Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3706-3716. doi:10.7150/jca.32092 This issue Cite
Research Paper
Co-Expression Network Analysis Identified Gene Signatures in Osteosarcoma as a Predictive Tool for Lung Metastasis and Survival
Honghua Zhang1*, Linwei Guo2,3*, Zheng Zhang1, Yunlong Sun1, Honglei Kang1, Chao Song1, Huiyong Liu1, Zhuowei Lei1, Jia Wang1, Baoguo Mi4, Qian Xu5, Hanfeng Guan1 ![]() , Feng Li1
, Feng Li1 ![]()
1. Department of Orthopedics, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, 1095#, Jiefang Ave, Wuhan, 430030, China
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
3. Key Laboratory of Breast Cancer in Shanghai, Department of Breast Surgery, Fudan University Shanghai Cancer Center, Shanghai, China
4. Department of Spine Surgery, Honghui Hospital, Xi'an Jiaotong University College of Medicine, No. 76 Nanguo Road, Xi'an, 710054, Shanxi, China
5. Department of Hematology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, 1095#, Jiefang Ave., Wuhan, 430030, China
*Equal contribution
Received 2018-12-8; Accepted 2019-5-4; Published 2019-6-9
Abstract
Osteosarcoma (OS) is the most common primary bone tumor, whose poor prognosis is mainly due to lung metastasis. The aim of this study is to build a practical and valid diagnostic test that can predict the risk of OS metastasis and progression. We performed weighted gene co-expression network analysis (WGCNA) on GSE21257 from the Gene Expression Omnibus (GEO) database, which contains microarray data of biopsies from OS patients. In these modules, the highest association was found between the blue module and metastasis stage (r = -0.52) by Pearson's correlation analysis. Based on Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression, we derived eight clinically significant genes and constructed an eight-gene signature for metastasis status. It showed great efficacy to distinguish metastasis from non-metastasis (AUC = 0.886) and the results were validated in The Cancer Genome Atlas (TCGA) database. Functional enrichment analysis of hub genes showed that their biological processes focused on immune-related pathways, suggesting the important roles of immune cells, immune pathways and the tumor microenvironment in metastasis development. In conclusion, we discovered an efficient gene signature with great efficacy to distinguish metastasis status, which may help improve early diagnosis and treatment, enhancing the clinical outcomes of OS patients. Besides we created an effective protocol to seek for several hub genes in high-throughput data by combining WGCNA and LASSO Cox regression.
Keywords: gene signature, LASSO Cox regression, lung metastasis, osteosarcoma, WGCNA
Introduction
Osteosarcoma (OS), which is characterized by a high propensity of lung metastasis, is the most common primary bone tumor in adolescents and young adults (incidence: 0.2 - 3/100 000/year overall and 0.8 - 11/100 000/year at age 15-19 years) [1-3]. Despite advances in surgical techniques, multi-agent systemic chemotherapy, precise radiotherapy and immunotherapy, the 5-year survival rate of a localized tumor remains at 60 - 70%, while that of metastasis and recurrence is less than 20% [4-7]. The poor prognosis (metastasis and recurrence) necessitates intensive seeking for the molecular mechanism of metastasis development and an effective method for early diagnosis. However, the extremely low incidence of OS presents an inevitable challenge to study the rare but deadly disease in depth.
Over the last decades, high-throughput technologies such as gene microarray and gene sequencing have been broadly applied to identify driver genes and to detect significant somatic nucleotide polymorphisms and gene fusions in the processes of tumor genesis, recurrence and metastasis [8-11]. Understanding these genetic alterations may help elucidate the molecular mechanism of OS, but the genetic and cytogenetic complexity intrinsic to OS is challenging since cancer biology is mediated by various factors, such as circulating immune cells, hypoxia state and the tumor microenvironment [12-14]. A practical and valid diagnostic test that can predict the risk of OS metastasis or progression is urgently needed.
Weighted gene co-expression network analysis (WGCNA) is a systematic biology method for describing the correlation patterns among genes across microarray samples [15-18]. Instead of screening out differentially expressed genes (DEGs), WGCNA clusters highly correlated genes into one module and relates it to clinical traits, which may be more beneficial in identifying clinical biomarkers for diagnosis and therapy. In the current study, we performed WGCNA on OS microarray data and clinical traits with the aims of identifying biomarkers that are significantly associated with metastasis development.
Material and Methods
Data Sources and Data Preprocessing
Preprocessed gene expression profiles of GSE42352, GSE21257 and GSE36001 were downloaded from the GEO database. GSE42352 is a microarray dataset containing 103 OS cells and 15 mesenchyme stem cells [19]. GSE21257 is a microarray dataset containing 53 pre-chemotherapy biopsies of OS patients, the clinical characteristics of which are also attached [11]. Among the 53 OS patients, 34 developed metastasis within 5 years, while 19 did not. GSE36001 is a microarray dataset containing gene expression patterns of 19 OS cell lines and 6 normal samples (osteoblasts and bones)[20]. The platforms of these datasets are the GPL10295 Illumina human-6 v2.0 expression beadchip and GPL6102 Illumina human-6 v2.0 expression beadchip. For multiple probes corresponding to one gene, their median expression values were taken as the gene expression value. After removing 6 samples without complete clinical characteristics in GSE21257, 47 samples were used for further analysis. The ratio of metastasis status in male OS patients was 74.2% (23/31) and that for female OS patients was 43.8% (7/16), showing no significant difference between gender (P = 0.057). The expression matrix and clinical matrix were acquired.
Screening for DEGs
Processed data of 24,998 mRNAs of 103 OS cells and 15 mesenchyme stem cells samples in GSE42352 were subjected to DEG analysis. The linear models for microarray data (limma) package in R (x64, version 3.4.3) was utilized [21]. Genes with |log2FC| value > 1 and false discovery rate (FDR) < 0.05 were identified as DEGs and selected to match the GSE21257 expression matrix for subsequent analysis.
Constructing Dynamic Weighted Gene Co-Expression Network
WGCNA is a systems biology method for describing the correlation patterns among genes across microarray samples [15,17]. WGCNA can be used to find clusters (modules) of highly correlated genes, to summarize such clusters using the module eigengene or an intramodular hub gene, to relate modules to one another and to external sample traits (using eigengene network methodology), and to calculate module membership measures.
In the current study, WGCNA was performed on DEG-matched GSE21257, and modules were identified with a dynamic tree-cutting algorithm with a minimum module size of 10 genes, a scale-free topology threshold of 0.9 and merged with a MEDissThres parameter of 0.25. After relating modules to clinical traits, modules with the highest Pearson's correlation coefficient were selected for subsequent analysis.
Lasso Cox Regression and Metastasis Signature
A metastasis signature was constructed according to the expression level and association with the metastasis status of genes. If a gene was positively associated with metastasis status (acting as risk factor), the score was assigned 1, 2, 3, or 4 from low- to high-quartered expression, which was reverse for negatively associated genes. The sum of the scores of selected genes represents the meta-score for an individual sample. Metastasis-free survival analysis was performed on the clinical traits of GSE21257 with R package “survival” based on the meta-score and expression values of each individual gene. Moreover, receiver operating characteristic (ROC) curves were drawn and the area under the curve (AUC) was computed using R package “pROC” [22] for further verification.
Least Absolute Shrinkage and Selection Operator (LASSO) is a popular method that has been extended and broadly applied to the Cox proportional hazard regression model for survival analysis with high-dimensional data [23-25]. In the current study, a LASSO Cox regression model was used to detect hub genes that were significantly associated with metastasis-free survival. Ten-fold cross-validation for tuning parameter selection was performed and the partial likelihood deviance met the minimum criteria.
Furthermore, survival analysis was performed on survival data downloaded from Oncolnc [26], based on The Cancer Genome Atlas (TCGA) database to validate the significance of LASSO Cox-derived signature. Meanwhile, the expression levels of these derived genes were compared between human OS cell lines and normal tissues based on the dataset GSE36001.
Functional Annotation and Gene Set Enrichment Analysis (GSEA)
FUNRICH software was used to conduct Gene Ontology (GO) and biological pathway enrichment analyses of selected genes [27]. The 47 OS samples in the GSE21257 dataset were used to conduct GSEA analysis [28] according to metastasis status (metastasis vs. non-metastasis). Differences of a nominal P value < 0.05 and an FDR less than 25% were defined as significant.
Drawing Protein-Protein Interactions (PPIs) and Detecting Hub Genes
After downloading PPIs data from the Search Tool for the Retrieval of Interacting Genes (STRING) database [29] with an interaction score threshold of 0.15, a plug-in cyto-Hubba in Cytoscape software [30,31] was performed to detect hub genes with the strongest interactions with other genes. Using 12 algorithms in cyto-Hubba, dozens of hub genes were selected for subsequent analysis.
Statistical Analysis
Univariate statistical analyses were performed using GraphPad Prism Software (Version 6.01). A T-test was used to compare continuous data with normal distribution between two groups. The difference between rates was tested by a chi-square test or Fisher's exact test. Cumulative survival time was calculated by the Kaplan-Meier method and analyzed by the log-rank test. A P-value < 0.05 or a corrected P-value < 0.05 was considered statistically significant. The version of R used in the current study was 3.4.3 (x64).
Results
Screening DEGs
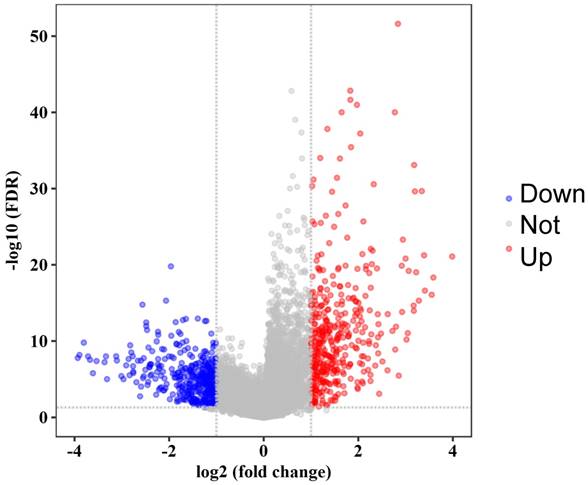
The flow diagram of our protocol is shown in Figure 1. With limma package in R performed on the preprocessed expression matrix of GSE42352 under the threshold of FDR < 0.05 and |log2FC| > 1, 814 DEGs (406 up-regulated and 408 down-regulated) were screened out (Figure 2) and selected for subsequent analysis. Specific information of these genes is shown in Supplementary Table S1.
A flow diagram of our study. First, we annotated gene probes to gene symbols and screened out differentially expressed genes between osteosarcoma cells and mesenchyme stem cells in GSE42352. Then we performed WGCNA on GSE21257 and identified clinically significant modules. LASSO Cox regression was performed on the genes in the module and several clinically significant genes were screened out, with which we constructed a multigene signature to predict metastasis risk and clinical outcome. The results were internally validated and externally validated in TCGA database. Meanwhile we performed functional enrichment on the module and it shed light on the in-depth mechanism of metastasis development. PPIs network also helped to identify hub genes.
Volcano plot of significance of gene expression difference between OS cells and mesenchyme stem cells. The x axis shows the gene expression difference by a log transformed fold change while the y axis shows significance by -log10 transformed p-value value. A gene is considered significantly differentially expressed if its |log(FC)| > 1 and p-value < 0.05. Red dot represents the up-regulated gene while blue dot represents the down-regulated gene.
Detecting Clinically Significant Modules
WGCNA was performed on the 814 DEGs of 47 samples in GSE21257 (Figure 3). There was no obvious outlier in the sample clustering (Figure 3A), and the connectivity between genes in the gene network met a scale-free network distribution with a soft threshold power of β = 4 (scale-free R2 = 0.9) (Figure 3B). After merging similar clusters, nine modules that contained groups of genes with similar patterns of connection strengths with other genes were identified (Figure 3C).
Weighted gene co-expression network analysis. (A) Sample clustering showed no evident outliers. (B) Analysis of network topology showed that it met the scale-free topology threshold of 0.9 when β = 4. The left panel shows the scale-free fit index as a function of the soft-threshold power. The right panel displays the mean connectivity as a function of the soft-threshold power. (C) Clustering dendrogram of genes based on topological overlap. Each module represents a cluster of co-related genes and was assigned a unique color. (D) Heatmap displaying the correlations and significant differences between gene modules and clinical traits. Correlations are displayed in the rectangle, while significant differences are displayed in brackets. (E) A scatterplot of Gene Significance (GS) for metastasis vs. Module Membership (MM) in the blue module. (F) A scatterplot of Gene Significance (GS) for metastasis speed vs. Module Membership (MM) in the blue module. Both (E) and (F) showed a highly significant correlation between GS and MM in the blue module.
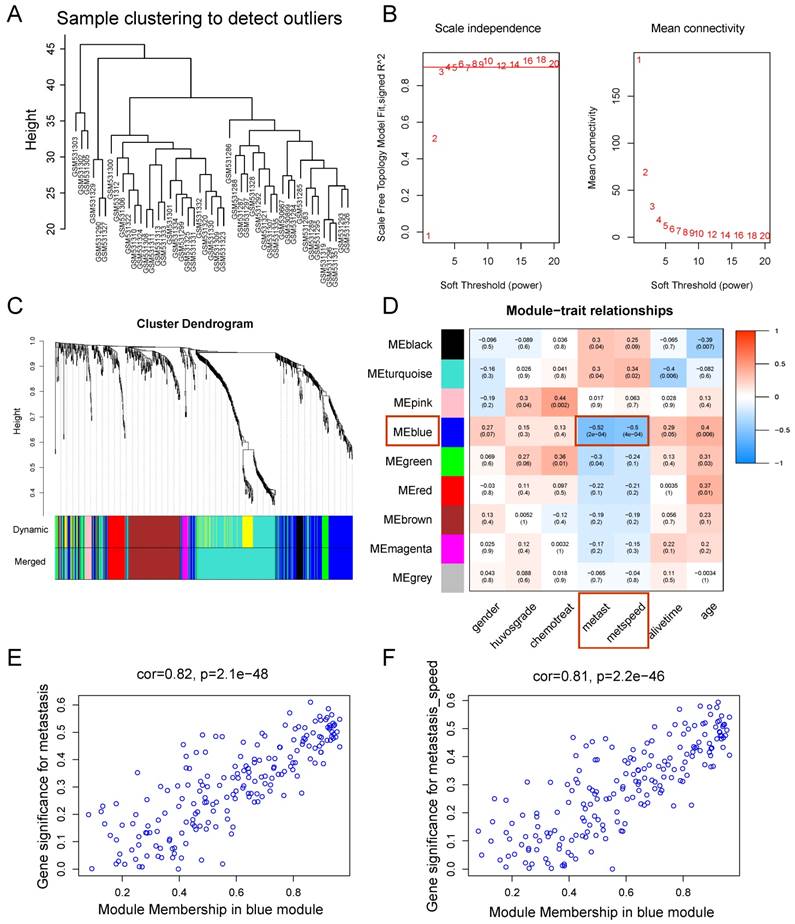
As shown in the module-feature relationship, the highest association was found between the blue module and metastasis stage (r = -0.52, P = 2e-4) and the speed of metastasis (r = -0.5, P = 4e-4) by Pearson's correlation analysis (Figure 3D). Thus, the blue module was selected as a module of interest and as a clinical feature to be studied in subsequent analyses (Supplementary Table S2). In addition, scatterplots of Gene Significance vs. Module Membership in the blue module showed that they were highly correlated (Figure 3E, 3F).
Metastasis Signatures
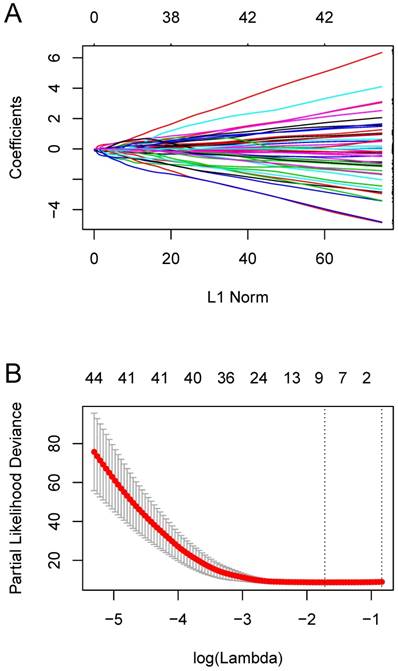
We constructed an eight-mRNA-based classifier using a LASSO Cox regression model with the tuning parameter meeting the criteria that partial likelihood deviance was minimal (Figure 4). The eight genes were HLA-DRA, HLA-DMA, LYZ, PEA15, NUPR1, C12orf75, ASPM and MATN2. Their coefficients are listed in Table 1. We validated the differentially expression of these genes between human OS cell lines and normal tissues in the dataset GSE36001 (Figure 5A), showing highly consistent results with LASSO Cox regression.
Discrimination ability and Survival analysis of genes in the LASSO Cox-derived signature.
| Gene Symbol | Coefficient | AUC | Metastasis-free Survival (GSE21257) | Overall Survival (GSE21257) | Overall Survival (TCGA) | |||
|---|---|---|---|---|---|---|---|---|
| correlation | P-value | correlation | P-value | correlation | P-value | |||
| HLA-DRA | 0.153 | 0.849 | +† | <0.001* | + | 0.013* | + | 0.016* |
| HLA-DMA | 0.073 | 0.828 | + | 0.001* | + | 0.141 | + | 0.020* |
| LYZ | 0.052 | 0.826 | + | <0.001* | + | 0.047* | + | 0.032* |
| PEA15 | 0.011 | 0.827 | + | 0.002* | + | 0.245 | + | 0.031* |
| NUPR1 | 0.241 | 0.731 | + | 0.010* | + | 0.061 | + | 0.039* |
| C12orf75 | 0.095 | 0.790 | + | <0.001* | + | 0.188 | - | 0.308 |
| ASPM | -0.004 | 0.785 | -‡ | 0.005* | - | 0.151 | - | 0.287 |
| MATN2 | -0.121 | 0.694 | - | 0.273 | - | 0.663 | - | 0.039* |
| Signature | 0.886 | - | <0.001* | - | 0.003* | - | 0.010* | |
† Addition sign '+' means positive relationship with clinical outcome (protective factor).
‡ Subtraction sign '-' means negative relationship with clinical outcome (risk factor).
* Means statistic significant difference (P < 0.05).
Constructing the eight-mRNA-based classifier by the LASSO Cox regression model. (A) LASSO coefficient profiles of the 194 metastasis-related genes in the blue module. (B) Ten-fold cross-validation for tuning parameter selection in the LASSO model. Partial likelihood deviance is plotted against log (λ), where λ is the tuning parameter. Dotted vertical lines were drawn at the optimal values by minimum criteria and 1-s.e. criteria.
Internal validation of LASSO Cox-derived signature. (A) Histograms showed gene expression level of LASSO Cox-derived genes between OS cell lines and normal tissue. (B) Box plots showed that the risk scores were significantly higher in metastasis patients compared to non-metastasis patients in both signatures. (C) ROC curves showed great classifying efficacy (AUC = 0.886). (D) Metastasis-free survival analysis showed that the signature could significantly predict poor metastasis-free survival (P < 1e-6). (E) Overall survival analysis showed that the LASSO Cox-derived signature could predict poor overall survival (P = 0.003). Data are presented as the means ± SDs. Two-tailed unpaired t test: *P < 0.05, **P < 0.01, ***P < 0.001 and ***P < 0.0001, n.s.: no significance.
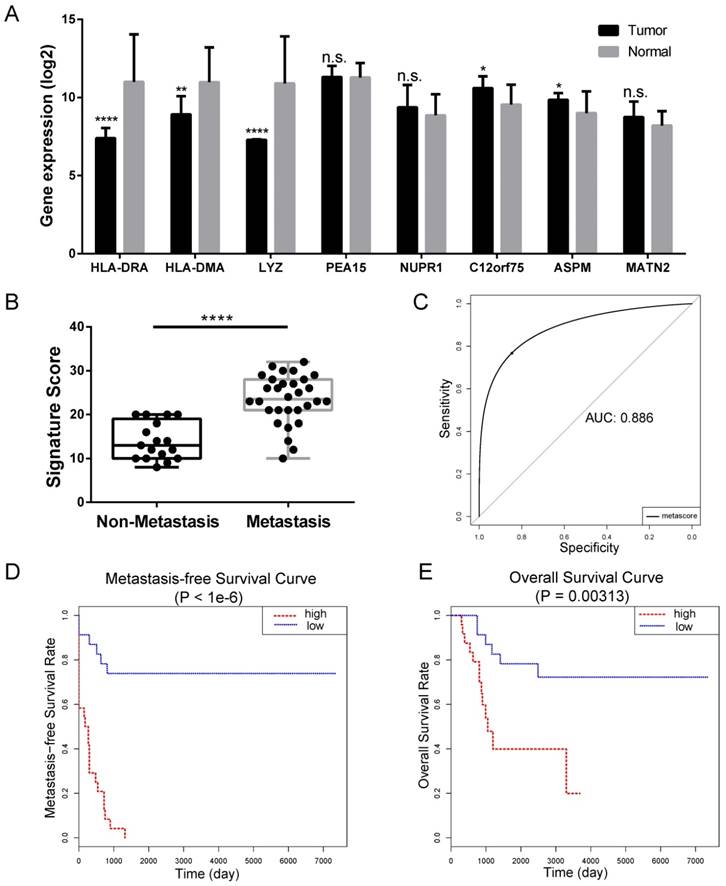
In the LASSO Cox-derived signature (Figure 5), the meta-score of the metastasis group was significantly higher than the non-metastasis group (P < 0.0001) (Figure 5B), while the ROC curves demonstrated great efficacy to distinguish metastasis from non-metastasis (AUC = 0.886, Figure 5C), whose AUCs were higher than that of any individual gene (Table 1 and Supplementary Figure S1). Metastasis-free survival analysis according to the meta-score (Figure 5D) and the expression value of an individual gene (Supplementary Figure S2) showed most of them were significantly associated with the clinical outcomes of OS patients. We also found the LASSO Cox-derived signature could significantly predict poor overall survival while some of the individual genes could not (Figure 5E and Supplementary Figure S3).
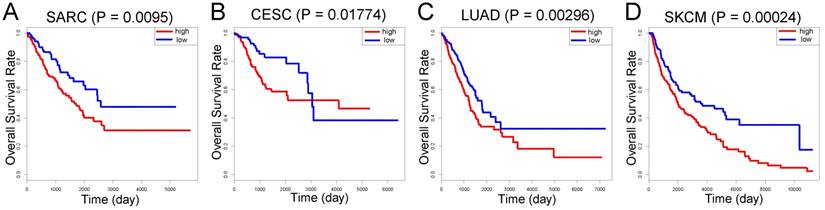
Compared with individual genes, the LASSO Cox-derived signature had a much greater efficacy to distinguish metastasis from non-metastasis patients and to predict clinical outcomes. In view of the fact that there are few microarray data of OS, we performed survival analysis on different cancers based on TCGA database (Figure 6). The signature score was negatively associated with overall survival in sarcoma (SARC) patients (n = 258) (Figure 6A). Interestingly, we also found that a high signature score could predict poor clinical outcomes in cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) (n = 264), lung adenocarcinoma (LUAD) (n = 592), and skin cutaneous melanoma (SKCM) (n = 458) despite their different histogeneses, which validated the important prognostic role of the LASSO Cox-derived signature (Figure 6B, 6C, 6D).
External validation of the LASSO Cox-derived signature. External validation cohort based on TCGA database showed that the LASSO Cox could significantly predict clinical outcomes in (A) sarcoma (SARC), (B) cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), (C) lung adenocarcinoma (LUAD), and (D) skin cutaneous melanoma (SKCM).
Functional Annotation and GSEA on Hub Genes
To obtain a primary understanding of the biological relevance of the blue module, GO enrichment and biological pathway analyses were conducted (Figure 7). The top GO terms and pathways are shown in Figure 7A. The most enriched GO terms were BP (biological process), such as Immune response, Signal transduction, Cell communication, and Regulation of cell growth, CC (cellular component) such as Plasma membrane, Extracellular, Lysosome, and Exosomes, and MF (molecular function) such as Receptor activity, GTPase activity, and Complement activity. Moreover, these genes were mainly enriched in pathways such as Immune System and Epithelial-to-mesenchyme transition, suggesting the importance of the tumor microenvironment in metastasis development (Figure 7B).
Functional annotation of genes in the blue module. (A) Enriched biological processes, cellular components and molecular functions of the blue module. (B) Enriched KEGG pathways of the blue module. (C) GSEA results on cancer-related pathways of the blue module.
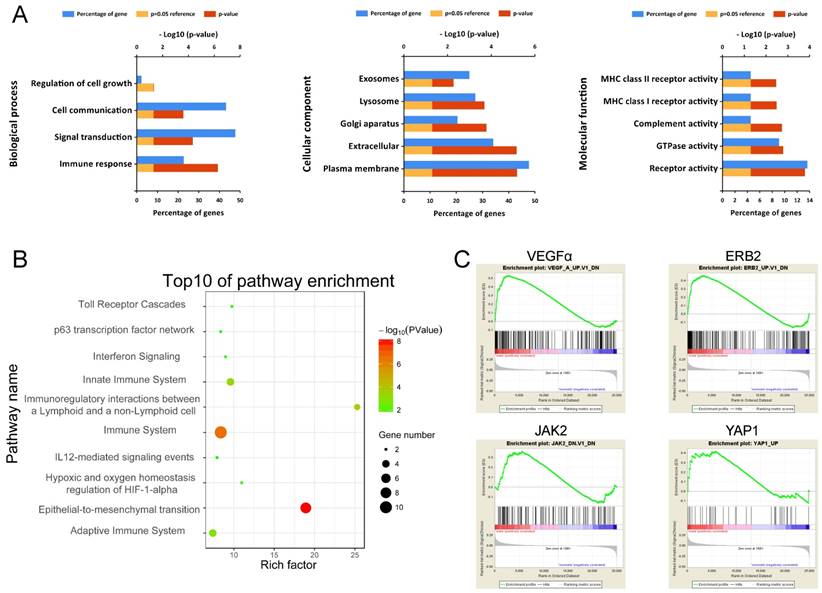
Moreover, GSEA results on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways revealed that most of the gene sets focused on immune-related pathways (Supplementary Figure S4), such as up-regulated ubiquitin-mediated proteolysis, cell cycle and down-regulated nod-like receptor signaling pathway, cytokine-cytokine receptor interaction, toll-like receptor signaling pathway, cell adhesion molecules (CAMs), and natural killer cell-mediated cytotoxicity. The GSEA results on cancer-related gene sets showed that metastasis samples were significantly enriched in several well-known cancer-related pathways, such as VEGF, ERB2, JAK2 and YAP1 (Figure 7C). The results provide clues into the in-depth mechanism of metastasis development.
Constructing PPIs Network
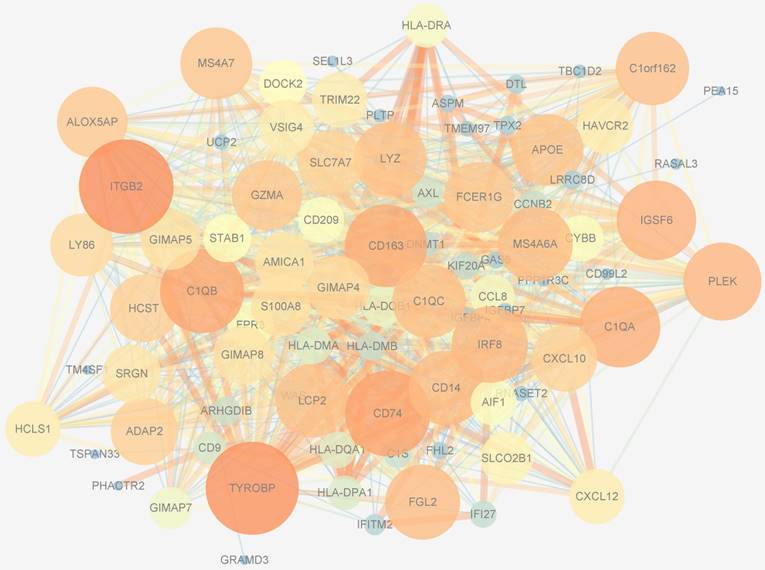
PPIs data, whose size and color were related to the number of interactions and the weighted score of the interactions, respectively, of 194 genes in the blue module were downloaded from the STRING database and visualized by Cytoscape software (Figure 8). After applying 12 algorithms in the plug-in cyto-Hubba, 44 hub genes were screened out and selected for subsequent analysis. Among the 44 hub genes, 23 had a frequency of at least 2, the most frequent of which were ITGB2, TYROBP, CD163, CD74, IGSF6, C1QB, LYZ, MS4A6A, C1QA, S100A8 and DNMT1.
Protein-protein interactions network of the blue module. The size of genes was related to the number of interactions and the color was related to the weighted score of the interactions.
Discussion
With the explosive development of microarray and sequencing technology and their decreasing costs, we can obtain vast information of genomics, proteomics, and metabolomics. However, most omics data are only used to identify DEGs, proteins or amino acids between diseased samples and normal samples or between metastasis and non-metastasis samples. A mass of information is ignored with simple screening, requiring deep data mining to make better use of it.
WGCNA is a systems biology method that is used to describe the correlation patterns among genes across transcriptome samples by a soft-threshold algorithm [17]. After clustering highly correlated genes into different modules, it correlates the modules to clinical traits of interest. In the current study, we performed WGCNA to identify 9 modules and found that the blue module was highly related to lung metastasis status (r = -0.52, P = 2e-04) and the speed of lung metastasis (r = -0.5, P = 4e-04) of OS. To identify clinically significant genes in the blue module, we then performed LASSO Cox regression, which is broadly used to construct a regression model of survival analysis with high-dimensional data, and finally eight genes were screened out.
With these genes, we constructed an eight-gene signature, which showed remarkable efficacy in distinguishing different metastasis status (AUC = 0.886, greater than any individual genes) and predicting clinical outcome. We also validated our results in independent external datasets from TCGA database, showing the clinical significance of the LASSO Cox-derived signature in SARC, CESC, LUAD, and SKCM. In clinical circumstances, if we apply the signature to OS patients, we can detect the expression levels of specific genes from biopsies or surgically procured samples and predict metastasis progression. For patients with a high score or at a high risk, more frequent follow-ups and active treatment may greatly improve their survival and quality of life, corresponding with the concept of precision medicine.
In the signature, some genes have been reported participating in tumor genesis and progression. ASPM (Abnormal Spindle Microtubule Assembly) participates in the self-renewal of gastric stem cells, revealing the role of ASPM as a biomarker for gastric carcinogenesis [32]. ASPM also supports postnatal cerebellar neurogenesis and maintains the growth of medulloblastoma [33]. HLA-DRA, α-chain of Major Histocompatibility Complex Class II-DR, binds to antigen-derived peptides from antigen presenting cells (APCs) and serves as an activation signal for CD4+ T-cells. Although few researchers have determined the in-depth mechanism of HLA-DRA in cancer metastasis, M1-type (CD14/HLA-DRA-positive) tumor-associated macrophages (TAMs) have been found in the OS microenvironment and are associated with angiogenesis [11]. Aaron J discovered that low expression of HLA-DRA could predict poor prognosis in colon adenocarcinoma [34]. LYZ, which encodes human lysozyme, is associated with the monocyte-macrophage system and enhances the activity of immune agents. LYZ is also associated with an immune-reactive microenvironment, and high expression of LYZ can predict good disease-specific survival in advanced classical Hodgkin's lymphoma [35,36]. Among other hub genes derived from PPIs, we also found some known oncogenes such as DNMT1, S100A8 and MATN2.
These researches verified that the hub genes screened out through WGCNA based on high-throughput data were indeed associated with tumor metastasis development, some of which even showed great efficacy in predicting chemotherapy response and prognosis in various cancers. Thus, the hub genes not mentioned above may also play important roles in metastasis development, calling for further experimental validation.
To obtain insights into the biological relevance of these genes in the blue module, we performed functional enrichment analysis and found that these genes are mainly enriched in immune-related pathways and epithelial-to-mesenchyme transition, which conformed to the classical process necessary for metastasis and suggests a potential mechanism for OS metastasis. Furthermore, the GSEA results on cancer-related gene sets showed that metastasis samples were significantly enriched in several cancer-related pathways, such as VEGF, ERB2, JAK2, and YAP1. These results shed light on the in-depth mechanism of metastasis development.
Some limitations exist in the present study. First, DEGs are not recommended for WGCNA because the diversity of high-throughput data may be lost. However, according to others' opinions, DEGs can be included in WGCNA if the expression difference between genes is small (15-17). As a matter of fact, how to prepare input data for WGCNA is not completely determined and what really matters is whether the results can be validated in future research in cell experiment, animal models and clinical practice. Second, we did not obtain independent external transcriptome and clinical data of OS patients since the incidence of OS is rather low. We look forward to cooperating with different hospitals and institutes to allow for the long-term follow-up of OS patients and to perform necessary gene detection in a subsequent study. Further experimental research on the biological function of the prognostic gene signature and hub genes should be conducted.
In conclusion, we identified an easy and practical eight-gene signature to distinguish different metastasis statuses of OS patients and predict clinical progression by integrating transcriptome and clinical data. WGCNA and LASSO Cox regression were combined in osteosarcoma for the first time. Validation was performed based on independent external data of different cancers from TCGA database. Further functional annotation revealed enriched immune-related pathways and epithelial-to-mesenchymal transition, suggesting a role for the tumor microenvironment in metastasis development, which could indicate the direction for further research. We not only identified an efficient multigene signature for predicting lung metastasis and prognosis in osteosarcoma, but also created an effective protocol to seek for several hub genes in high-throughput data by combining WGCNA and LASSO Cox regression.
Abbreviations
OS: osteosarcoma; GEO: Gene Expression Omnibus; TCGA: The Cancer Genome Atlas; WGCNA: weighted gene co-expression network analysis; LASSO: Least Absolute Shrinkage and Selection Operator; DEGs: differentially expressed genes; GSEA: Gene Set Enrichment Analysis; PPIs: protein-protein interactions; ROC curves: receiver operating characteristic curves; AUC: area under the curve.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was funded by the National Key R&D program of China (No. 2016YFB1101305 to FL) and the National Natural Science Foundation of China (No. 81472133 to FL, No. 81572857 to HG, and No. 81401762 to WW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author's Contributions
ZH, GL and ZZ wrote the manuscript. ZH and GL designed the original research. SY, KH, SC, LH, LZ and WJ analyzed and interpreted the data. SY, MB and XQ collected and preprocessed the data. LF and GH are in charge of the whole research conduction and paper writing. All of the authors reviewed the manuscript before submission and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer-Am Cancer Soc. 2009;115:1531-1543
2. Bacci G, Longhi A, Versari M. et al. Prognostic factors for osteosarcoma of the extremity treated with neoadjuvant chemotherapy - 15-year experience in 789 patients treated at a single institution. Cancer-Am Cancer Soc. 2006;106:1154-1161
3. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;217:320-325
4. Lewis IJ, Nooij MA, Whelan J. et al. Improvement in histologic response but not survival in osteosarcoma patients treated with intensified chemotherapy: A randomized phase III trial of the European Osteosarcoma Intergroup. Journal of The National Cancer Institute. 2007;99:112-128
5. Ta HT, Dass CR, Choong PFM, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metast Rev. 2009;28:247-263
6. Daw NC, Chou AJ, Jaffe N. et al. Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br J Cancer. 2015;112:278-282
7. Bielack S, Carrle D, Casali PG. Osteosarcoma: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20:137-139
8. Moriarity BS, Otto GM, Rahrmann EP. et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet. 2015;47:615-624
9. Smida J, Xu H, Zhang Y. et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int J Cancer. 2017;141:816-828
10. Kuijjer ML, Rydbeck H, Kresse SH. et al. Identification of osteosarcoma driver genes by integrative analysis of copy number and gene expression data. Genes, Chromosomes and Cancer. 2012;51:696-706
11. Buddingh EP, Kuijjer ML, Duim RA. et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res. 2011;17:2110-2119
12. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-322
13. Qiu GZ, Jin MZ, Dai JX. et al. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci. 2017;38:669-686
14. Wang Z, Li B, Ren Y, Ye Z. T-Cell-Based Immunotherapy for Osteosarcoma: Challenges and Opportunities. Front Immunol. 2016;7:353
15. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:e17
16. Zhao W, Langfelder P, Fuller T. et al. Weighted gene coexpression network analysis: state of the art. J Biopharm Stat. 2010;20:281-300
17. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
18. Chen L, Yuan L, Qian K. et al. Identification of Biomarkers Associated with Pathological Stage and Prognosis of Clear Cell Renal Cell Carcinoma by Co-expression Network Analysis. Front Physiol. 2018:9
19. Kuijjer ML, Peterse EF, van den Akker BE. et al. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer. 2013;13:245
20. Kresse SH, Rydbeck H, Skarn M. et al. Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. Plos One. 2012;7:e48262
21. Ritchie ME, Phipson B, Wu D. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47
22. Robin X, Turck N, Hainard A. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77
23. Liu C, Liang Y, Luan X. et al. The L1/2 regularization method for variable selection in the Cox model. Appl Soft Comput. 2014;14:498-503
24. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33:1-22
25. Simon N, Friedman J, Hastie T, Tibshirani R. Regularization Paths for Cox's Proportional Hazards Model via Coordinate Descent. J Stat Softw. 2011;39:1-13
26. Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. Peerj Computer Science. 2016;2:e67
27. Pathan M, Keerthikumar S, Ang CS. et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15:2597-2601
28. Subramanian A, Tamayo P, Mootha VK. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545-15550
29. Szklarczyk D, Morris JH, Cook H. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362-D368
30. Shannon P, Markiel A, Ozier O. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-2504
31. Chin CH, Chen SH, Wu HH. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11
32. Vange P, Bruland T, Beisvag V. et al. Genome-wide analysis of the oxyntic proliferative isthmus zone reveals ASPM as a possible gastric stem/progenitor cell marker over-expressed in cancer. J Pathol. 2015;237:447-459
33. Williams SE, Garcia I, Crowther AJ. et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Development (Cambridge, England). 2015;142:3921-3932
34. Schetter AJ, Nguyen GH, Bowman ED. et al. Association of inflammation-related and microRNA gene expression with cancer-specific mortality of colon adenocarcinoma. Clin Cancer Res. 2009;15:5878-5887
35. Sanchez-Espiridion B, Martin-Moreno AM, Montalban C. et al. Immunohistochemical markers for tumor associated macrophages and survival in advanced classical Hodgkin's lymphoma. Haematologica. 2012;97:1080-1084
36. Mahanta S, Paul S, Srivastava A. et al. Stable self-assembled nanostructured hen egg white lysozyme exhibits strong anti-proliferative activity against breast cancer cells. Colloids Surf B Biointerfaces. 2015;130:237-245
Author contact
![]() Corresponding authors: Dr. Feng Li, lifengmdedu.cn. Dr. Hanfeng Guan, hguanedu.cn.
Corresponding authors: Dr. Feng Li, lifengmdedu.cn. Dr. Hanfeng Guan, hguanedu.cn.