Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(25):6327-6340. doi:10.7150/jca.34171 This issue Cite
Review
Molecular Mechanisms and Anticancer Therapeutic Strategies in Vasculogenic Mimicry
Xue Zhang*, Jigang Zhang*, Heming Zhou, Guorong Fan, Qin Li ![]()
Department of Clinical Pharmacy, Shanghai General Hospital, Shanghai Jiao Tong University School of medicine, No.100 Haining Road, Shanghai, 200080, P.R. China.
* These authors contributed equally to this paper.
Received 2019-7-4; Accepted 2019-8-31; Published 2019-10-18
Abstract
Vasculogenic mimicry (VM) is a vascular formation mechanism used by aggressive tumor cells. VM provides an alternative pathway for adequate blood perfusion and challenges the traditional angiogenesis mechanism that depends only on endothelial cells (ECs), as VM-forming tumor cells express a mixed endothelial/tumor phenotype. VM is closely correlated with tumor invasion, migration, and progression. Hence, anticancer therapeutic strategies targeting VM biogenesis are essential. It is widely acknowledged that the VM formation mechanism involves multiple pathways. The purpose of this review is to describe the potential molecular mechanisms related to different pathways and discuss the involvement of microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) in VM formation. Moreover, we discuss the significance of VM in clinical practice and present new anticancer therapeutic strategies that target VM.
Keywords: vasculogenic mimicry, molecular mechanisms, miRNAs, lncRNAs, circRNAs
Introduction
The tumor formation mechanisms are complex and diverse. Tumors characterized by a rapid growth speed have unfavorable prognosis, and high mortality, as they are difficult to diagnose early and there are no effective measures to treat them. Angiogenesis is one of the mechanisms through which tumor cells have an adequate blood supply and nutrition for their growth. However, many conventional antiangiogenic drugs adopted clinically produce disappointing results. The discovery of vasculogenic mimicry (VM) provides a new therapeutic opportunity for patients battling aggressive tumors for which treatment with conventional antiangiogenic agents is limited. Since VM was first discovered, it has been consistently found in different cancer types, including hepatocellular carcinoma (HCC) [1], breast carcinomas [2], ovarian carcinoma [3], lung cancer [4], glioma [5], and renal carcinoma [6]. Despite the high amount of literature on VM, the biogenesis mechanisms of VM are still not fully elucidated, and further research on VM base biology is critically important. The purpose of this review is to discuss the potential molecular mechanisms hypothesized for VM, and the involvement of miRNAs, lncRNAs and circRNAs as well as discuss the clinical significance of VM and report new anticancer drugs that target VM.
Conceptual progress on VM biology
Definition of Vasculogenic mimicry. VM is the de novo formation of a perfused, matrix-rich, vasculogenic-like network of blood vessels by aggressive tumor cells. VM mimics the embryonic vascular network pattern to provide sufficient blood supply for the growth of the tumor. The initial morphologic and molecular characterization of VM was by the Maniotis group, which revealed that human melanoma cells formed channels, networks, and tubular structures that are rich in laminin, collagens IV and VI, and heparin sulfate proteoglycans. The newly formed network contained plasma and red blood cells to facilitate tumor perfusion, remold the extracellular matrix, and change the cell phenotype [7].
Plasticity and perfusion ability of VM. Cancer cells capable of VM present multipotent, stem cell-like phenotypes, including both a tumor and endothelial phenotype, indicating a remarkable degree of plasticity. A seminal example of VM functional plasticity was the transplantation of fluorescently labeled metastatic melanoma cells into a surgically induced ischemic microenvironment in the hind limbs of nude mice, which demonstrated the powerful influence of the tumor microenvironment on the transendothelial differentiation of aggressive melanoma cells and provided a new perspective on tumor cell plasticity [8]. A previous study investigated the plasticity of tumor cells in melanoma VM, reporting that the hypoxic microenvironment in metastases promotes to a phenotype switch that allows melanoma cells to physically contribute to the blood vessel formation [9]. A recent study revealed that the Epstein-Barr virus (EBV) induced tumor cell plasticity by promoting VM formation [10]. VM facilitates perfusion in rapidly growing tumors by transferring fluid from leaky vessels and/or by linking the VM network with the endothelial-lined vasculature. This was demonstrated by Doppler imaging of microbeads circulation, showing physiologic perfusion of blood between mouse endothelial-lined neovasculature and VM networks in human melanoma xenografts [11].
Types of VM. In aggressive malignant tumors, two distinctive VM patterns have been identified: matrix VM and tubular VM. Matrix VM is composed of a basement membrane that is surrounded by tumor cells rich in fibronectin, collagens, and laminin. The presence of matrix VM is an unfavorable prognostic factor compared to tubular VM in HCC patients [12]. Tubular VM is composed of tumor cells that mimic the normal endothelium to form perfused channels. However, in many tumors, it is common to have both angiogenic and non-angiogenic areas. Interestingly, in the absence of angiogenesis and normal blood vessels exploitation, VM can act in a non-angiogenic way to provide oxygen and nutrients to the tumor [13].
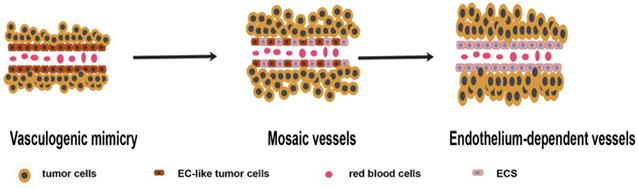
Microcirculation patterns associated with VM. Different studies have proposed three microcirculation patterns: VM, mosaic vessels (MVs), and endothelium-dependent vessels (EVs), representing different stages of tumor growth. In the early stages, VM plays a major role in providing blood supply. With the increase in tumor size, tumor cells lining the wall of VM vessels are replaced by endothelium cells. At this point, MVs represent a transitional state between EVs and VM. Finally, EVs become the major blood supply pattern [14] (Figure 1). A recent research showed that VM acts as a part of the functional microcirculation, cancer cells within the tumor-lined vascular channels can easily transfer into endothelial-lined blood vessels in VM angiogenesis junction, consequently, contributing to tumor invasion and metastasis [15].
Schematic illustration showing the three microcirculation patterns associated with VM. In the early stages, VM play a major role in providing blood supply. With the increase of tumor size, tumor cells lining the wall of VM vessels are replaced by endothelium. MVs is the transitional state between EVs and VM. Finally, EVs become the major pattern of blood supply.
VM assessment. A positive staining pattern with Periodic Acid-Schiff stain (PAS) along with the absence of CD31 or CD34, two classical markers, indicates the existence of matrix-associated vascular channels. Thus, VM can be diagnosed by performing immunohistochemical analysis (IHC) in tumor samples. VM positive samples have a positive PAS staining pattern and a negative CD31 staining pattern [7]. Interestingly, a recent study found that VM channels also exist in CD31/CD34-positive gastric adenocarcinoma cells, probably because the genetically deregulated tumor cells express angiogenic and vasculogenic markers [16].
Potential molecular mechanisms involved in VM
Relationship among EMT, CSCs, and VM. The mechanism of VM biogenesis is closely related to the epithelial-to-mesenchymal transition (EMT) and to cancer stem cells (CSCs).
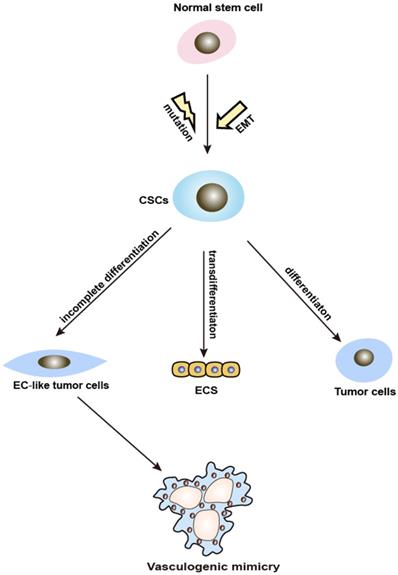
CSC and VM. CSCs are a small proportion of tumor possessing reversible self-renewal capabilities, which can differentiate into multiple cell types. It is widely accepted that normal stem cells differentiate into CSCs through a mutation process. However, recent findings indicate that CSCs could have originated and be maintained through EMT [17]. The increased knowledge about CSCs revealed a close connection to VM. It has been found that CSCs stimulate VM in the tumorigenic microenvironment by differentiating or transdifferentiating tumor and ECs, lining up to form branching tubes and lumens resembling a vascular network which provides nutrition for the tumor mass. Ultimately, the tubes extend, merge, and start transferring blood cells [18] (Figure 2). It has also been demonstrated that because of the loss of stem cell capacity, VM gradually skews toward a vascular phenotype [19]. Additionally, a handful of studies suggested that CSCs markers are also associated with VM. VM-forming cells contributing to the tumorigenicity are characterized by the expression of the CSCs markers CD133 and aldehyde dehydrogenase 1 (ALDH1) [20]. Liu et.al revealed that USP44+ CSCs subclones with an ALDH1+/USP44+/IL6+/IL8+ phenotype may promote VM and tumor aggressiveness [21]. In conclusion, CSCs can induce VM formation in two ways: incomplete differentiation and the up-regulation of CSCs-associated molecules. Further investigations of CSCs markers to target VM and the link between CSCs and VM may contribute to the discovery of new anticancer treatments to prevent VM formation.
Schematic illustration showing the involvement of CSCs and EMT in VM formation. VM: vasculogenic mimicry; CSCs: cancer stem cells; EMT: epithelial-mesenchymal transition.
EMT and VM. The involvement of EMT in cancer is currently widely accepted. EMT is a biological process that plays a central physiological role during embryogenesis and a pathological role in cancer progression. The activation of EMT triggers tumor cell invasion and the formation of metastasis in distant organs. The high expression of EMT-associated adhesion molecules can contribute to the VM-forming process. The molecular mechanisms of ZEB1-induced VM formation were confirmed to be dependent on mediation of the Src signaling pathway, which also plays an important role in EMT as well as in maintaining CSCs properties [22]. Notably, Langer et.al demonstrated that miRNA clusters repressed by ZEB1 stimulated VM through autocrine signaling in breast cancer cells [23]. Further studies showed that EMT regulators such as Twist, Snail, and Slug were closely related to VM [24, 25]. Meng et.al found that Twist1 interacts with Hsp90β, activating VE-cadherin transcription to induce EMT and promote VM in HCC [26].
Both EMT and CSCs confer resistance to chemotherapy and are supposed to be the underlying causes of low survival rates in patients with aggressive tumors. Therefore, given the relationship between EMT, CSCs, and VM, it is plausible to hypothesize therapeutic strategies targeting EMT and CSCs as a promising candidate for VM-related therapies.
Tumor Microenvironment. The extracellular microenvironment (ECM) is an important structural element for tumor cells. ECM can be changed by cellular processes and in turn can exert an influence on cellular activities. This mutual interaction plays an integral role in VM. ECM composition regulates the acquisition of the VM phenotype in CSCs. In the early tumor ECM, CSCs can secrete a high amount of angiopoietin factors such as IGFBP1/2/3, MCP1, IL8, EGF, and VEGF, to stimulate CSCs growth/self-renewal and start the VM process [27]. Distinct collagen architectures in the ECM affect tumor cell motility behaviors linked to VM, and various ECM molecules, such as COL4A1, JAG1, and THBS1, may facilitate the emergence of VM [28]. A study reported that highly aggressive melanoma cells can alter their ECM to form VM tubular networks and that the cooperative interaction of Matrix metalloproteinase-2 (MMP-2) -14 or Ln5γ2 chains is required for VM formation [29].
This discovery opened up a new avenue of research on the role of other MMPs in VM formation. It has been found that, in renal carcinoma cells, downregulation of MMP-9 leads to the decrease of VM formation, revealing that MMP-9 is necessary for VM formation in this cell type [6]. Another study demonstrated that, in melanoma cells, Myoferlin has an effect on VM formation by mediating the expression of MMP-2 and inducing EMT [30]. A further report showed that MMP-2 activates the epidermal growth factor receptor (EGFR), enhances cytoskeletal rearrangement and facilitates VM formation. However, MMP-13 lowers the EGFR/F-actin expression, degrades ECM components and hinders VM formation. Although both MMP-2 and MMP-13 promote the activity of Ln-5 cleavage and degrade ECM components, they exert distinct influences on large cell lung cancer cells [31]. Further research is needed to better understand the role of the distinct MMPs in VM formation. MMP-2, MMP-13, and MMP-9 could be used as therapeutic targets to inhibit VM in anti-tumor therapy.
VE-cadherin. VM-forming tumor cells can express both endothelial and tumor phenotypes. Vascular endothelial-cadherin (VE-cadherin), a calcium-dependent protein, is the key factor regulating cell-cell adhesion in ECs, and it is the most important molecular determinant for the acquisition of VM capabilities [32]. A previous study attempted to elucidate the molecular mechanism of VE-cadherin involvement in VM, showing that the downregulation of VE-cadherin impaired the ability to form VM and the plasticity of aggressive human melanoma. They also showed that VE-cadherin is often overexpressed in highly aggressive tumor cells compared with non-aggressive ones [33]. Interestingly, VE-cadherin could co-localize with phosphorylated Eph receptor tyrosine kinase A2 (EphA2), which is an important factor promoting the formation of a vessel-like network [34]. A recent study reported that VE-cadherin-positive small cell lung cancer (SCLC) cells were able to show VM and were more resistant to cisplatin than VE-cadherin negative cells [4]. Another study revealed that in melanoma cells the activation of focal adhesion kinase (FAK) increased the expression of VE-cadherin and was positively correlated to VM formation [35].
EphA2. The main role of VE-cadherin in VM is to mediate EphA2, an epithelial cell-associated kinase that is phosphorylated when bound to its ligand, ephrin-A1 [36]. Knockdown of VE-cadherin reduced the phosphorylation of EphA2 on the cell surface, while down-regulation of EphA2 expression had no effect on VE-cadherin, suggesting that EphA2 may be a downstream regulator of VE-cadherin expression [37]. Furthermore, it has been demonstrated in vivo that the reduction of EphA2 expression significantly inhibits VM formation and suppresses the invasion, proliferation and clonogenicity capabilities of melanoma tumor cells [38]. Additionally, a previous study showed that EphA2 may be an EMT mediator, contributing to VM formation in head and neck squamous cell carcinoma [39]. Current report revealed that serum activated EphA2 and up-regulated Twist/VE-cadherin, which in turn activated AKT that up-regulated MMP-2 and LAMC2, thereby inducing the invasion and VM of PC-3 human prostate cancer cells [40].
PI3K. Phosphoinositide 3-Kinase (PI3K) is a family of intermediate signaling molecules that are involved in various cell responses, particularly in the signal transmission from the cell surface to the cytoplasm pathway. PI3K proteins are commonly found in a wide variety of cancers and have been recognized as a diagnostic marker of cancer [41]. High PI3K-mediated phosphorylation levels of EphA2 and VE-cadherin increase MMP-14 and MMP-2 activity, which in turn promotes the cleavage of the Ln5γ2-chain into γ2′ and γ2 fragments, ultimately leading to VM formation [42, 43]. It has been shown that AKT, also known as protein kinase B, is a downstream effector of PI3K proteins and is a critical vasculogenesis regulator [44]. In HCC, the PI3K/AKT signaling also regulate the MMP-9 levels and activity, contributing to ECM remodeling towards VM [45]. Hence, inhibiting the PI3K/AKT pathway may provide a new target for anti-VM therapy.
ERK1/2. Similar to the PI3K-mediated signaling pathway, the extracellular signal-regulated kinase (ERK) is another key signaling pathway involved in the cell signal transduction process, participating in various physiological and pathological cellular processes, such as tumor cell invasion, proliferation, migration, and apoptosis [46]. It has been showed that when human hepatoma cells are exposed to hypoxic conditions, the activation of ERK1/2 mediated by mitogen extracellular kinase (MEK) promotes the expression of VE-cadherin, ultimately contributing to VM formation [1].
FAK. FAK is a key molecule in the process of VM formation, through its interaction with PI3K proteins [47]. Additionally, FAK is a downstream effector of EphA2 and plays a critical role in highly aggressive GBC-SD cell growth. It has been showed that upregulation of the EphA2/FAK/Paxillin signaling pathway promoted VM formation [48]. FAK is also a pivotal mediator of the aggressive melanoma phenotype, which is characterized by an increased expression of ERK1/2 to regulate the levels of urokinase activity or to enhance the expression of MMP-2 and MMP-14 activity, both signaling pathways contribute to VM formation [49]. Another recent study reported that in NSCLC, the FAK/AKT signaling pathway is involved in the cyclin-dependent kinase 5-mediated VM formation [50].
VEGF-A/VEGFR. Vascular endothelial growth factor-A (VEGF-A) is a major regulator of vascularization and is critical for vascular EC proliferation, migration, and survival, especially when it is bound to vascular endothelial growth factor receptor 1 (VEGFR1). Activation of the PI3K/AKT pathway by VEGFR1 is involved in endothelial angiogenesis, whereas activation of the Src and ERK1/2 pathways results in tumor cell invasion and proliferation [51]. In addition, in melanoma cells, the integrin-mediated signaling pathway involving VEGF-A/VEGFR1/PI3K/PKCα, is required for VM formation [52]. Moreover, it has been established that in ovarian carcinoma cells, VEGF-A takes part in the formation of VM via indirectly upregulating the expression of EphA2, MMP-2, MMP-9, and VE-cadherin [3]. The increased expression of VEGFR-2, another type of vascular endothelial growth factor receptor, in CSCs-derived tumors, influences the formation of VM networks [27]. A recent study reported that in glioma stem cells, autophagy-induced phosphorylation of the kinase insert domain receptor of VEGFR-2 contributes to VM formation [53]. Other studies supported the evidence that the Hippo pathway is a key regulator of VM and angiogenesis through the VEGF-Induced PI3K/MAPK signaling [54]. VEGF binding to semaphorin4D (SEMA4D) had a synergistic effect on VM formation [55]. However, the role of VEGF signaling in mediating VM is controversial. A recent study reported that VEGF-A silencing upregulated the expression of MMP-2 and VM marker VE-cadherin, leading to VM formation. Thus, VEGF-A inhibition may have a dual biological role that could confound its clinical effectiveness [56].
PEDF. Various evidences suggest a role for pigment epithelium-derived factor (PEDF), a serpin protease inhibitor, in suppressing angiogenesis by inhibiting VEGF-induced phosphorylation of VEGFR-1 [57]. Furthermore, a study showed that PEDF silencing enhances the capability of forming VM in poorly aggressive melanoma cells lines, implying that the expression of PEDF is negatively associated with VM formation [58].
TF, TFPI-1, and TFPI-2. Tissue factor (TF), TF pathway inhibitor 1 (TFPI-1), and TFPI-2 are overexpressed in aggressive melanoma cells. All these three genes play a critical role in mediating the coagulation pathway. TFPI-1 was shown to regulate the anticoagulant function of TF, which is associated with perfusion of VM, whereas TFPI-2 seems to contribute to VM plasticity through increasing the MMP-2 activity and influencing the extracellular matrix remodeling [11].
Nodal. Nodal is a member of the superfamily of the transforming growth factor β (TGF-β) family. It plays a critical role as embryonic morphogen. Nodal regulates tumor cell plasticity, the transendothelial phenotype, and VM formation by binding to cripto-1, ALK4/5/7 and type 2 (ACTR-IIB) proteins to phosphorylate SMAD2/3, which in turn translocates to the nucleus where it mediates gene expression [59, 60]. In situ hybridization found that the expression of Nodal mRNA is consistent with the formation of vasculogenic networks and that the downregulation of Nodal contributes to the reduction of VE-cadherin, thereby influencing VM formation [61]. Recently, it was found that in MCF-7 cells, Nodal, through the Smad2/3 pathway, regulates the transcription factors Snail and Slug and increases MMPs expression, thereby inducing EMT and VM formation [25].
Notch. Similar to Nodal, Notch is essential for embryonic development. It is well known that four transmembrane Notch receptors (Notch1, 2, 3, 4), coupled with five ligands, participate in the vertebrate embryogenesis process. Emerging evidence showed molecular cross-talk between Nodal and Notch. Nodal signaling is initiated through a series of proteolytic cleavages that release the Notch intracellular domain (NICD). Then, NICD translocates to the nucleus, activating the transcription of Nodal [62]. Consistent with this, the co-expression of Nodal and Notch4 is required for tumor cell proliferation and survival. A study showed that the inhibition of Notch4 reduces the expression of VE-cadherin and blocks VM in a Nodal-dependent manner, implying that the Notch4-N odal signaling axis may be a key mediator of vasculogenic networks [63]. In addition, a recent study found that in HCC, Notch1 expression is associated with VM formation by mediating the EMT pathway, while in gastric cancer by increasing VEGF secretion [64, 65]. Furthermore, Notch3 silencing using lentiviral shRNA attenuated both tumor growth and VM in melanoma stem-like cells, suggesting that Notch3 is closely associated with tumor angiogenesis [66].
TGF-β. TGF-β superfamily plays essential roles in cell growth, apoptosis, motility, and invasion. Various studies showed that members of the TGF-β superfamily have both negative and positive effects on carcinoma cells. The relationship between various TGF-β proteins and VM is well established. Experimental evidence in aggressive tumor cells indicates that the binding between Endoglin (ENG) and TGF-β leads to neoangiogenesis and VM [67]. TGF-β inhibition in U251MG cells can also reduce the expression of MMP-14, resulting in a significant decrease in VM formation [5]. Furthermore, blocking TGF-β signaling by silencing TGF-ΒR1 in HCC cells attenuates VE-cadherin/MMP-2/LAMC2 expression and inhibits cancer-associated fibroblast (CM-CAF)-promoted VM formation [68]. Thus, TGF-β-related signaling pathways could be potential targets for anti-VM cancer therapy.
Hypoxia. A large body of evidence supports the role of hypoxia in maintaining the stem cell-like phenotype of tumor cells and in promoting tumor invasion, metastasis, and VM. In melanoma, HCC, glioblastoma, and breast cancer, hypoxia is capable of inducing VM channel formation [7, 69-71]. Recently, the relationship between hypoxia and VEGFA has been further investigated. VEGFA is a critical downstream effector in hypoxia-induced VM in human salivary adenoid cystic carcinoma (SACC) tissues. This process is mediated by EMT and CSC [72]. Moreover, hypoxia-inducible factor-1 (HIF-1) is involved in VM formation either by directly regulating VEGF-A, VEGFR1, EphA2, Twist, Nodal, and COX2 expression or by indirectly regulating VE-cadherin and TF expression. The hypoxia-induced regulation of Nodal expression occurs via a combinatorial mechanism mediated by HIF-1α and stabilized by the Notch protein NICD, which activates the Notch signaling pathway [73, 74]. Another study supported the relationship between HIF-1α, EMT, and VM. They showed that, under hypoxic conditions, HIF-1α affects VM formation by mediating EMT in HCT-116 [75]. Additionally, it has been shown that HIF-1α targets LOXL2, which in turn mediates VE-cadherin, E-cadherin, and vimentin expression, thus contributing to EMT and VM formation [76]. Both HIF and Twist are transcription factors with similar functions, in the VM formation process. Other than HIF-1α, a recent report showed that in pancreatic cancer cells, also HIF-2α can interact with Twist1 mediating the VM process [77]. Furthermore, a strong positive correlation was demonstrated in glioblastoma cells between hypoxia-induced VM, macrophage migration inhibitory factor (MIF) and C-X-C motif chemokine receptor 4 (CXCR4) co-localization, and HIF-1α levels [70].
Twist1/2. Twist1/2, two transcription factors who play a key role in EMT, are correlated with angiogenesis and VM formation. Upregulation of Twist1 in HCC cells enhances the expression of VE-cadherin and MMPs, which is ultimately critical to VM formation. Moreover, the EMT marker E-cadherin is suppressed following the upregulation of Twist1, suggesting a connection between Twist1, EMT, and VM [24]. Furthermore, Bcl-2 can enhance the expression of Twist-1 to promote VM formation through EMT [78]. Likewise, the HMGA2-regulated Twist-1/VE-cadherin pathway enhances the expression of MMP-2, thereby inducing VM [79]. Additionally, a recent study found that in HCC cells Protease-activated receptor-1 (PAR1) increases the Twist1 transcription activity both in vitro and in vivo, thereby promoting epithelial-endothelial transition (EET) and facilitating VM formation [80].
COX-2. Cyclooxygenases-2 (COX-2) is a key enzyme in prostaglandin E2 (PGE2) synthesis and has been found to increase tumor-associated VEGF expression through the protein kinase C (PKC)-mediated pathway in non-small cell lung cancer [81]. PEG2 binding to Prostanoid receptors (EP1, -2, -3, -4) activates EGF receptor (EGFR) signaling and the PKC-mediated ERK1/2 pathway, which promotes tumor cell invasion, metastasis, and proliferation [82]. Another study showed that in breast cancer cells, the overexpression of COX-2 promoted the formation of vascular channels, whereas low levels of COX-2 did not, implying that COX-2 is essential for VM. Furthermore, the COX-2/PEG2/EP3 signaling pathway regulates the expression of MMP-2 to form vasculogenic structures [83, 84]. Interestingly, a recent study found that both M2 macrophages and the PEG2/EP1/PKC signal transduction pathway participated in the process of VM formation by activating COX-2 [85].
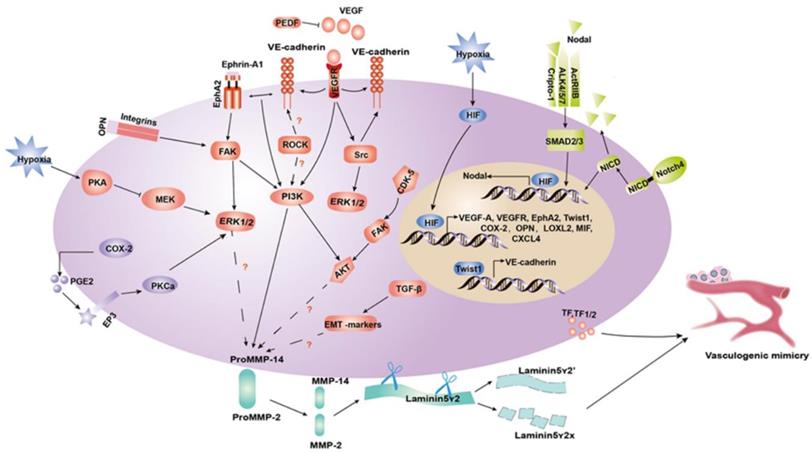
RhoA/ROCKs. The Rho kinases (ROCKs) family, which includes the two isoforms ROCK1 and ROCK2, are serine/threonine kinases acting downstream of the Rho GTPases (RhoA, RhoB and RhoC). ROCKs play a major role in regulating actin dynamics such as actin-myosin-mediated contractile processes, by phosphorylating the myosin light chain (MLC) and LIMK1/2. This process influences cell adhesion, cell motility, and invasiveness [86]. A growing number of studies reported a correlation between Rho GTPases and VM-associated markers such as VE-cadherin and MMPs in various cancer cells, implying that Rho GTPases are involved in the VM formation process [87, 88]. In our previous study, blocking the ROCKs pathway in MHCC97H cells inhibits the expression of VM-related factors such as, EphA2, VE-cadherin, PI3K, MMPs and Ln5γ2. Furthermore, we suggested that ROCKs, rather than RhoA, participate in the formation of VM channels [89]. In the subsequent study, we demonstrated that the activated RhoC/ROCK2 promotes VE-cadherin and MMP-2 expression, increasing the EMT occurrence by upregulating the ERK/MMPs signaling, and ultimately promoting VM formation. Furthermore, our results showed for the first time that RhoC/FAK/paxillin is involved in VM formation [90]. Moreover, another recent study showed that in NSCLC cells, Sema4D activates the RhoA/ROCK pathway to regulate tumor cell plasticity, migration, and VM formation (Figure 3) [91].
Schematic model of a molecular mechanisms implicated in tumor cell VM. (1) Tumor microenvironment components, including MMP-2 and MMP-14, facilitate the cleavage of Ln5γ2 into γ2 and γ2' fragments to contribute to ECM remodeling in VM. (2) PI3K, ERK1/2 and AKT are in intermediate signaling pathways that can influence the tumor microenvironment and take part in the process of VM formation through the VE-cadherin/EphA2/FAK/ERK1/2, VEGF-A/VEGFR1/PI3K/PKCα, COX-2/PEG2/EP3/PKCα/ERK1/2, and CDK5/FAK/AKT axis. (3) Nodal regulates VM formation by binding to cripto-1, ALK4/5/7 and ACTR-IIB, and then phosphorylating SMAD2/3, which translocates to the nucleus where it mediates gene expression. (4) HIF-1 induced by hypoxia directly regulates the gene expression of VEGF-A, VEGFR, EphA2, Twist, COX-2, OPN, LOXL2, MIF, and CXCL4. The hypoxia-induced regulation of Nodal expression occurs via a combinatorial mechanism that mediates HIF-1α and stabilizes the Notch protein NICD, activating Notch signaling.
MiRNAs involved in the formation of VM
A large number of studies support a role for miRNAs in influencing tumor cell invasion, proliferation and metastasis by targeting different genes, such as many classical markers of VM. However, the full spectrum of miRNAs activity in regulating the tumor VM remains to be elucidated. The majority of miRNAs regulating the VM process have been identified as VM suppressors, suggesting that this class of molecules could be potential antitumor therapeutics. Hsa-miR-299-5p is involved in the regulation of breast cancer cells, its downregulation increases the expression of osteopontin (OPN), which is a protein secreted by a sub-population of cells, called SFCs, which is required for tumorigenicity and the VM forming ability [92]. Other functional studies revealed that both miR-26b and miR-200a are key downregulators of EphA2 in glioma and ovarian cancer cells, suppressing VM and invasion [93, 94]. In HCC, miR-1236 downregulates the PTEN/PI3K/AKT pathway by targeting the 3'UTR of AFP mRNA, causing the reduction of VM [95]. In human bladder cancer cells, MiR-124 competitively binds to the 3'UTR of UHRF1 mRNA, contributing to the reduction of UHRF1. MiR-124 levels are inversely correlated with the expression of MMP-2, MMP-9, and VEGF, ultimately attenuating cellular migration, invasion, angiogenesis, and VM formation [96]. Likewise, in cervical cancer cells, a study showed that miR-124 exerts a negative effect on angiomotin-like protein 1 (AmotL1), which regulates the EMT phenotype, leading to vasculogenic network suppression [97]. Additionally, miR186 acts as a Twist1 mediator, dramatically repressing VM formation capacity, EMT, tumorigenesis, and metastasis ability of prostate cancer cells [98]. Furthermore, MiR-158-3p plays a suppressive role in VM formation of malignant glioma cells by inhibiting the ROCK1-dependent stress fiber formation [99]. Another study elucidated that in breast cancer cells, miR-193b has a downstream effect on dimethylarginine dimethylaminohydrolase 1, which is a newly-discovered mediator of VM [100]. In glioma cells, miR-9 and miR-Let-7f were found to be tumor suppressors. MiR-9 was identified due to its negative effect on Stathmin, and leads to a VM-forming failure [101]. MiR-Let-7f instead represses periostin expression, directly inhibiting VM formation [102]. MiR-27a-3p is considered a key mediator of Twist 1 in HCC cells, where it decreases the expression of VE-cadherin and suppresses EMT signaling, reducing tumor invasion and VM levels [103]. Similarly to miR-27a-3p, in ovarian cancer cells, miR-27b decreases angiogenesis and VM formation by binding to the 3'UTR of VE-cadherin mRNA [104]. MiR-101 inhibits cancer-associated fibroblast (CAF)-promoted VM in HCC cells through a novel regulatory network, which involves the TGF-β and SDF1-mediated VE-cadherin/MMP-2/LAMC2 signaling pathway [68]. A recent study showed that miR-204 in breast cancer cells exerted a positive effect on VM by directly and indirectly regulating the expression levels of 13 proteins involved in multiple signaling pathways including PI3K/AKT, RAF1/MAPK, VEGF, and FAK/SRC [105]. Moreover, Yarely et.al demonstrated the role of miR-765 in VM formation in the SKOV3 ovarian cancer cell line, through the modulation of the VEGFA/AKT1/SRC-α axis [106]. Additionally, recent findings suggested that in the MDA-MB 231 breast cancer cell line two miRNAs, miR-125a and let-7e, which are highly expressed in ECs, inhibit the activation of IL-6 signaling to suppress VM formation [107] (Table 1).
MiRNA, lncRNA and CircRNA involved in VM.
| Genes | Targets | Effects on VM | Cancer Type | References | |
|---|---|---|---|---|---|
| miRNA | Hsa-miR-299-5p | OPN | Promote | Breast cancer | [92] |
| MiR-26b | EphA2 | Suppress | Glioma | [93] | |
| MiR-200a | EphA2 | Suppress | Ovarian cancer | [94] | |
| MiR-1236 | PTEN/PI3K/AKT | Suppress | HCC | [95] | |
| MiR-124 | UHRF1, MMP-2, MMP9, VEGF AmotL1 | Suppress | Bladder cancer Cervical cancer | [96,97] | |
| MiR186 | Twist1 | Suppress | Prostate cancer | [98] | |
| MiR-158-3p | ROCK1 | Suppress | Glioma | [99] | |
| MiR-193b | DDAH1 | Suppress | Breast cancer | [100] | |
| MiR-9 | STMN1 | Suppress | Glioma | [101] | |
| MiR-Let-7f | POSTN | Suppress | Glioma | [102] | |
| MiR-27a-3p | Twist1, VE-cadherin | Suppress | HCC | [103] | |
| MiR-27b | VE-cadherin | Suppress | Ovarian cancer | [104] | |
| MiR-101 | TGF-β, SDF1 VE-cadherin/MMP2/LAMC2 | Suppress | HCC | [68] | |
| MiR-204 | PI3K/AKT, RAF1/MAPK, VEGF, and FAK/SRC | promote | Breast cancer | [105] | |
| MiR-765 | VEGFA/AKT1/SRC-α | Suppress | Ovarian cancer | [106] | |
| MiR-125a MiRlet-7e | IL-6 | Suppress | breast cancer | [107] | |
| lncRNA | MALAT1 | VE-cadherin, β-catenin, MMPs, p-ERK, p-FAK, p-paxillin | Promote | Gastric cancer | [108] |
| miR145-5p/NEDD9 | Promote | Non-small cell lung cancer | [109] | ||
| LNC00339 | miR-539-5p/TWIST1/MMPs | Promote | Glioma | [110] | |
| HOXA-AS2 | miR-373, EGFR VE-cadherin, MMP-2, MMP-9 , PI3K/AKT | Suppress | Glioma | [111] | |
| LNC00312 | YBX1 | Promote | lung adenocarcinoma | [13] | |
| lncRNAn340532 | TGF-β | Promote | Osteosarcoma | [112] | |
| CircRNA | cZNF292 | hypoxia | Promote | HCC | [113] |
LncRNAs involvement in VM formation
LncRNAs were found to serve a similar function to miRNAs in regulating the VM forming process. A recent study showed that in gastric cancer, knockdown of the lncRNA MALAT1 reduced the expression of VE-cadherin, β-catenin, MMP-2, -9, -14, p-ERK, p-FAK, and p-paxillin and impaired VM formation, suggesting that MALAT1 contributes to angiogenesis and VM [108]. A further study reported that in NSCLC the MALAT1/miR145-5p/NEDD9 signaling pathway mediated by the estrogen receptor β promoted VM formation and cell invasion [109]. The LNC00339 RNA was reported to promote glioma VM formation by targeting the miR-539-5p/TWIST1/MMPs pathway [110]. Recent evidence showed that in glioma cells, the HOXA cluster antisense RNA 2 (HOXA-AS2) lncRNA played a negative role in VM formation. Knockdown of HOXA-AS2 in glioma cells upregulates miR-373, which targets EGFR regulating the expression of VE-cadherin, MMP-2, MMP-9, and PI3K/AKT pathway proteins [111]. The LNC00312 RNA promoted VM formation in lung adenocarcinoma by directly binding to the transcription factor Y-Box Binding Protein 1 [13]. Ke et.al reported that in Osteosarcoma, the lncRNA n340532 facilitated VM formation through the TGF-β signaling pathway [112] (Table 1).
CircRNAs involvement in VM formation
Circular RNAs (circRNAs) are novel RNA molecules with a covalently closed circular structure, which are highly expressed in eukaryotic transcriptomes. A study showed that knockdown of circRNA ZNF292 in HCC resulted in the suppression of cell proliferation and VM formation [113] (Table 1).
An increasing amount of evidence showed that cell viability migration, invasion, and VM formation can be affected by miRNA, lncRNA and circRNA. Thus, novel therapies targeting these three molecules are needed the effective treatment of advanced cancer. Further investigations validating the functions of miRNA, lncRNA and circRNAs in VM formation are necessary.
VM significance in clinical practice
Routinely assessing the presence of VM is critical for clinical practice. For some malignant cancer biopsies, VM can be diagnosed with IHC staining. The current golden standard for the detection of VM is the positive PAS and negative CD31 staining of vessel-like structures. In a study on non-functioning Pituitary Adenomas (NFPAs), the presence of VM was confirmed by histological staining in 22/49 (44.9%) of the analyzed specimens, but the possible link between VM and NFPAs has not been further investigated [114]. Zhang et al. [115] showed that 12 of 17 (70.6%) intracranial hemangiopericytoma samples were VM-positive and associated with tumor recurrence. Other than IHC staining, VM presence in a clinical setting can be detected, using novel molecular imaging technologies, thanks to the availability of contrasting agents that can enter inside the VM tubular structure. A study using the dynamic micro-MRI technique in the WIBC-9 breast cancer xenograft showed a significant blood flow through the tumor, demonstrating the tumor tissue perfusion, which is consistent VM histological features [116]. Likewise, Yamamoto et al. used MRI to prove the VM presence in malignant gliomas, describing the radiological features of VM structures [117]. Additionally, Doppler imaging of microbeads circulation in human melanoma xenografts showed the physiologic perfusion of blood between the endothelial-lined mouse vasculature and VM networks [11]. Furthermore, confocal Indocyanine Green Angiography was used in uveal melanoma to detect the blood circulation in VM patterns [118] (Table 2). A correct VM identification with the help of these techniques is critical for clinical practice and it is necessary for researchers to understand the biologic processes governing VM in living organisms at a cellular and molecular level in living organisms.
Identification methods of VM
| Identification methods | Cancer Type | References |
|---|---|---|
| IHC | Non-functioning Pituitary Adenomas | [114] |
| Intracranial hemangiopericytoma | [115] | |
| MRI | Breast cancer | [116] |
| Gliomas | [117] | |
| Doppler imaging | Melanoma | [11] |
| Confocal Indocyanine Green Angiography | Uveal Melanoma | [118] |
Numerous studies showed that VM is closely associated with distant metastasis, a higher recurrence rate, and a shorter survival rate. A study by Lv et al. showed that gastric carcinoma patients with VM have a higher histological grade, more hematogenous metastasis, and a shorter overall and disease-free survival compared to non-VM patients [119]. A meta-analysis summarizing the results of 36 clinical studies representing 3609 patients affected by malignant cancers showed that a positive VM status significantly predicted lower overall survival [120]. Similarly, another study demonstrated that high grade gliomas had a higher incidence of VM than low grade gliomas, with VM positivity being correlated to a poor prognosis for gliomas patients [121]. A meta-analysis including 22 studies representing 2411 patients showed that VM was a poor prognosis factor for digestive cancer patients and positively correlated with tumor differentiation, lymph node metastasis, and TNM stage [122]. In addition, Stuart et al. showed that the presence of VM in SCLC specimens decreases tumor latency and negatively affects cisplatin efficacy [4]. In contrast, a study reported that VM has no prognostic impact in pT3 and pT4 cutaneous melanomas [123].
VM and cancer therapeutic
The purpose of tumor angiogenesis is to provide adequate blood supply and nutrition for the cancer cells growth. Therefore, many conventional antiangiogenic drugs that attenuate the EC growth or accelerate EC death are widely used in clinical practice, even if their therapeutic effect is partially limited. One of the causes that could explain the limited efficacy of antiangiogenic drugs is the presence of VM, which is endothelial cells-independent. Furthermore, many antiangiogenic drugs could generate hypoxia due to the blood supply blocking, thus inadvertently contributing to VM formation and tumor proliferation. Thus, anti-VM therapies to treat different tumors type should be considered.
Encouragingly, different studies focusing on targeting VM-related molecules with novel anticancer agents to inhibit VM formation have demonstrated the viability of this strategy. Cilengitide, an inhibitor of αvβ5 integrins, represses VM in aggressive melanoma by reducing the expression of VEGFR-2 and NRP-1 [124]. Doxycycline have an anti-VM potential in HCC through EMT process inhibition [125]. Curcumin restrains VM channel formation in HCC cells by mediating the STAT3 and the PI3K/AKT signaling pathways, and in laryngeal squamous cell carcinoma by mediating the JAK-2/STAT-3 pathway [45, 126]. Galunisertib, a TGF-β1 inhibitor, is currently under clinical trials in glioma patients, where it exhibited an inhibitory effect on VM activity by regulating the astrocytes cells, which relies on TGF-β1 secretion, and by decreasing the expression of VE-cadherin and smooth muscle actin-α, reducing the phosphorylation of AKT and FLK [127]. Verteporfin, an FDA-approved photosensitizer, that has been clinically used for the treatment of age-related macular degeneration, was recently found to suppress VM in pancreatic ductal adenocarcinoma by inhibiting MMP-2, VE-cadherin, and a-SMA expression [128]. Norcantharidin (NICD) suppresses the VM network formation both in human gallbladder carcinoma and in melanoma by downregulating the expression of PI3K, MMP-2, MTI-MMP, and Ln-5γ2 [129, 130]. Furthermore, a study found that Niclosamide, an oral anti-helminthic drug, has a wide application in oral cancer, where it downregulates the expression of VEGFA, MMP-2, ROCK1, Cdc42, and STAT3 and upregulates the levels of miR-124, ultimately preventing VM formation [131]. Additionally, several traditional Chinese medicines also exert an anti-VM effect in various cancer types. A study reported that Hinokitiol, also known as a tropolone-related natural compound, has anti-VM activity in breast cancer cells by decreasing the EGFR protein expression [132]. Paris polyphylla was able to block VM in human osteosarcoma, by reducing the expression of FAK, Mig-7, MMP-2, and MMP-9 [133]. Triptonide is a novel VM inhibitor that in pancreatic cancer cells decreases the expression of VE-cadherin and chemokine ligand 2 (CXCL2) genes [134]. Celastrus orbiculatus extract (COE), a mixture of terpenoids, can effectively suppress the angiogenesis and VM formation in HCC cells by suppressing Notch1 and Hes1 expression [135]. Luteolin is a flavonoid extracted from green plants that was found to inhibit VM tube formation in gastric cancer through downregulating the Notch1-VEGF signaling pathway [64]. Polyphyllin I, isolated from Rhizoma paridis saponins, impaired VM formation in HCC cells by blocking the PI3k-Akt-Twist1-VE-cadherin pathway [136]. A novel peptide, KVEPQDPSEW, isolated from abalone (Haliotis discus hannai), effectively inhibited VM formation in HT1080 cells by negatively regulating MMPs, VEGF, and AKT/mTOR signaling pathways [137]. In vitro and in vivo experiment revealed that JQ1, a bromodomain and extraterminal domain inhibitor, suppressed VM in pancreatic ductal adenocarcinoma cells by inhibiting the ERK1/2-MMP2/9 signal pathway. SCH772984, an ERK1/2 inhibitor, strongly suppressed VM formation in the PDAC cell line, implying a positive correlation between VM and p-ERK1/2 expression [138]. Ethoxy-erianin phosphate (EBTP) is an erianin analog that blocks VM in indoleamine 2,3-dioxygenase -induced Lewis lung cancer cells by regulating the levels of MMP-2, MMP-9, and STAT3 [139]. In addition, our previous study reported that Incarvine C restrains the vessel-like structure formation in HCC by blocking ROCK expression [140] (Table 3).
Therapeutic agents targeting VM
| Therapeutic agents | Molecular targets or function | Cancer Type | References |
|---|---|---|---|
| Cilengitide | αvβ5 integrins,VEGFR-2, NRP-1 | Melanoma | [124] |
| Doxycycline | EMT inhibition | HCC | [125] |
| Curcumin | STAT3, PI3K/AKT | HCC | [45] |
| JAK-2/STAT-3 | Laryngeal squamous cell carcinoma | [126] | |
| Galunisertib | Astrocytes, SMα, Akt,Flk | Glioma | [127] |
| Verteporfin | MMP-2,VE-cadherin, a-SMA | Pancreatic ductal adenocarcinoma | [128] |
| Norcantharidin | PI3K, MMP-2, MTI-MMP,Ln-5γ2 | Gallbladder carcinoma, melanoma | [129,130] |
| Niclosamide | VEGFA, MMP-2, ROCK1, Cdc42, STAT3, MiR-124 | Oral cancer | [131] |
| Hinokitiol | EGFR | Breast cancer | [132] |
| Paris polyphylla | FAK, Mig-7, MMP-2,MMP9 | Osteosarcoma | [133] |
| Triptonide | VE-cadherin, CXCL2 | Pancreatic cancer | [134] |
| Celastrus orbiculatus extract | Notch1, Hes1 | HCC | [135] |
| Luteolin | Notch1-VEGF | Gastric cancer | [64] |
| Polyphyllin I | PI3k-Akt-Twist1-VE-cadherin | HCC | [136] |
| KVEPQDPSEW | MMPs , VEGF AKT/mTOR | Fibrosarcoma | [137] |
| JQ1 | ERK1/2-MMP2/9 | Pancreatic ductal adenocarcinoma | [138] |
| SCH772984 | ERK1/2 | Pancreatic ductal adenocarcinoma | [138] |
| Ethoxy‐erianin phosphate | MMP‐2, MMP‐9, and STAT3 | 2,3‐dioxygenase -induced Lewis lung cancer | [139] |
| Incarvine C | ROCK | HCC | [140] |
Conclusions
VM is a biological process closely correlated with tumor invasion, migration, and progression. The discovery of VM provides new therapeutic strategies for patients battling with cancer, which are currently limited by treatment with conventional antiangiogenic agents. Hence, the combination of VM inhibitors and anti-angiogenic therapies may be promising therapeutic strategies. This review examined different VM-related markers, including miRNAs, IncRNAs, and circRNAs which can provide to researchers a deeper understanding of the underlying molecular mechanisms. In addition, further studies focusing on the clinical applications of novel agents targeting VM can contribute to the development of more effective therapies.
Abbreviations
VM: Vasculogenic mimicry; ECs: endothelial cells; miRNAs: microRNAs; LncRNAs: long non-coding RNAs; HCC: hepatocellular carcinoma; MVs: mosaic vessels; EVs: endothelium-dependent vessels; EMT: epithelial-to-mesenchymal Transition; CSCs: cancer stem cells; ALDH1: Aldehyde dehydrogenase 1; ZEB1: Zinc finger E-box binding homeobox 1; MMP: Matrix metalloproteinase; EGFR: epidermal growth factor receptor; VE-cadherin: Vascular endothelial-cadherin; EphA2: Eph receptor tyrosine kinase A2; SCLC: small cell lung cancer; FAK: focal adhesion kinase; PI3K: Phosphoinositide 3-Kinase; ERK: extracellular signal-regulated kinase; MEK: mitogen extracellular kinase; CDK-5: cyclin-dependent kinase 5; VEGF-A: Vascular endothelial growth factor-A; VEGFR1: vascular endothelial growth factor receptor 1; SEMA4D: semaphorin4D; PEDF: pigment epithelium-derived factor; TF: Tissue factor; TFPI: TF pathway inhibitor; TGF-β: transforming growth factor β; NICD: Notch intracellular domain; CAF: Cancer-associated fibroblast; HIF-1: hypoxia-inducible factor-1; MIF: migration inhibitory factor; CXCR4: C-X-C motif chemokine receptor 4; COX-2: Cyclooxygenases-2; PGE2: prostaglandin E2; PKC: protein kinase C; ROCKs: Rho kinases; MLC: myosin light chain; OPN: osteopontin; AmotL1: angiomotin-like protein 1; DDAH1: dimethylarginine dimethylaminohydrolase 1; STMN1: Stathmin1; POSTN: periostin; ERβ: estrogen receptor β; YBX1: Y-Box Binding Protein 1; NICD: Norcantharidin; CXCL2: chemokine ligand 2; COE: Celastrus orbiculatus extract; PPI: Polyphyllin I; AATP: KVEPQDPSEW; BET: bromodomain and extraterminal domain; PDAC: pancreatic ductal adenocarcinoma; EBTP: Ethoxy‐erianin phosphate.
Acknowledgements
This study was funded by grants from the National Natural Science Foundation of China (NSFC, No.81602524); sponsored by the Interdisciplinary Program of Shanghai Jiao Tong University (No.YG2015QN18 and No.YG2017MS29).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huang B, Xiao E, Huang M. MEK/ERK pathway is positively involved in hypoxia-induced vasculogenic mimicry formation in hepatocellular carcinoma which is regulated negatively by protein kinase A. Medical oncology. 2015;32:408
2. Karroum A, Mirshahi P, Benabbou N, Faussat AM, Soria J, Therwath A. et al. Matrix metalloproteinase-9 is required for tubular network formation and migration of resistant breast cancer cells MCF-7 through PKC and ERK1/2 signalling pathways. Cancer letters. 2010;295:242-51
3. Wang JY, Sun T, Zhao XL, Zhang SW, Zhang DF, Gu Q. et al. Functional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicry. Cancer biology & therapy. 2008;7:758-66
4. Williamson SC, Metcalf RL, Trapani F, Mohan S, Antonello J, Abbott B. et al. Vasculogenic mimicry in small cell lung cancer. Nature communications. 2016;7:13322
5. Ling G, Wang S, Song Z, Sun X, Liu Y, Jiang X. et al. Transforming growth factor-beta is required for vasculogenic mimicry formation in glioma cell line U251MG. Cancer biology & therapy. 2011;12:978-88
6. Lin H, Pan JC, Zhang FM, Huang B, Chen X, Zhuang JT. et al. Matrix metalloproteinase-9 is required for vasculogenic mimicry by clear cell renal carcinoma cells. Urologic oncology. 2015;33:168 e9-16
7. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J. et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. The American journal of pathology. 1999;155:739-52
8. Hendrix MJ, Seftor RE, Seftor EA, Gruman LM, Lee LM, Nickoloff BJ. et al. Transendothelial function of human metastatic melanoma cells: role of the microenvironment in cell-fate determination. Cancer research. 2002;62:665-8
9. Mihic-Probst D, Ikenberg K, Tinguely M, Schraml P, Behnke S, Seifert B. et al. Tumor cell plasticity and angiogenesis in human melanomas. PloS one. 2012;7:e33571
10. Xiang T, Lin YX, Ma W, Zhang HJ, Chen KM, He GP. et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nature communications. 2018;9:5009
11. Ruf W, Seftor EA, Petrovan RJ, Weiss RM, Gruman LM, Margaryan NV. et al. Differential role of tissue factor pathway inhibitors 1 and 2 in melanoma vasculogenic mimicry. Cancer research. 2003;63:5381-9
12. Liu WB, Xu GL, Jia WD, Li JS, Ma JL, Chen K. et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Medical oncology. 2011;28(Suppl 1):S228-38
13. Peng Z, Wang J, Shan B, Li B, Peng W, Dong Y. et al. The long noncoding RNA LINC00312 induces lung adenocarcinoma migration and vasculogenic mimicry through directly binding YBX1. Molecular cancer. 2018;17:167
14. Yang Z, Yao H, Fei F, Li Y, Qu J, Li C. et al. Generation of erythroid cells from polyploid giant cancer cells: re-thinking about tumor blood supply. Journal of cancer research and clinical oncology. 2018;144:617-627
15. Ge H, Luo H. Overview of advances in vasculogenic mimicry - a potential target for tumor therapy. Cancer management and research. 2018;10:2429-2437
16. Kim HS, Won YJ, Shim JH, Kim HJ, Kim J, Hong HN. et al. Morphological characteristics of vasculogenic mimicry and its correlation with EphA2 expression in gastric adenocarcinoma. Scientific reports. 2019;9:3414
17. Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H. et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer research. 2011;71:5317-26
18. Liu Q, Qiao L, Liang N, Xie J, Zhang J, Deng G. et al. The relationship between vasculogenic mimicry and epithelial-mesenchymal transitions. Journal of cellular and molecular medicine. 2016;20:1761-9
19. Donnem T, Reynolds AR, Kuczynski EA, Gatter K, Vermeulen PB, Kerbel RS. et al. Non-angiogenic tumours and their influence on cancer biology. Nature reviews Cancer. 2018;18:323-336
20. Lai CY, Schwartz BE, Hsu MY. CD133+ melanoma subpopulations contribute to perivascular niche morphogenesis and tumorigenicity through vasculogenic mimicry. Cancer research. 2012;72:5111-8
21. Liu T, Sun B, Zhao X, Li Y, Zhao X, Liu Y. et al. USP44+ Cancer Stem Cell Subclones Contribute to Breast Cancer Aggressiveness by Promoting Vasculogenic Mimicry. Molecular cancer therapeutics. 2015;14:2121-31
22. Wang H, Huang B, Li BM, Cao KY, Mo CQ, Jiang SJ. et al. ZEB1-mediated vasculogenic mimicry formation associates with epithelial-mesenchymal transition and cancer stem cell phenotypes in prostate cancer. Journal of cellular and molecular medicine. 2018;22:3768-3781
23. Langer EM, Kendsersky ND, Daniel CJ, Kuziel GM, Pelz C, Murphy KM. et al. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene. 2018;37:1005-19
24. Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N. et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology (Baltimore, Md). 2010;51:545-56
25. Gong W, Sun B, Zhao X, Zhang D, Sun J, Liu T. et al. Nodal signaling promotes vasculogenic mimicry formation in breast cancer via the Smad2/3 pathway. Oncotarget. 2016;7:70152-70167
26. Meng J, Chen S, Lei Y-y, Han J-x, Zhong W-l, Wang X-r. et al. Hsp90β promotes aggressive vasculogenic mimicry via epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene. 2018;38:228-243
27. Biondani G, Zeeberg K, Greco MR, Cannone S, Dando I, Dalla Pozza E. et al. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. The FEBS journal. 2018;285:2104-2124
28. Velez DO, Tsui B, Goshia T, Chute CL, Han A, Carter H. et al. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nature communications. 2017;8:1651
29. Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M. et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer research. 2001;61:6322-7
30. Zhang W, Zhou P, Meng A, Zhang R, Zhou Y. Down-regulating Myoferlin inhibits the vasculogenic mimicry of melanoma via decreasing MMP-2 and inducing mesenchymal-to-epithelial transition. Journal of cellular and molecular medicine. 2018;22:1743-1754
31. Li Y, Sun B. MMP-2 and MMP-13 affect vasculogenic mimicry formation in large cell lung cancer. Journal of cellular and molecular medicine. 2017;21:3741-3751
32. Delgado-Bellido D, Serrano-Saenz S, Fernandez-Cortes M, Oliver FJ. Vasculogenic mimicry signaling revisited: focus on non-vascular VE-cadherin. Molecular cancer. 2017;16:65
33. Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA. et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8018-23
34. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nature reviews Cancer. 2003;3:411-21
35. Delgado-Bellido D, Fernandez-Cortes M, Rodriguez MI, Serrano-Saenz S, Carracedo A, Garcia-Diaz A. et al. VE-cadherin promotes vasculogenic mimicry by modulating kaiso-dependent gene expression. Cell death and differentiation. 2019;26:348-361
36. Hess AR, Seftor EA, Gardner LM, Carles-Kinch K, Schneider GB, Seftor RE. et al. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer research. 2001;61:3250-5
37. Hess AR, Margaryan NV, Seftor EA, Hendrix MJ. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Developmental dynamics: an official publication of the American Association of Anatomists. 2007;236:3283-96
38. Margaryan NV, Strizzi L, Abbott DE, Seftor EA, Rao MS, Hendrix MJ. et al. EphA2 as a promoter of melanoma tumorigenicity. Cancer biology & therapy. 2009;8:279-88
39. Wang W, Lin P, Sun B, Zhang S, Cai W, Han C. et al. Epithelial-mesenchymal transition regulated by EphA2 contributes to vasculogenic mimicry formation of head and neck squamous cell carcinoma. BioMed research international. 2014. 2014 803914
40. Yeo C, Lee HJ, Lee EO. Serum promotes vasculogenic mimicry through the EphA2/VE-cadherin/AKT pathway in PC-3 human prostate cancer cells. Life sciences. 2019;221:267-273
41. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017;170:605-635
42. Wang H, Lin H, Pan J, Mo C, Zhang F, Huang B. et al. Vasculogenic Mimicry in Prostate Cancer: The Roles of EphA2 and PI3K. Journal of Cancer. 2016;7:1114-24
43. Hess AR, Seftor EA, Seftor RE, Hendrix MJ. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer research. 2003;63:4757-62
44. Jiang BH, Liu LZ. PI3K/PTEN Signaling in Angiogenesis and Tumorigenesis. Adv Cancer Res. 2009;102:19-65
45. Chiablaem K, Lirdprapamongkol K, Keeratichamroen S, Surarit R, Svasti J. Curcumin suppresses vasculogenic mimicry capacity of hepatocellular carcinoma cells through STAT3 and PI3K/AKT inhibition. Anticancer research. 2014;34:1857-64
46. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. Journal of receptor and signal transduction research. 2015;35:600-4
47. Seftor EA, Meltzer PS, Schatteman GC, Gruman LM, Hess AR, Kirschmann DA. et al. Expression of multiple molecular phenotypes by aggressive melanoma tumor cells: role in vasculogenic mimicry. Critical reviews in oncology/hematology. 2002;44:17-27
48. Lu XS, Sun W, Ge CY, Zhang WZ, Fan YZ. Contribution of the PI3K/MMPs/Ln-5gamma2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. International journal of oncology. 2013;42:2103-15
49. Hess AR, Postovit LM, Margaryan NV, Seftor EA, Schneider GB, Seftor RE. et al. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer research. 2005;65:9851-60
50. Zhou X, Gu R, Han X, Wu G, Liu J. Cyclin-dependent kinase 5 controls vasculogenic mimicry formation in non-small cell lung cancer via the FAK-AKT signaling pathway. Biochemical and biophysical research communications. 2017;492:447-452
51. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. The Biochemical journal. 2011;437:169-83
52. Vartanian A, Stepanova E, Grigorieva I, Solomko E, Baryshnikov A, Lichinitser M. VEGFR1 and PKCalpha signaling control melanoma vasculogenic mimicry in a VEGFR2 kinase-independent manner. Melanoma research. 2011;21:91-8
53. Wu HB, Yang S, Weng HY, Chen Q, Zhao XL, Fu WJ. et al. Autophagy-induced KDR/VEGFR-2 activation promotes the formation of vasculogenic mimicry by glioma stem cells. Autophagy. 2017;13:1528-1542
54. Azad T, Janse van Rensburg HJ, Lightbody ED, Neveu B, Champagne A, Ghaffari A. et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nature communications. 2018;9:1061
55. Chen Y, Zhang L, Liu WX, Wang K. VEGF and SEMA4D have synergistic effects on the promotion of angiogenesis in epithelial ovarian cancer. Cellular & molecular biology letters. 2018;23:2
56. Schnegg CI, Yang MH, Ghosh SK, Hsu MY. Induction of Vasculogenic Mimicry Overrides VEGF-A Silencing and Enriches Stem-like Cancer Cells in Melanoma. Cancer research. 2015;75:1682-90
57. Hoshina D, Abe R, Yamagishi SI, Shimizu H. The role of PEDF in tumor growth and metastasis. Current molecular medicine. 2010;10:292-5
58. Orgaz JL, Ladhani O, Hoek KS, Fernandez-Barral A, Mihic D, Aguilera O. et al. 'Loss of pigment epithelium-derived factor enables migration, invasion and metastatic spread of human melanoma'. Oncogene. 2009;28:4147-61
59. Strizzi L, Postovit LM, Margaryan NV, Lipavsky A, Gadiot J, Blank C. et al. Nodal as a biomarker for melanoma progression and a new therapeutic target for clinical intervention. Expert review of dermatology. 2009;4:67-78
60. Khalkhali-Ellis Z, Kirschmann DA, Seftor EA, Gilgur A, Bodenstine TM, Hinck AP. et al. Divergence(s) in nodal signaling between aggressive melanoma and embryonic stem cells. International journal of cancer. 2015;136:E242-51
61. McAllister JC, Zhan Q, Weishaupt C, Hsu MY, Murphy GF. The embryonic morphogen, Nodal, is associated with channel-like structures in human malignant melanoma xenografts. Journal of cutaneous pathology. 2010;37(Suppl 1):19-25
62. Strizzi L, Hardy KM, Seftor EA, Costa FF, Kirschmann DA, Seftor RE. et al. Development and cancer: at the crossroads of Nodal and Notch signaling. Cancer research. 2009;69:7131-4
63. Hardy KM, Kirschmann DA, Seftor EA, Margaryan NV, Postovit LM, Strizzi L. et al. Regulation of the embryonic morphogen Nodal by Notch4 facilitates manifestation of the aggressive melanoma phenotype. Cancer research. 2010;70:10340-50
64. Zang M, Hu L, Zhang B, Zhu Z, Li J, Zhu Z. et al. Luteolin suppresses angiogenesis and vasculogenic mimicry formation through inhibiting Notch1-VEGF signaling in gastric cancer. Biochemical and biophysical research communications. 2017;490:913-919
65. Jue C, Lin C, Zhisheng Z, Yayun Q, Feng J, Min Z. et al. Notch1 promotes vasculogenic mimicry in hepatocellular carcinoma by inducing EMT signaling. Oncotarget. 2017;8:2501-2513
66. Hsu MY, Yang MH, Schnegg CI, Hwang S, Ryu B, Alani RM. Notch3 signaling-mediated melanoma-endothelial crosstalk regulates melanoma stem-like cell homeostasis and niche morphogenesis. Laboratory investigation; a journal of technical methods and pathology. 2017;97:725-736
67. Puerto-Camacho P, Amaral AT, Lamhamedi-Cherradi SE, Menegaz BA, Castillo-Ecija H, Ordonez JL. et al. Preclinical Efficacy of Endoglin-Targeting Antibody-Drug Conjugates for the Treatment of Ewing Sarcoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2019;25:2228-2240
68. Yang J, Lu Y, Lin Y-Y, Zheng Z-Y, Fang J-H, He S. et al. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer letters. 2016;383:18-27
69. Liu WB, Xu GL, Jia WD, Li JS, Ma JL, Chen K. et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Medical oncology. 2011;28(Suppl 1):S228-38
70. Guo X, Xu S, Gao X, Wang J, Xue H, Chen Z. et al. Macrophage migration inhibitory factor promotes vasculogenic mimicry formation induced by hypoxia via CXCR4/AKT/EMT pathway in human glioblastoma cells. Oncotarget. 2017;8:80358-80372
71. Li S, Zhang Q, Zhou L, Guan Y, Chen S, Zhang Y. et al. Inhibitory effects of compound DMBT on hypoxia-induced vasculogenic mimicry in human breast cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;96:982-992
72. Wang HF, Wang SS, Zheng M, Dai LL, Wang K, Gao XL. et al. Hypoxia promotes vasculogenic mimicry formation by vascular endothelial growth factor A mediating epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Cell proliferation. 2019 e12600
73. Quail DF, Taylor MJ, Walsh LA, Dieters-Castator D, Das P, Jewer M. et al. Low oxygen levels induce the expression of the embryonic morphogen Nodal. Molecular biology of the cell. 2011;22:4809-21
74. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Developmental cell. 2005;9:617-28
75. Li W, Zong S, Shi Q, Li H, Xu J, Hou F. Hypoxia-induced vasculogenic mimicry formation in human colorectal cancer cells: Involvement of HIF-1a, Claudin-4, and E-cadherin and Vimentin. Scientific reports. 2016;6:37534
76. Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F. et al. HIF-1alpha promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. Journal of experimental & clinical cancer research: CR. 2017;36:60
77. Yang J, Zhu DM, Zhou XG, Yin N, Zhang Y, Zhang ZX. et al. HIF-2alpha promotes the formation of vasculogenic mimicry in pancreatic cancer by regulating the binding of Twist1 to the VE-cadherin promoter. Oncotarget. 2017;8:47801-47815
78. Zhao N, Sun BC, Zhao XL, Wang Y, Sun HZ, Dong XY. et al. Changes in microRNAs associated with Twist-1 and Bcl-2 overexpression identify signaling pathways. Experimental and molecular pathology. 2015;99:524-32
79. Sun J, Sun B, Sun R, Zhu D, Zhao X, Zhang Y. et al. HMGA2 promotes vasculogenic mimicry and tumor aggressiveness by upregulating Twist1 in gastric carcinoma. Scientific reports. 2017;7:2229
80. Xiao T, Zhang Q, Zong S, Zhong WL, Qin Y, Bi Z. et al. Protease-activated receptor-1 (PAR1) promotes epithelial-endothelial transition through Twist1 in hepatocellular carcinoma. Journal of experimental & clinical cancer research: CR. 2018;37:185
81. Luo H, Chen Z, Jin H, Zhuang M, Wang T, Su C. et al. Cyclooxygenase-2 up-regulates vascular endothelial growth factor via a protein kinase C pathway in non-small cell lung cancer. Journal of experimental & clinical cancer research. 2011;30:6
82. Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer letters. 2010;295:7-16
83. Basu GD, Liang WS, Stephan DA, Wegener LT, Conley CR, Pockaj BA. et al. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells. Breast cancer research. 2006;8:R69
84. Robertson FM, Simeone AM, Lucci A, McMurray JS, Ghosh S, Cristofanilli M. Differential regulation of the aggressive phenotype of inflammatory breast cancer cells by prostanoid receptors EP3 and EP4. Cancer. 2010;116:2806-14
85. Rong X, Huang B, Qiu S, Li X, He L, Peng Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget. 2016;7:83976-83986
86. Grise F, Bidaud A, Moreau V. Rho GTPases in hepatocellular carcinoma. Biochimica et biophysica acta. 2009;1795:137-51
87. Vishnubhotla R, Sun S, Huq J, Bulic M, Ramesh A, Guzman G. et al. ROCK-II mediates colon cancer invasion via regulation of MMP-2 and MMP-13 at the site of invadopodia as revealed by multiphoton imaging. Laboratory investigation; a journal of technical methods and pathology. 2007;87:1149-58
88. Sun K, Duan X, Cai H, Liu X, Yang Y, Li M. et al. Curcumin inhibits LPA-induced invasion by attenuating RhoA/ROCK/MMPs pathway in MCF7 breast cancer cells. Clinical and experimental medicine. 2016;16:37-47
89. Zhang JG, Li XY, Wang YZ, Zhang QD, Gu SY, Wu X. et al. ROCK is involved in vasculogenic mimicry formation in hepatocellular carcinoma cell line. PloS one. 2014;9:e107661
90. Zhang JG, Zhang DD, Liu Y, Hu JN, Zhang X, Li L. et al. RhoC/ROCK2 promotes vasculogenic mimicry formation primarily through ERK/MMPs in hepatocellular carcinoma. Biochimica et biophysica acta Molecular basis of disease. 2019;1865:1113-1125
91. Xia Y, Cai XY, Fan JQ, Zhang LL, Ren JH, Li ZY. et al. The role of sema4D in vasculogenic mimicry formation in non-small cell lung cancer and the underlying mechanisms. International journal of cancer. 2019;144:2227-2238
92. Shevde LA, Metge BJ, Mitra A, Xi Y, Ju J, King JA. et al. Spheroid-forming subpopulation of breast cancer cells demonstrates vasculogenic mimicry via hsa-miR-299-5p regulated de novo expression of osteopontin. Journal of cellular and molecular medicine. 2010;14:1693-706
93. Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y. et al. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PloS one. 2011;6:e16264
94. Sun Q, Zou X, Zhang T, Shen J, Yin Y, Xiang J. The role of miR-200a in vasculogenic mimicry and its clinical significance in ovarian cancer. Gynecologic oncology. 2014;132:730-8
95. Gao R, Cai C, Gan J, Yang X, Shuang Z, Liu M. et al. miR-1236 down-regulates alpha-fetoprotein, thus causing PTEN accumulation, which inhibits the PI3K/Akt pathway and malignant phenotype in hepatoma cells. Oncotarget. 2015;6:6014-28
96. Wang X, Wu Q, Xu B, Wang P, Fan W, Cai Y. et al. MiR-124 exerts tumor suppressive functions on the cell proliferation, motility and angiogenesis of bladder cancer by fine-tuning UHRF1. The FEBS journal. 2015;282:4376-88
97. Wan HY, Li QQ, Zhang Y, Tian W, Li YN, Liu M. et al. MiR-124 represses vasculogenic mimicry and cell motility by targeting amotL1 in cervical cancer cells. Cancer letters. 2014;355:148-58
98. Zhao X, Wang Y, Deng R, Zhang H, Dou J, Yuan H. et al. miR186 suppresses prostate cancer progression by targeting Twist1. Oncotarget. 2016;7:33136-51
99. Xu S, Zhang J, Xue H, Guo X, Han X, Li T. et al. MicroRNA-584-3p reduces the vasculogenic mimicry of human glioma cells by regulating hypoxia-induced ROCK1 dependent stress fiber formation. Neoplasma. 2017;64:13-21
100. Hulin JA, Tommasi S, Elliot D, Hu DG, Lewis BC, Mangoni AA. MiR-193b regulates breast cancer cell migration and vasculogenic mimicry by targeting dimethylarginine dimethylaminohydrolase 1. Scientific reports. 2017;7:13996
101. Song Y, Mu L, Han X, Li Q, Dong B, Li H. et al. MicroRNA-9 inhibits vasculogenic mimicry of glioma cell lines by suppressing Stathmin expression. Journal of neuro-oncology. 2013;115:381-90
102. Xue H, Gao X, Xu S, Zhang J, Guo X, Yan S. et al. MicroRNA-Let-7f reduces the vasculogenic mimicry of human glioma cells by regulating periostin-dependent migration. Oncology reports. 2016;35:1771-7
103. Zhao N, Sun H, Sun B, Zhu D, Zhao X, Wang Y. et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: an essential role for Twist-1 in HCC. Scientific reports. 2016;6:23091
104. Liu W, Lv C, Zhang B, Zhou Q, Cao Z. MicroRNA-27b functions as a new inhibitor of ovarian cancer-mediated vasculogenic mimicry through suppression of VE-cadherin expression. RNA. 2017;23:1019-1027
105. Salinas-Vera YM, Marchat LA, Garcia-Vazquez R, Gonzalez de la Rosa CH, Castaneda-Saucedo E, Tito NN. et al. Cooperative multi-targeting of signaling networks by angiomiR-204 inhibits vasculogenic mimicry in breast cancer cells. Cancer letters. 2018;432:17-27
106. Salinas-Vera YM, Gallardo-Rincon D, Garcia-Vazquez R, Hernandez-de la Cruz ON, Marchat LA, Gonzalez-Barrios JA. et al. HypoxamiRs Profiling Identify miR-745 as a Regulator of the Early Stages of Vasculogenic Mimicry in SKOV3 Ovarian Cancer Cells. Frontiers in oncology. 2019;9:381
107. Park Y, Kim J. Regulation of IL-6 signaling by miR-125a and let-7e in endothelial cells controls vasculogenic mimicry formation of breast cancer cells. BMB Reports. 2019;52:214-219
108. Li Y, Wu Z, Yuan J, Sun L, Lin L, Huang N. et al. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer letters. 2017;395:31-44
109. Yu W, Ding J, He M, Chen Y, Wang R, Han Z. et al. Estrogen receptor beta promotes the vasculogenic mimicry (VM) and cell invasion via altering the lncRNA-MALAT1/miR-145-5p/NEDD9 signals in lung cancer. Oncogene. 2019;38:1225-1238
110. Guo J, Cai H, Liu X, Zheng J, Liu Y, Gong W. et al. Long Non-coding RNA LINC00339 Stimulates Glioma Vasculogenic Mimicry Formation by Regulating the miR-539-5p/TWIST1/MMPs Axis. Molecular therapy Nucleic acids. 2018;10:170-186
111. Gao Y, Yu H, Liu Y, Liu X, Zheng J, Ma J. et al. Long Non-Coding RNA HOXA-AS2 Regulates Malignant Glioma Behaviors and Vasculogenic Mimicry Formation via the MiR-373/EGFR Axis. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2018;45:131-147
112. Ren K, Ni Y, Li X, Wang C, Chang Q, Li Y. et al. Expression profiling of long noncoding RNAs associated with vasculogenic mimicry in osteosarcoma. Journal of cellular biochemistry. 2019;120:12473-12488
113. Yang W, Liu Y, Gao R, Xiu Z, Sun T. Knockdown of cZNF292 suppressed hypoxic human hepatoma SMMC7721 cell proliferation, vasculogenic mimicry, and radioresistance. Cellular signalling. 2019;60:122-135
114. Di Michele J, Rotondo F, Kovacs K, Syro LV, Yousef GM, Cusimano MD. et al. Vasculogenic Mimicry in Clinically Non-functioning Pituitary Adenomas: a Histologic Study. Pathology oncology research: POR. 2017;23:803-809
115. Zhang Z, Han Y, Zhang K, Teng L. Investigation of vasculogenic mimicry in intracranial hemangiopericytoma. Molecular medicine reports. 2011;4:1295-8
116. Shirakawa K, Kobayashi H, Heike Y, Kawamoto S, Brechbiel MW, Kasumi F. et al. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer research. 2002;62:560-6
117. Yamamoto J, Shimajiri S, Miyaoka R, Nishizawa S. Pitfalls of conservative treatments of multiple probable cerebral cavernous malformations (CCMs): clinicopathological features of CCMs coexisting with vasculogenic mimicry in an anaplastic oligodendroglioma. Brain tumor pathology. 2014;31:215-21
118. Frenkel S, Barzel I, Levy J, Lin AY, Bartsch DU, Majumdar D. et al. Demonstrating circulation in vasculogenic mimicry patterns of uveal melanoma by confocal indocyanine green angiography. Eye (London, England). 2008;22:948-52
119. Lv J, Sun B, Sun H, Zhang Y, Sun J, Zhao X. et al. Significance of Vasculogenic Mimicry Formation in Gastric Carcinoma. Oncology research and treatment. 2017;40:35-41
120. Yang JP, Liao YD, Mai DM, Xie P, Qiang YY, Zheng LS. et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: a meta-analysis. Angiogenesis. 2016;19:191-200
121. Liu XM, Zhang QP, Mu YG, Zhang XH, Sai K, Pang JC. et al. Clinical significance of vasculogenic mimicry in human gliomas. Journal of neuro-oncology. 2011;105:173-9
122. Ren HY, Shen JX, Mao XM, Zhang XY, Zhou P, Li SY. et al. Correlation Between Tumor Vasculogenic Mimicry and Poor Prognosis of Human Digestive Cancer Patients: A Systematic Review and Meta-Analysis. Pathology oncology research. 2019;25:849-858
123. Massi D, Franchi A, Paglierani M, Ketabchi S, Borgognoni L, Reali UM. et al. Vasculogenic mimicry has no prognostic significance in pT3 and pT4 cutaneous melanoma. Human pathology. 2004;35:496-502
124. Ruffini F, Graziani G, Levati L, Tentori L, D'Atri S, Lacal PM. Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of alphavbeta5 integrin. International journal of cancer. 2015;136:E545-58
125. Meng J, Sun B, Zhao X, Zhang D, Zhao X, Gu Q. et al. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Molecular cancer therapeutics. 2014;13:3107-22
126. Hu A, Huang JJ, Jin XJ, Li JP, Tang YJ, Huang XF. et al. Curcumin suppresses invasiveness and vasculogenic mimicry of squamous cell carcinoma of the larynx through the inhibition of JAK-2/STAT-3 signaling pathway. American journal of cancer research. 2015;5:278-88
127. Zhang C, Chen W, Zhang X, Huang B, Chen A, He Y. et al. Galunisertib inhibits glioma vasculogenic mimicry formation induced by astrocytes. Scientific reports. 2016;6:23056
128. Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong J. et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer science. 2017;108:478-487
129. Wang Z, You D, Lu M, He Y, Yan S. Inhibitory effect of norcantharidin on melanoma tumor growth and vasculogenic mimicry by suppressing MMP-2 expression. Oncology letters. 2017;13:1660-1664
130. Zhang JT, Sun W, Zhang WZ, Ge CY, Liu ZY, Zhao ZM. et al. Norcantharidin inhibits tumor growth and vasculogenic mimicry of human gallbladder carcinomas by suppression of the PI3-K/MMPs/Ln-5gamma2 signaling pathway. BMC cancer. 2014;14:193
131. Li X, Yang Z, Han Z, Wen Y, Ma Z, Wang Y. Niclosamide acts as a new inhibitor of vasculogenic mimicry in oral cancer through upregulation of miR-124 and downregulation of STAT3. Oncology reports. 2018;39:827-833
132. Tu DG, Yu Y, Lee CH, Kuo YL, Lu YC, Tu CW. et al. Hinokitiol inhibits vasculogenic mimicry activity of breast cancer stem/progenitor cells through proteasome-mediated degradation of epidermal growth factor receptor. Oncology letters. 2016;11:2934-2940
133. Yao N, Ren K, Wang Y, Jin Q, Lu X, Lu Y. et al. Paris polyphylla Suppresses Proliferation and Vasculogenic Mimicry of Human Osteosarcoma Cells and Inhibits Tumor Growth In Vivo. The American journal of Chinese medicine. 2017;45:575-598
134. Han H, Du L, Cao Z, Zhang B, Zhou Q. Triptonide potently suppresses pancreatic cancer cell-mediated vasculogenic mimicry by inhibiting expression of VE-cadherin and chemokine ligand 2 genes. European Journal of Pharmacology. 2018;818:593-603
135. Jue C, Min Z, Zhisheng Z, Lin C, Yayun Q, Xuanyi W. et al. COE inhibits vasculogenic mimicry in hepatocellular carcinoma via suppressing Notch1 signaling. Journal of ethnopharmacology. 2017;208:165-173
136. Xiao T, Zhong W, Zhao J, Qian B, Liu H, Chen S. et al. Polyphyllin I suppresses the formation of vasculogenic mimicry via Twist1/VE-cadherin pathway. Cell death & disease. 2018;9:906
137. Gong F, Chen MF, Zhang YY, Li CY, Zhou CX, Hong PZ. et al. A Novel Peptide from Abalone (Haliotis discus hannai) to Suppress Metastasis and Vasculogenic Mimicry of Tumor Cells and Enhance Anti-Tumor Effect In Vitro. Marine drugs. 2019;17:244
138. Zhuo M, Yuan C, Han T, Hu H, Cui J, Jiao F. et al. JQ1 effectively inhibits vasculogenic mimicry of pancreatic ductal adenocarcinoma cells via the ERK1/2-MMP-2/9 signaling pathway both in vitro and in vivo. American journal of translational research. 2019;11:1030-1039
139. Yuan W, Su C, Yang X, Li Y, Cao Y, Liang X. et al. Biological and anti-vascular activity evaluation of ethoxy-erianin phosphate as a vascular disrupting agent. Journal of cellular biochemistry. 2019;120:16978-16989
140. Zhang JG, Zhang DD, Wu X, Wang YZ, Gu SY, Zhu GH. et al. Incarvine C suppresses proliferation and vasculogenic mimicry of hepatocellular carcinoma cells via targeting ROCK inhibition. BMC cancer. 2015;15:814
Author contact
![]() Corresponding author: Qin Li, Department of Clinical Pharmacy, Shanghai General Hospital, Shanghai Jiao Tong University School of medicine, Shanghai, China. Tel: 86-37797957 E-mail: liqin0626com (Q. Li)
Corresponding author: Qin Li, Department of Clinical Pharmacy, Shanghai General Hospital, Shanghai Jiao Tong University School of medicine, Shanghai, China. Tel: 86-37797957 E-mail: liqin0626com (Q. Li)