC” and “Hepatitis B virus or HBV” and “DNA methylation or gene methylation” from PubMed, Web of Science, Embase, CNKI, Wanfang and VIP literature database. After a series of selection procedure shown in Figure 1, we excluded 458 irrelevant studies, 401 non-case control studies, 216 studies without methylation frequency data and four studies less than three articles. Thus, a total of 69 eligible studies were included in the final meta-analysis (Supplementary Document 2). The 69 case-control studies (from 1999 to 2016) included 2,423 HBV-positive carcinoma tissues, 681 HBV-negative carcinoma tissues, 401 HBV-positive adjacent tissues, 188 HBV-negative adjacent tissues, 690 HBV-positive carcinoma serums and 172 HBV-negative carcinoma serums within 13 genes. Among these 13 genes, the meta-analysis of p16, RASSF1A and APC genes methylation was performed between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues, HBV-positive adjacent tissues and HBV-negative adjacent tissues and finally between HBV-positive carcinoma serums and HBV-negative carcinoma serums. The meta-analysis of GSTP1, RUNX3, SOCS1, CDH1 and SFRP1 genes methylation was performed between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues and between HBV-positive adjacent tissues and HBV-negative adjacent tissues. The meta-analysis of p14, WIF1, PRDM2, p15 and MGMT gene methylation was performed between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues.
For 13 genes reported in at least three studies between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues (Table 1), no evidence of statistical heterogeneity was observed for 12 genes, including p16 (I2 = 49%), RASSF1A (I2 = 46%), GSTP1 (I2 = 0%), APC (I2 = 0%), RUNX3 (I2 = 0%), p14 (I2 = 39%), WIF1 (I2 = 0%), PRDM2 (I2 = 15%), p15 (I2 = 0%), SOCS1 (I2 = 8%), SFRP1 (I2 = 0%) and MGMT (I2 = 0%). No visual bias was shown in the meta-analysis of the above 12 genes (Figure 2). Our data also demonstrated a significant heterogeneity of the remaining CDH1 gene (I2 = 56%). Therefore, the random effect test was applied for the meta-analysis of CDH1 gene. The funnel plot was shown in Figure 2.
Characteristics of 13 genes methylation between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues in HCC
| Gene | Studies (n) | Overall OR (95% CI) | I2 | P value | HBV-positive carcinoma tissues/HBV-negative carcinoma tissues |
|---|---|---|---|---|---|
| p16 | 24 | 2.87 [2.05, 4.01] | 49% | <0.00001 | 708/224 |
| RASSF1A | 18 | 2.05 [1.44, 2.92] | 46% | <0.0001 | 914/228 |
| GSTP1 | 12 | 2.50 [1.52, 4.12] | 0% | 0.0003 | 234/129 |
| APC | 10 | 1.87 [1.11, 3.14] | 0% | 0.02 | 342/120 |
| RUNX3 | 8 | 1.32 [0.81, 2.16] | 0% | 0.27 | 292/104 |
| p14 | 6 | 1.75 [0.93, 3.29] | 39% | 0.08 | 191/93 |
| WIF1 | 6 | 0.73 [0.43, 1.25] | 0% | 0.25 | 430/62 |
| CDH1 | 6 | 1.01 [0.61, 1.67] | 56% | 0.96 | 255/111 |
| PRDM2 | 6 | 1.61 [0.81, 3.20] | 15% | 0.17 | 156/51 |
| p15 | 6 | 2.66 [1.25, 5.64] | 0% | 0.01 | 103/55 |
| SOCS1 | 5 | 1.60 [0.75, 3.45] | 8% | 0.23 | 58/52 |
| SFRP1 | 4 | 3.43 [1.19, 9.88] | 0% | 0.02 | 65/60 |
| MGMT | 3 | 1.46 [0.62, 3.45] | 0% | 0.39 | 114/41 |
(A) Funnel plots of 13 genes methylation between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues in HCC in the meta-analysis. (B) Funnel plots of eight genes methylation between HBV-positive adjacent tissues and HBV-negative adjacent tissues in HCC in the meta-analysis. (C) Funnel plots of three genes methylation between HBV-positive carcinoma serums and HBV-negative carcinoma serums in HCC in the meta-analysis.

For eight genes reported in at least three studies between HBV-positive adjacent tissues and HBV-negative adjacent tissues (Table 2), no evidence of statistical heterogeneity was observed for six genes, including p16 (I2 = 0%), GSTP1 (I2 = 0%), APC (I2 = 0%), RUNX3 (I2 = 0%), CDH1 (I2 = 11%) and SFRP1 (I2 = 0%). No visual bias was shown in the meta-analysis of the above six genes (Figure 2). Our data also demonstrated a significant heterogeneity of the remaining two genes that included RASSF1A (I2 = 63%) and SOCS1 (I2 = 72%). Therefore, random effect tests were applied for the meta-analysis of the above two genes. Their funnel plots were shown in Figure 2.
Characteristics of eight genes methylation between HBV-positive adjacent tissues and HBV-negative adjacent tissues in HCC
| Gene | Studies (n) | Overall OR (95% CI) | I2 | P value | HBV-positive adjacent tissues/HBV-negative adjacent tissues |
|---|---|---|---|---|---|
| p16 | 10 | 1.39 [0.71, 2.71] | 0% | 0.33 | 274/103 |
| GSTP1 | 6 | 2.54 [1.04, 6.21] | 0% | 0.04 | 84/63 |
| RASSF1A | 3 | 0.94 [0.08, 11.22] | 63% | 0.96 | 63/29 |
| APC | 3 | 0.62 [0.07, 5.28] | 0% | 0.66 | 21/24 |
| RUNX3 | 3 | 3.07 [0.40, 23.77] | 0% | 0.28 | 21/24 |
| SOCS1 | 3 | 1.79 [0.20, 16.36] | 72% | 0.61 | 39/36 |
| CDH1 | 3 | 0.91 [0.35, 2.36] | 11% | 0.85 | 78/44 |
| SFRP1 | 3 | 1.86 [0.40, 8.62] | 0% | 0.43 | 40/55 |
For three genes reported in at least three studies between HBV-positive carcinoma serums and HBV-negative carcinoma serums (Table 3), no evidence of statistical heterogeneity was observed for two genes, including RASSF1A (I2 = 0%) and APC (I2 = 0%). No visual bias was shown in the meta-analysis of the above two genes and their funnel plots were shown in Figure 2. Our data also demonstrated a significant heterogeneity of the remaining p16 gene (I2 = 55%). Therefore, the random effect test was applied for the meta-analysis of p16 gene. The funnel plot was shown in Figure 2.
Characteristics of three genes methylation between HBV-positive carcinoma serums and HBV-negative carcinoma serums in HCC
| Gene | Studies (n) | Overall OR (95% CI) | I2 | P value | HBV-positive carcinoma serums/HBV-negative carcinoma serums |
|---|---|---|---|---|---|
| p16 | 10 | 2.51 [1.16, 5.44] | 55% | 0.02 | 460/122 |
| RASSF1A | 7 | 1.30 [0.76, 2.23] | 0% | 0.34 | 371/69 |
| APC | 3 | 5.11 [2.09, 12.52] | 0% | 0.0004 | 228/46 |
As shown in Table 1, the meta-analysis of p16 gene was involved with 24 studies between 708 HBV-positive carcinoma tissues and 224 HBV-negative carcinoma tissues. Our results revealed that the frequency of p16 gene methylation in HBV-positive carcinoma tissues was significantly higher than HBV-negative carcinoma tissues (the overall OR = 2.87, 95% CI = 2.05-4.01, p < 0.00001). The meta-analysis of RASSF1A methylation between 914 HBV-positive carcinoma tissues and 228 HBV-negative carcinoma tissues indicated a statistical difference (the overall OR = 2.05, 95% CI = 1.44-2.92, p < 0.0001). The same consequence was also found in the other four genes including GSTP1 (the overall OR = 2.50, 95% CI = 1.52-4.12, p = 0.0003), APC (the overall OR = 1.87, 95% CI = 1.11-3.14, p = 0.02), p15 (the overall OR = 2.66, 95% CI = 1.25-5.64, p = 0.01) and SFRP1 (the overall OR = 3.43, 95% CI = 1.19-9.88, p = 0.02). Our meta-analysis was unable to find any statistical significance between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues for the methylation of the remaining seven genes, including RUNX3, p14, WIF1, CDH1, PRDM2, SOCS1 and MGMT.
As shown in Table 2, the meta-analysis of GSTP1 gene was involved with six studies between 84 HBV-positive adjacent tissues and 63 HBV-negative adjacent tissues. Our results revealed that the frequency of GSTP1 gene methylation in HBV-positive adjacent tissues was significantly higher than HBV-negative adjacent tissues (the overall OR = 2.54, 95% CI = 1.04-6.21, p = 0.04). However, our meta-analysis was unable to find any statistical significance between HBV-positive adjacent tissues and HBV-negative adjacent tissues for the methylation of the remaining seven genes, including p16, RASSF1A, APC, RUNX3, SOCS1, CDH1 and SFRP1.
As shown in Table 3, our meta-analysis showed statistical significance between HBV-positive carcinoma serums and HBV-negative carcinoma serums for the methylation of two genes, including p16 (the overall OR = 2.51, 95% CI = 1.16-5.44, p = 0.02) and APC (the overall OR = 5.11, 95% CI = 2.09-12.52, p = 0.0004). However, our meta-analysis was unable to find any statistical significance between HBV-positive carcinoma serums and HBV-negative carcinoma serums for the methylation of the remaining RASSF1A gene.
In this meta-analysis, we selected various geographical populations to analyze any source of heterogeneity. We didn't find any significance in 13 methylated genes between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues (Supplementary Table 2), eight methylated genes between HBV-positive adjacent tissues and HBV-negative adjacent tissues (Supplementary Table 3) and three methylated genes between HBV-positive carcinoma serums and HBV-negative carcinoma serums (Supplementary Table 4) in geographical populations. Our meta-analysis indicated that the heterogeneity was not contributed by geographical populations.
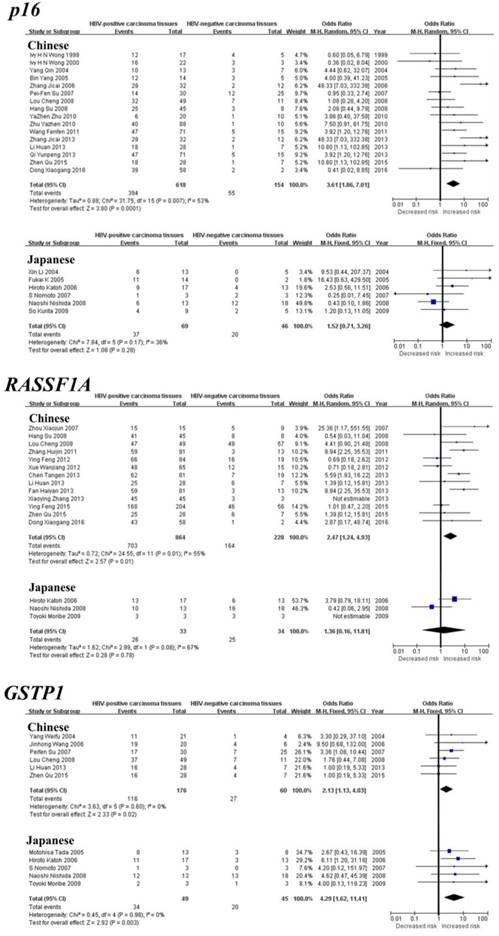
Subgroup meta-analysis by geographical populations was performed for p16, RASSF1A, GSTP1, APC and RUNX3 between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues. As shown in Figure 3, we found a statistical difference between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues for GSTP1 methylation in China from six studies (OR = 2.13, 95% CI = 1.13-4.03, I2 = 0%, p = 0.02) and in Japan from five studies (OR = 4.29, 95% CI = 1.62-11.41, I2 = 0%, p = 0.003). In addition, there was a significant geographical difference in the meta-analysis of p16 methylation in China (OR = 3.61, 95% CI = 1.86-7.01, I2 = 53%, p = 0.0001), but not in Japan (OR = 1.52, 95% CI = 0.71-3.26, I2 = 36%, p = 0.28). A similar result was also found in RASSF1A (China: OR = 2.47, 95% CI = 1.24-4.93, I2 = 55%, p = 0.01; Japan: OR = 1.36, 95% CI = 0.16-11.81, I2 = 67%, p = 0.78). The subgroup meta-analysis of APC and RUNX3 was unable to observe any significant result in each geographical population (Supplementary Figure 1).
Forest plots of p16, RASSF1A and GSTP1 methylation between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues in HCC in the meta-analysis.
Discussion
Our meta-analysis included numerous studies that evaluated the contribution of HBV infection to DNA methylation in HCC. The meta-analysis focused on 13 genes between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues, eight genes between HBV-positive adjacent tissues and HBV-negative adjacent tissues and three genes between HBV-positive carcinoma serums and HBV-negative carcinoma serums. In six genes (p16, RASSF1A, GSTP1, APC, p15 and SFRP1), there was significant difference in the level of hypermethylation between HBV-positive carcinoma tissues and HBV-negative carcinoma tissues, revealing that HBV infection could induce methylation of these genes in HCC. One gene (GSTP1) hypermethylation showed significant difference between HBV-positive adjacent tissues and HBV-negative adjacent tissues, revealing that HBV infection could induce methylation of this gene in the early stage of hepatocarcinogenesis.
This meta-analysis showed that methylation of GSTP1 gene in HBV-positive carcinoma tissues was significantly higher than in HBV-negative carcinoma tissues, and the same consequence was also found in adjacent tissues. This revealed that methylation of GSTP1 gene could contribute to the whole process of the hepatocarcinogenesis of HBV-related HCC. In addition, methylation of p16, RASSF1A, APC, p15 and SFRP1 genes in HBV-positive carcinoma tissues was significantly higher than HBV-negative carcinoma tissues but not adjacent tissues, revealing that methylation of these five genes may play a significant role in the middle-late stage of the hepatocarcinogenesis process.
On the other hand, this meta-analysis showed that methylation of RASSF1A gene in HBV-positive carcinoma tissues was significantly higher than HBV-negative carcinoma tissues, while the same phenomenon was not found in carcinoma serums. The effects of this gene methylation in two sample types are different, due to the diversities of specificity and sensitivity in different sample types [25]. Also, subgroup meta-analysis by geographical populations found that methylation of GSTP1 was significant in both China and Japan. However, methylation of p16 and RASSF1A genes was significant only in China, but not in Japan. The effects of these genes methylation are different, due to the diversities of hereditary in the different geographic regions [19, 26]. In addition, the lack of association for these genes in Japanese population might be due to the small size of samples. For example, the size of carcinoma tissue samples in the meta-analysis of p16 methylation in Chinese population was larger than the sample size of Japanese population, which might lead to the different associations.
Some studies showed that DNA methylation was associated with HBV-related HCC [21, 27-29]. However, the mechanism of HBV infection in DNA methylation to induce hepatocarcinogenesis has not been clarified. Some studies found that hepatitis B virus X protein (HBx) could play a crucial role in epigenetic tumorigenesis of HBV-related HCC [30-32]. HBx could increase the expressions of DNA-methyltransferase1 (DNMT1) and DNMT3b to induce hypermethylation of tumor suppressor genes [28, 33-35]. DNA hypermethylation could silence the expressions of TSGs and lead to hepatocarcinogenesis. In this meta-analysis, methylation of some TSGs (p16, RASSF1A, GSTP1, APC, p15 and SFRP1) could be induced by HBV infection, while other TSGs methylation could not be induced. We speculated that interactions of HBx and DNMTs of these TSGs caused the different effects. In the future, we need to explore the biological mechanisms of HBx-induced DNA methylation in HCC.
However, there were several limitations in this meta-analysis. Firstly, selection bias might exist because literatures only in English and Chinese were included in the meta-analysis. Secondly, this study focused on the genes with at least three independent studies and excluded those two studies within large size of samples. Thirdly, the main geographical populations of the meta-analysis were Chinese and Japanese. In the future, studies in other geographical populations were needed to explore the contribution of DNA methylation to HBV-related HCC.
In summary, this meta-analysis showed that DNA methylation and HBV infection were associated with the development of HCC. HBV infection could induce DNA methylation in HCC and DNA methylation contributed to the development of HBV-related HCC. Our results provided new clues for epigenetic therapy of HBV-related HCC.
Abbreviations
PLC: Primary liver cancer; HCC: Hepatocellular carcinoma; HBV: Hepatitis B virus; HBx: Hepatitis B virus X protein; CNKI: China National Knowledge Infrastructure; OR: Odds ratio; CI: Confidence interval; RASSF1A: Ras association domain family member1A; GSTP1: Glutathione S-transferase pi 1; APC: Adenomatous polyposis coli; RUNX3: Runt-related transcription factor 3; WIF1: WNT inhibitory factor 1; CDH1: E-cadherin; PRDM2: PR domain containing 2; SOCS1: Suppressor of the cytokine signalling 1; SFRP1: Secreted frizzled-related protein 1; MGMT: O6-methylguanine-DNA methyltransferase; DNMT1: DNA-methyltransferase 1; DNMT3b: DNA-methyltransferase 3b; TSGs: Tumor suppressor genes.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the grants from: National Natural Science Foundation of China (31100919, 81371469 and 81301891), Natural Science Foundation of Zhejiang Province (LY15H160015 and LQ19H030002), the Specialized Research Fund for the Social Development of Hangzhou (20160533B21) and the Scientific Innovation Fund of the Affiliated Hospital of Hangzhou Normal University.
Author contributions
CZ, CH and YL conceived and designed the research. CZ, CH, XS and WY analyzed the data. All authors made substantial contributions towards drafting the manuscript, reviewed the final manuscript for intellectual content, and authorized the submission. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34
3. Chu YJ, Yang HI, Wu HC. et al. Aflatoxin B1 exposure increases the risk of hepatocellular carcinoma associated with hepatitis C virus infection or alcohol consumption. Eur J Cancer. 2018;94:37-46
4. Stickel F, Buch S, Nischalke HD. et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol-related cirrhosis. Am J Gastroenterol. 2018;113(10):1475-1483
5. Ng CKY, Di Costanzo GG, Tosti N. et al. Genetic profiling using plasma-derived cell-free DNA in therapy-naive hepatocellular carcinoma patients: a pilot study. Ann Oncol. 2018;29(5):1286-1291
6. Lim CJ, Lee YH, Pan L. et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut. 2019;68(5):916-927
7. Dhanasekaran R, Nault JC, Roberts LR. et al. Genomic Medicine and Implications for Hepatocellular Carcinoma Prevention and Therapy. Gastroenterology. 2019;156(2):492-509
8. Bertoletti A, Brunetto M, Maini MK. et al. T cell receptor-therapy in HBV-related hepatocellularcarcinoma. Oncoimmunology. 2015;4(6):e1008354
9. Wong CM, Tsang FH, Ng IO. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 2018;15(3):137-151
10. Xu F, Li CH, Wong CH. et al. Genome-Wide Screening and Functional Analysis Identifies Tumor Suppressor Long Noncoding RNAs Epigenetically Silenced in Hepatocellular Carcinoma. Cancer Res. 2019;79(7):1305-1317
11. Liu KY, Wang LT, Hsu SH. Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma. Cancers (Basel). 2018:10 (1)
12. Zheng YF, Lu X, Zhang XY. et al. The landscape of DNA methylation in hepatocellular carcinoma. J Cell Physiol. 2019;234(3):2631-2638
13. Cai C, Wang W, Tu Z. Aberrantly DNA Methylated-Differentially Expressed Genes and Pathways in Hepatocellular Carcinoma. J Cancer. 2019;10(2):355-366
14. Harris CJ, Scheibe M, Wongpalee SP. et al. A DNA methylation reader complex that enhances gene transcription. Science. 2018;362(6419):1182-1186
15. Greenfield R, Tabib A, Keshet I. et al. Role of transcription complexes in the formation of the basal methylation pattern in early development. Proc Natl Acad Sci U S A. 2018;115(41):10387-10391
16. Kinde B, Wu DY, Greenberg ME. et al. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc Natl Acad Sci U S A. 2016;113(52):15114-15119
17. Ramakrishnan S, Pokhrel S, Palani S. et al. Counteracting H3K4 methylation modulators Set1 and Jhd2 co-regulate chromatin dynamics and gene transcription. Nat Commun. 2016;7:11949
18. Black JC, Whetstine JR. Chromatin landscape: methylation beyond transcription. Epigenetics. 2011;6(1):9-15
19. Zhang C, Li J, Huang T. et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget. 2016;7(49):81255-81267
20. Huang Y, Wei L, Zhao RC. et al. Predicting hepatocellular carcinoma development for cirrhosis patients via methylation detection of heparocarcinogenesis-related genes. J Cancer. 2018;9(12):2203-2210
21. Kao WY, Yang SH, Liu WJ. et al. Genome-wide identification of blood DNA methylation patterns associated with early-onset hepatocellular carcinoma development in hepatitis B carriers. Mol Carcinog. 2017;56(2):425-435
22. Fan H, Cui Z, Zhang H. et al. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene. 2017;36(17):2435-2445
23. Kanda T, Goto T, Hirotsu Y. et al. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int J Mol Sci. 2019:20 (6)
24. Konstantinou D, Deutsch M. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viralinteractions and management. Ann Gastroenterol. 2015;28(2):221-228
25. Ji X, Guan C, Jiang X. et al. Diagnostic accuracy of DNA methylation for head and neck cancer varies by sample type and number of markers tested. Oncotarget. 2016;7(48):80019-80032
26. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13(2):123-135
27. Ye C, Tao R, Cao Q. et al. Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma. Int J Oncol. 2016;49(2):589-602
28. Charostad J, Astani A, Goudarzi H. et al. DNA methyltransferases in virus-associated cancers. Rev Med Virol. 2019;29(2):e2022
29. Zhao Y, Xue F, Sun J. et al. Genome-wide methylation profiling of the different stages of hepatitis B virus-related hepatocellular carcinoma development in plasma cell-free DNA reveals potential biomarkers for early detection and high-risk monitoring of hepatocellular carcinoma. Clin Epigenetics. 2014;6(1):30
30. Zhao Z, Hu Y, Shen X. et al. HBx represses RIZ1 expression by DNA methyltransferase 1 involvement in decreased miR-152 in hepatocellular carcinoma. Oncol Rep. 2017;37(5):2811-2818
31. Zhang B, Han S, Feng B. et al. Hepatitis B virus X protein-mediated non-coding RNA aberrations in the development of human hepatocellular carcinoma. Exp Mol Med. 2017;49(2):e293
32. Dang S, Zhou J, Chen Y. et al. Dynamic expression of ZNF382 and its tumor-suppressor role in hepatitis B virus-related hepatocellular carcinogenesis. Oncogene. 2019;38(24):4804-4819
33. Arzumanyan A, Friedman T, Kotei E. et al. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31(5):563-572
34. Lee H, Sohn JB, Kim SS. et al. DNA methyltransferase inhibitor assay system based on the HBx-induced DNA methylation of E-cadherin. Biochem Biophys Res Commun. 2013;438(3):540-545
35. Lee H, Jeong H, Lee SY. et al. Hepatitis B Virus X Protein Stimulates Virus Replication Via DNA Methylation of the C-1619 in Covalently Closed Circular DNA. Mol Cells. 2019;42(1):67-78
Author contact
![]() Corresponding author: Yongqiang Li, e-mail: liyongqiang19701217com; Cheng Zhang, e-mail: zhangcheng666emailnet
Corresponding author: Yongqiang Li, e-mail: liyongqiang19701217com; Cheng Zhang, e-mail: zhangcheng666emailnet