Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(13):2942-2952. doi:10.7150/jca.31055 This issue Cite
Research Paper
Sorting Nexin 5 Controls Head and Neck Squamous Cell Carcinoma Progression by Modulating FBW7
Jinyang Cai1, Ming Sun1, Bin Hu2, Brad Windle1, Xin Ge3, Guoping Li4, Yue Sun1 ![]()
1. Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA
2. Cancer Mouse Models Developing Shared Resource Core, Virginia Commonwealth University, Richmond, VA 23298, USA
3. Department of Clinical Laboratory Medicine, Shanghai Tenth People's Hospital of Tongji University, Shanghai, China
4. The MOH Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing, China
Received 2018-10-29; Accepted 2019-5-6; Published 2019-6-2
Abstract
Head and neck squamous cell carcinoma (HNSCC) is the sixth most prevalent cancer worldwide. Long-term survival rates in patients with HNSCC have not increased significantly in the past 30 years. Therefore, looking for novel molecular targets that control HNSCC progression is urgently required to improve the treatment of HNSCC. Here, we identified Sorting Nexin 5 (SNX5) as a new regulator that plays an oncogenic function in HNSCC progression. Analyzing HNSCC patients' data from the Cancer Genome Atlas (TCGA) indicates that the expression levels of SNX5 in HNSCC are significantly elevated compared to normal tissues. Furthermore, higher SNX5 expression correlates with a worse prognosis for HNSCC patients. These results suggest that SNX5 has an oncogenic role. Consistently, loss of SNX5 in HNSCC cells dramatically reduces colony formation and significantly decreases tumor growth in xenograft mouse models. SNX5 interacts with the tumor suppressor F-box/WD repeat-containing protein 7 (FBW7), an E3 ubiquitin ligase that mediates ubiquitination and degradation of oncoproteins such as c-Myc, NOTCH1, and Cyclin E1. By interacting with FBW7, SNX5 inhibits FBW7-mediated oncoproteins ubiquitination. In this way, SNX5 decreases the FBW7-mediated oncoproteins degradation to promote HNSCC progression.
Keywords: SNX5, HNSCC, FBW7, ubiquitination, oncoprotein
Introduction
HNSCC comprises a heterogeneous group of tumors that arise from the squamous epithelium of the oral cavity, oropharynx, larynx and hypopharynx. In the United States, 63,000 patients are diagnosed annually, and about 13,000 die from the disease [1]. Moreover, the progress in HNSCC treatment in recent years is still very limited. Compared with other types of cancers such as ErbB2+ breast cancer and Non-Small Cell Lung Cancer (NSCLC) with EGFR mutants, it is difficult to develop efficient targeted-therapies in HNSCC for the lacking of effective drug targets. Conventional treatments for locally advanced disease are multimodal, utilizing combinations of surgery, radiotherapy, and chemotherapy, which result in significant short term and long term morbidity and are curative in only about 50% of patients [2]. Therefore, searching for novel modulators that effectively affect HNSCC progression is critical for the identification of new drug targets and improving HNSCC treatment.
SNX5 belongs to the Sorting Nexins (SNXs) family of proteins. The SNXs are a large family of proteins that are defined by the presence of a SNX phox homology (PX) domain (SNX-PX) [3-5]. The SNX-PX domains act as phosphoinositide-binding motifs that aid in the targeting of the SNX protein to phosphoinositide-enriched membranes [4]. SNXs play multiple roles in different aspects of endosomal trafficking, including endosome to Trans-Golgi Network (TGN) trafficking, recycling to the cell surface, and endocytosis [6]. The correlation of SNXs expression with tumorigenesis has been reported. Levels of SNX1 are significantly down-regulated in 75% of human colon cancers and this SNX1 down-regulation promotes colon tumorigenesis [7]. SNX5 is reported to be highly expressed in papillary thyroid carcinomas [8, 9]. However, the mechanisms by which SNXs control tumorigenesis have not been clearly demonstrated.
FBW7 is an ubiquitin ligase substrate receptor as a component of the SCFs (Skp1, Cullin-1, F box protein) E3 ubiquitin ligases [10]. FBW7 mediates the ubiquitination and degradation of critical oncoproteins such as c-Myc, NOTCH1, and Cyclin E1 [11-15]. Therefore, FBW7 down-regulates oncogenic signaling and functions as a critical tumor suppressor. Furthermore, the anti-apoptotic protein Mcl-1 is a substrate of FBW7 [16]. Loss of FBW7 function leads to dramatically enhanced expression of Mcl-1, which causes cancer cell resistance to chemotherapies and resistance to Bcl-2 inhibitors by protecting cancer cells from apoptosis [17-20]. The FBXW7 gene, which encodes FBW7, is one of the most frequently mutated genes in human cancers [21-23]. Data from cBioPortal indicates a 6.45% frequency of FBXW7 gene mutation in HNSCC patients (mutation found in 33 cases of total 512 cases, TCGA). However, the mechanisms modulating FBW7 function in HNSCC are still not clear.
Here, we show that SNX5 interacts with FBW7 and blocks FBW7-mediated ubiquitination of oncoproteins. In this way, SNX5 inhibits the degradation of FBW7 substrates such as c-Myc, NOTCH1, and Cyclin E1. Therefore, SNX5 enhances these oncoproteins expression and promotes HNSCC progression. Our results uncover a novel oncogenic function of SNX5 in cancer progression.
Materials and Methods
Cell cultures and transfection. The HNSCC cell lines UMSCC1 and FaDu were cultured using Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). For plasmid transfection, cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instructions.
Clinical samples. 5 pairs of frozen surgical HNSCC tumor and matched non-cancerous tissues were obtained from the Virginia Commonwealth University (VCU) Massey Cancer Center Tissue and Data Acquisition and Analysis Core.
Reagents. Antibodies to c-Myc (cat#: 9402), NOTCH1 (cat#: 3608), cleaved NOTCH1 (cat#: 4147) Cyclin E1 (cat#: 20808), and Mcl-1 (cat#: 5453) were from Cell Signaling Technology (Danvers, MA). Anti-SNX5 antibody (cat#: ab5983) was from Abcam (Cambridge, MA). Anti-tubulin antibody (cat#: sc-5286) was from Santa Cruz (Dallas, TX). Anti-HA antibody (cat#: 901502) was from BioLegend (San Diego, CA). Anti-Flag antibody (cat#: F3165) was from Sigma-Aldrich (St. Louis, MO). Secondary antibodies were obtained from Jackson Immuno Research Laboratories (West Grove, PA).
SNX5 plasmids and shRNA. Human SNX5 was sub-cloned into pcDNA3.0 vector (Invitrogen) or pWPT lentivirus-based vector (Addgene) with a Myc-tag at N-terminus. Lentivirus-based control shRNA (sc-108060) and SNX5 shRNA (sc-41355-SH) constructs were purchased from Santa Cruz (Dallas, TX).
TCGA SNX5 Gene Expression. TCGA data for gene expression for HNSCC was used to assess the expression levels of SNX5, based on RNA-Seq and normalized by Fragments per Kilobase per Million (FPKM) and Upper Quartile (UQ) [24, 25].
CRISPR-cas9 genome editing. Genomic disruptions were created in UMSCC1 cells using clustered regularly-interspaced short palindromic repeats (CRISPR)/Cas9 system as previously described [26]. LentiCRISPR v2 plasmid (Addgene) was used to mediate the expression of the single guide RNA (sgRNA) and cas9 protein by lentivirus infection. The sgRNAs targeting SNX5 specific sequence were designed using the online software from the website: http://crispr.mit.edu. Three different sgRNA-targeting sequences were chosen: 5'-CAAATTTACAGTGCACACAA-3' (sgRNA1), 5'-TGCGGTGTGGATATAGTCAT-3' (sgRNA2), and 5'-GCTCTGAAACGTGGGCAGTG-3' (sgRNA3). Lentivirus-infected cells were cultured in medium containing puromycin (2µg/ml) for 2 days for selection. Surviving cells were then trypsinized and a single cell was isolated by limited dilution of the selected cells in 96-well plates. Each single clone was passaged and expanded. The total DNA of each colony was extracted and genotyped. The genomic region surrounding the CRISPR/Cas9 target site for SNX5 was PCR amplified. The amplicons were cloned into the pCRTM 2.1 Vector (Invitrogen) and validated by sequencing.
Cell proliferation assay. Cells (1,000) were seeded in 48-well plates. After indicated culture times, the cell viability was then tested by using WST-1 cell viability assay kits according to the manufacturer's instructions. Briefly, the cells were incubated with WST-1 (10 μl) in the cell culture medium (100 μl) for 2hr. The absorbance of each sample solution was then measured at 450 nm against a background control as blank using a microplate reader (BioTekTM SynergyTM H1 Hybrid Multi-Mode). The wavelength of 650 nm was used as the reference wavelength.
Soft agar colony formation assay. Complete 2X DMEM cell culture media was mixed with 1% agarose at 1:1 ratio to achieve a final concentration of 0.5% agar and plated into 6-well plate to create the bottom layer. Trypsinized single cell suspensions containing 5,000 cells per well were mixed with 0.7% agarose to generate a final concentration of 0.35% agar as top layer and dispensed over the solidified bottom layer. After solidified, 1 ml 1X full media was added to each well and incubate at 37°C in 5% CO2 humidified incubator for 30 days. To stain the colonies, 200 ul nitrotetrazolium blue chloride (1mg/ml) was added and incubated overnight. Each well was photographed and the colony number and size were determined using ImageJ software.
Orthotopic Xenograft mouse models. Control or SNX5-knockdown UMSCC1 cells (5 × 105 cells for each injection) were injected separately into the lateral tongue of 6-week-old NOD scid gamma (NSG) mice (n = 10 per group) [27]. After 4 weeks, mice were euthanized by CO2 inhalation and the tumors in the tongue were removed and the size was measured using calipers. Tumor volume was calculated as V = AB2 (π/6), where A is the longest dimension of the tumor and B is the dimension of the tumor perpendicular to A.
Immunoprecipitation and immunoblotting. Immunoprecipitation was performed as described [28]. Briefly, cells were harvested and lysed in 25 mM HEPES, pH 7.2, 150 mM NaCl, 0.25% NP-40, 1 mM MgCl2, and protease inhibitor cocktail, then centrifuged and incubated with protein G-Sepharose and 2 µg antibody as indicated at 4 °C for 4 hours. The immunocomplexes were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed as indicated.
In vivo ubiquitination assay. Flag-c-Myc and HA-Ubiquitin were co-transfected with or without HA-FBW7. The cells were harvested and lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP40, 10% Glycerol, 0.1% SDS, and protease inhibitor cocktail, then centrifuged and incubated with protein G-Sepharose and 2 µg anti-Flag antibody at 4 °C overnight. The immunocomplexes were separated by SDS-PAGE and the ubiquitination of purified Flag-c-Myc was detected by anti-Ubiquitin antibody.
Statistics. All data analysis was performed using SigmaPlot, except for survival analysis that used JMP, with p-value based on the log-rank test. Bar graphs represent means ± SEM., as indicated. Statistical significance was assessed using the Student t-test.
Results
SNX5 expression in HNSCC is correlated with worse prognosis of patients. The HNSCC patients' data from TCGA was analyzed to compare the SNX5 mRNA expression level in normal tissues (n = 44) and primary tumors (n = 520). As shown in Fig. 1A, the expression of SNX5 in HNSCC primary tumors is significantly elevated compared with normal tissues. The elevated SNX5 expression in HNSCC may suggest a role of SNX5 in the development of HNSCC. SNX5 protein expression levels were further detected in 5 pairs of primary HNSCC tumor (T) and their matched non-cancerous tissues (N). As shown in Fig. 1B and 1C, SNX5 protein expression in HNSCC tumor (T) is significantly elevated compared to matched non-cancerous tissues (N) in these N/T pairs tested. This further supports that SNX5 expression is elevated in many HNSCC.
SNX5 Expression in HNSCC and normal tissues. (A) SNX5 transcript levels were compared in normal and HNSCC primary tumors based on analysis from TCGA data. (B) SNX5 expression was determined by Western blot analysis in 5 pairs of primary HNSCC tumors (T) and their matched non-cancerous tissues (N). Tubulin was used as a loading control. (C) Quantification of relative SNX5 protein expression levels in (B). (D) Kaplan-Meier plots indicate that higher SNX5 expression correlates with worse overall survival.
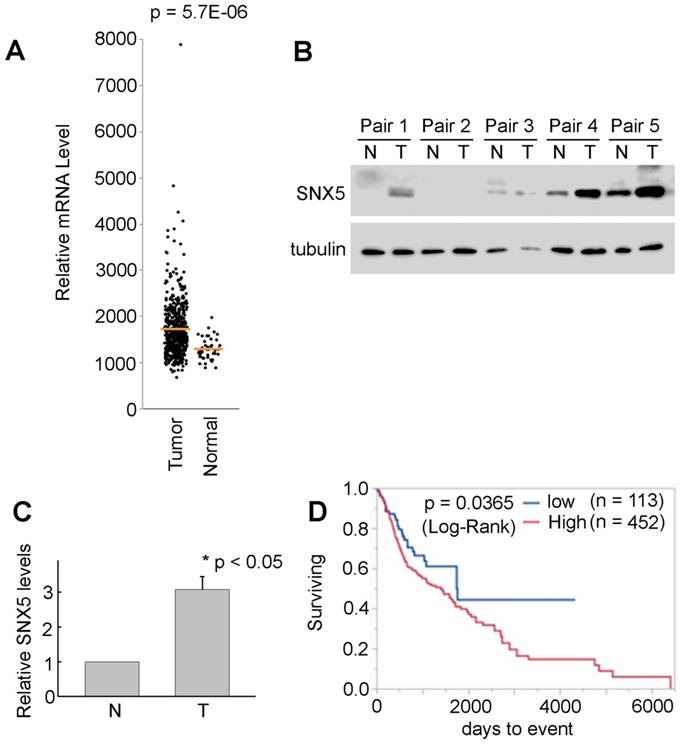
To explore the correlation of SNX5 expression with the prognosis of HNSCC patients, a Kaplan-Meier plot analysis was done using the HNSCC patients' data from TCGA. The patients were separated to two groups as SNX5-high expression (80%, n = 452) and SNX5-low expression (20%, n = 113). As shown in Fig. 1D, there is a significant difference of prognosis between these two patients groups. The higher expression of SNX5 in HNSCC correlates with worse overall survival rates. This suggests an oncogenic function of SNX5 in HNSCC.
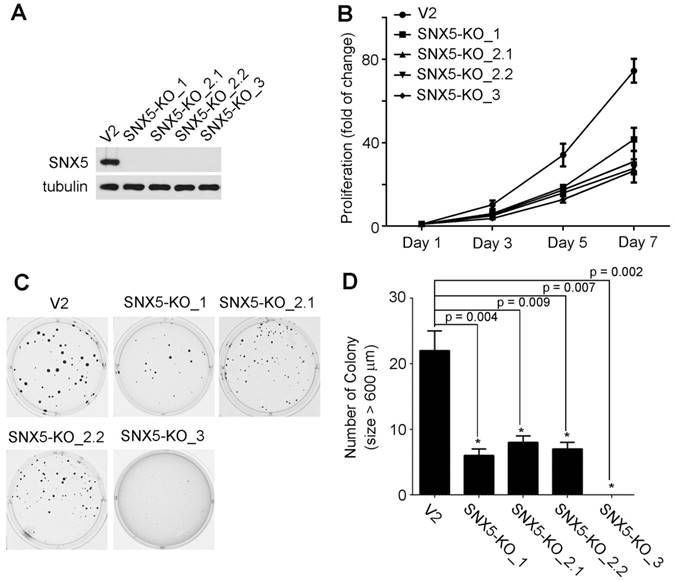
Loss of SNX5 decreased the growth and colony formation of HNSCC. To determine the effect of SNX5 in HNSCC progression, SNX5-knockout HNSCC cell lines were established using UMSCC1 cells by using the RNA-guided clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 nuclease system [29, 30]. To decrease the risk of off-targeting effects of CRISPR, three different sgRNA-targeting sequences were designed. Four single-clone SNX5-knockout cell lines were selected. One single-clone (SNX5-KO_1) was generated by sgRNA1, two single-clones (SNX5-KO_2.1 and SNX5-KO_2.2) were made by sgRNA2, and another single-clone (SNX5-KO_3) was generated by sgRNA3. The genome editing of SNX5 gene was confirmed via sequencing the targeted loci (Table 1). The loss of SNX5 protein expression was further validated by Western blot (Fig. 2A). The effects of SNX5 loss on cell growth was detected by cell proliferation assay. As shown in Fig. 2B, loss of SNX5 decreased the proliferation of UMSCC1 cells. The effects of SNX5 loss on HNSCC colony formation were tested by soft agar assays. As shown in Fig. 2C and 2D, the loss of SNX5 significantly attenuated the colony formation in all SNX5-knockout UMSCC1 cell lines. Compared with control cells (generated by LentiCRISPR v2 empty vector), the number of total colonies and number of big colonies (diameter > 600 µm) in SNX5-KO_1 and SNX5-KO_3 cells were both dramatically decreased. In SNX5-KO_2.1 and SNX5-KO_2.2 cells, the number of total colonies did not change significantly but the number of big colonies (diameter > 600 µm) was significantly decreased as compared with control cells. Although the results suggest that the use of different sgRNAs leads to some variations, the general effects of SNX5-knockout on soft agar assay is attenuating colony formation. The variations in these SNX5-knockout single clone cells may be related to the different editing of SNX5 gene in these cells (Table 1). The CRISPR-mediated SNX5 gene editing caused frameshift in all the four single clone cell lines and introduced premature stop codon at different sites. In SNX5-KO_1 and SNX5-KO_3 cells, the premature stop codon is introduced after 69 amino acids. In SNX5-KO_2.1 and SNX5-KO_2.2 cells, the premature stop codon is introduced after 251 amino acids or 244 amino acids. It is possible the different premature stop codon sites may be related to the variations in soft agar assay.
The Sequence of mutant alleles in SNX5-knockout single cell clones. PAM sequences are labeled by underlines.
| WT: 5'-GAGAGACAAAGTCAAATTTACAGTGCACA• CAAAGGTAAGTAACATGCAGTGGTAATACAT-3' |
|---|
| SNX5-KO_1: GAGAGACAAAGTCAAATTTACAGTGCACAACAAAGGTAAGTAACATGCAGTGGTAATACAT +1 bp |
| GAGAGACAAAGTCAAATTTACAGTGCACAACAAAGGTAAGTAACATGCAGTGGTAATACAT +1 bp |
| WT: 5'-CCTTGTTTCTGCAGATGTTGCCGATG• ACTATATCCACACCGCAGCCTGCTTACATAGCC-3' |
| SNX5-KO_2.1: CCTTGTTTCTGCAGATGTTGCCGATG• • • TATATCCACACCGCAGCCTGCTTACATAGCC -2 bp |
| CCTTGTTTCTGCAGATGTTGCCGATGAACTATATCCACACCGCAGCCTGCTTACATAGCC +1 bp |
| SNX5-KO_2.2: CCTTGTTTCTGCAGATGTTGCCGATGAACTATATCCACACCGCAGCCTGCTTACATAGCC +1 bp |
| CCTTGTTTCTGCAG • • • • • • • • • • • • • • • • • • • • • • • ACCGCAGCCTGCTTACATAGCC -23 bp |
| WT: 5'-AGTTAACTTCTTTCTTTCTTTACAGACCACAC• TGCCCACGTTTCAGAGCCCAGAGTTTT-3' |
| SNX5-KO_3: AGTTAACTTCTTTCTTTCTTTACAGACCACACTTGCCCACGTTTCAGAGCCCAGAGTTTT +1 bp |
| AGTTAACTTCTTTCTTTCTTTACAGACCACACTTGCCCACGTTTCAGAGCCCAGAGTTTT +1 bp |
Knockout of SNX5 attenuated cell growth and colony formation in UMSCC1 cells. (A) Loss of SNX5 protein expression in SNX5-Knockout UMSCC1 cell lines (SNX5-KO_1, SNX5-KO_2.1, SNX5-KO_2.2, and SNX5-KO_3) was confirmed by Western blot. Tubulin was used as a loading control. (B) The growth of control or SNX5-Knockout UMSCC1 cell lines was detected by cell proliferation assay. (C) Colony formation in control or SNX5-Knockout UMSCC1 cell lines via soft agar assay. (D) Quantification of colony numbers (size > 600 µm). Error bars indicate mean ± SEM from three independent experiments.
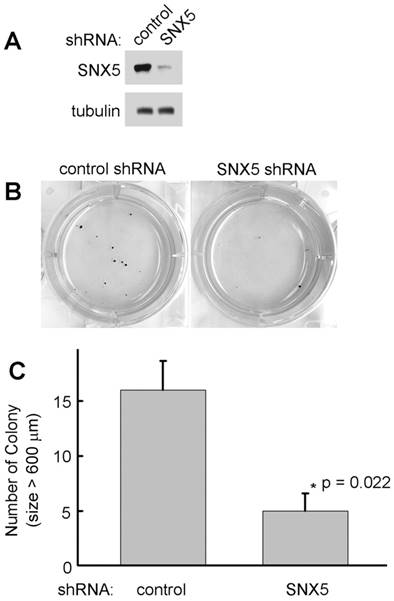
To determine whether SNX5 can modulate colony formation in other HNSCC cell lines, SNX5 expression was knocked down by shRNA in FaDu cells. As shown in Fig. 3, loss of SNX5 in FaDu cells significantly decreases colony formation, a similar effect as in UMSCC1 cells. These results support an oncogenic function of SNX5 in HNSCC.
Knockdown of SNX5 decreased colony formation in FaDu cells. (A) Loss of SNX5 protein expression in SNX5-Knockdown FaDu cells was confirmed by Western blot. Tubulin was used as a loading control. (B) Colony formation in control or SNX5-Knockdown FaDu cells via soft agar assay. (C) Quantification of colony numbers (size > 600 µm). Error bars indicate mean ± SEM from three independent experiments.
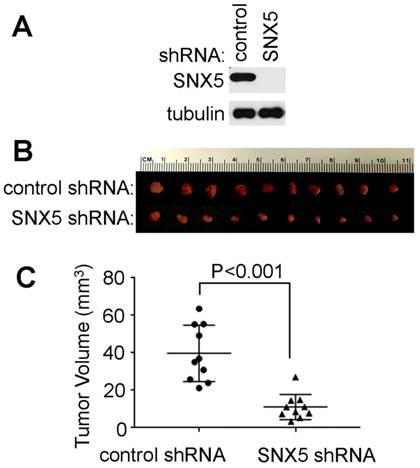
Loss of SNX5 decreased in vivo tumor growth of HNSCC. To further validate the effect of SNX5 in HNSCC tumor formation, an orthotopic xenograft mouse model was tested. The SNX5-knockdown UMSCC1 cell line was made by SNX5 shRNA. The loss of SNX5 protein expression was confirmed by Western blot (Fig. 4A). Control or SNX5-knockout cells were injected orthotopically into the lateral tongue of immune-deficient NSG mice. After 4 weeks, the mice were euthanized and tumors in the tongue were removed and the tumor volume was measured. As shown in Fig. 4B and 4C, the loss of SNX5 significantly decreased tumor volume in the orthotopic xenograft mouse model. This is consistent with the result that loss of SNX5 decreased colony formation in soft agar assay (Fig. 2 and 3). These results indicate that loss of SNX5 can attenuate HNSCC tumor growth in both in vitro and in vivo systems.
Loss of SNX5 decreased tumor growth in orthotopic xenograft mouse models. (A) Loss of SNX5 protein expression in SNX5-Knockdown UMSCC1 cell lines was confirmed by Western blot. (B) Same amount of control shRNA- or SNX5 shRNA-transfected UMSCC1 cells (5 × 105) were separately injected into the lateral tongue of 6-week-old NSG mice (n = 10 per group). Tumor volume was measured after 4 weeks. (C) Quantification of tumor volume.
SNX5 interacts with FBW7 and modulates the expression levels of FBW7 substrates. In the screening for HNSCC progression-related cell signaling that is modulated by SNX5, we first identified NOTCH1 and c-Myc. NOTCH1 is a single-pass transmembrane receptor that is essential for the control of cell proliferation and differentiation [31]. Upon activation, NOTCH1 receptors undergo a cascade of proteolytic cleavage by metalloprotease tumor necrosis factor-α-converting enzyme (TACE) and γ-secretase complex, releasing the cleaved NOTCH1 intracellular domain. Subsequently, the cleaved NOTCH1 translocates into the nucleus and regulates target gene expression to modulate cancer progression [32-34]. C-Myc is a transcription factor and plays fundamental roles in cell cycle progression, apoptosis and cellular transformation [35, 36]. Amplification of the Myc gene has been found in HNSCC [37]. As shown in Fig. 5A and 5B, the expression levels of cleaved NOTCH1 and c-Myc in SNX5-knockout cells were significantly decreased compared with control cells. Consistently, SNX5-knockdown by shRNA has similar effects on down-regulating cleaved NOTCH1 and c-Myc expression (Fig. 5C). To exclude the possible shRNA off-targeting effects, a SNX5 construct containing siRNA resistant silent mutations was used to re-express SNX5 in SNX5-knockdown cells. The re-expression of SNX5 can rescue cleaved NOTCH1 and c-Myc expression in SNX5-knockdown cells (Fig. 5C). This further supports the role of SNX5 in modulating NOTCH1 and c-Myc.
SNX5 interacts with FBW7 and controls the expression of FBW7 substrates. (A) In control (V2) and four single clones of SNX5-knockout UMSCC1 cell lines (SNX5-KO_1, SNX5-KO_2.1, SNX5-KO_2.2, and SNX5-KO_3), expression of SNX5 and indicated FBW7 substrates proteins were detected. (B) Quantification of protein expression levels. Error bars indicate mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) SNX5-knockdown UMSCC1 cells were transfected with or without Myc-SNX5, and then the expression of SNX5 and indicated FBW7 substrates proteins were detected. (D) HA-FBW7 was co-expressed with Myc-SNX5, Myc-SNX6, or Myc-SNX1, and then Myc antibody was used for immunoprecipitation from cell lysates. The immunocomplexes were separated by SDS-PAGE, and analyzed as indicated.
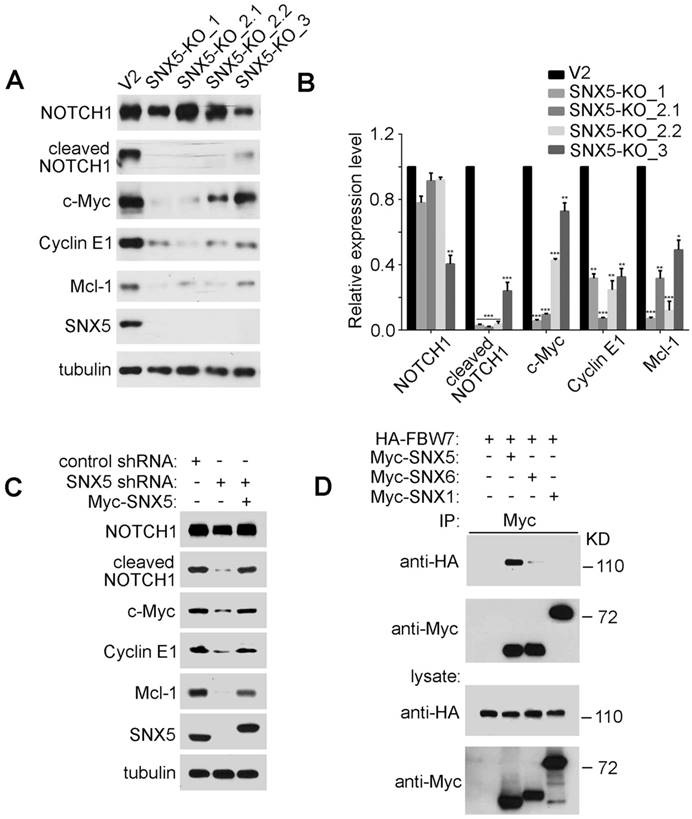
The expression of cleaved NOTCH1 and c-Myc can be modulated by an E3 ubiquitin ligase FBW7. FBW7 binds to cleaved NOTCH1 and c-Myc to down-regulate their protein levels [12, 38, 39]. To determine whether SNX5 can modulate cleaved NOTCH1 and c-Myc by FBW7, an immunoprecipitation assay was performed to test the interaction of SNX5 with FBW7. As shown in Fig. 5D, FBW7 can be co-immunoprecipitated with SNX5, which indicates that SNX5 interacts with FBW7. Other members of Sorting Nexin family proteins such as SNX1 and SNX6 cannot interact with FBW7 (Fig. 5D). This suggests that the SNX5-FBW7 interaction is specific. SNX5 may modulate HNSCC progression by regulating FBW7 function.
FBW7 is a well-known tumor suppressor. Including cleaved NOTCH1 and c-Myc, FBW7 also controls the ubiquitination and degradation of other important oncoproteins such as Cyclin E1. Cyclin E1 regulates cell-cycle entry and progression [40, 41]. The frequent cyclin E1 amplifications in solid tumors suggest its function as oncogenic driver [42, 43]. FBW7 binds to Cyclin E1 to mediate its degradation [44]. Loss of SNX5 significantly decreased Cyclin E1 levels (Fig. 5A, 5B, and 5C). Mcl-1 is an anti-apoptotic protein and is another substrate of FBW7 [16]. Loss of SNX5 also decreased Mcl-1 expression (Fig. 5A, 5B, and 5C). These results indicate that SNX5 can modulate the expression levels of FBW7 substrates.
SNX5 can inhibit FBW7-mediated substrates ubiquitination. To further determine whether SNX5 can modulate FBW7-induced down-regulation of oncoproteins, c-Myc was co-expressed with or without FBW7 or SNX5. As shown in Fig. 6A, the expression of FBW7 decreased c-Myc levels. The down-regulation of c-Myc by FBW7 could be rescued by treatment of proteasome inhibitor MG132. This indicates that the FBW7-mediated c-Myc down-regulation is dependent on proteasomal degradation. Expression of SNX5 also can rescue FBW7-mediated c-Myc down-regulation. This result is consistent with that loss of SNX5 decreased c-Myc expression (Fig. 5). This result suggests that SNX5 regulates FBW7 function.
SNX5 blocked FBW7-mediated c-Myc ubiquitination. (A) Flag-c-Myc was co-expressed with or without HA-FBW7 or Myc-SNX5. The effect on Flag-c-Myc expression was detected by Western blot. (B) Flag-c-Myc was co-expressed with HA-Ubiquitin, HA-FBW7, with or without Myc-SNX5 as indicated. Then c-Myc was immunoprecipitated by anti-Flag antibody and the levels of its ubiquitination was detected by anti-Ubiquitin antibody. Ub, Ubiquitin.
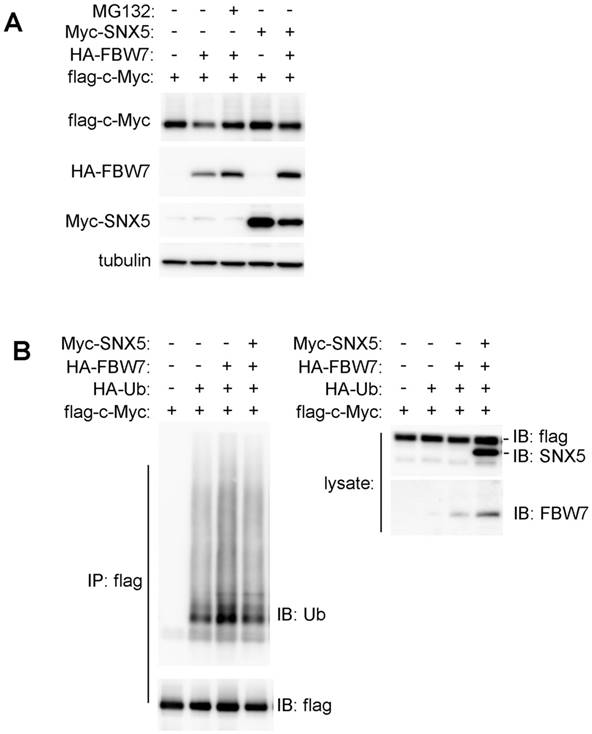
To further determine how SNX5 modulates FBW7-mediated c-Myc down-regulation, the effect of SNX5 on FBW7-mediated c-Myc ubiquitination was detected. To avoid the degradation of ubiquitinated c-Myc by proteasome, cells were pre-treated with proteasome inhibitor MG132. As shown in Fig. 6B, the expression of FBW7 dramatically enhanced c-Myc ubiquitination levels. The expression of SNX5 decreased FBW7-mediated c-Myc ubiquitination. This indicates that SNX5 can block FBW7-mediated c-Myc ubiquitination.
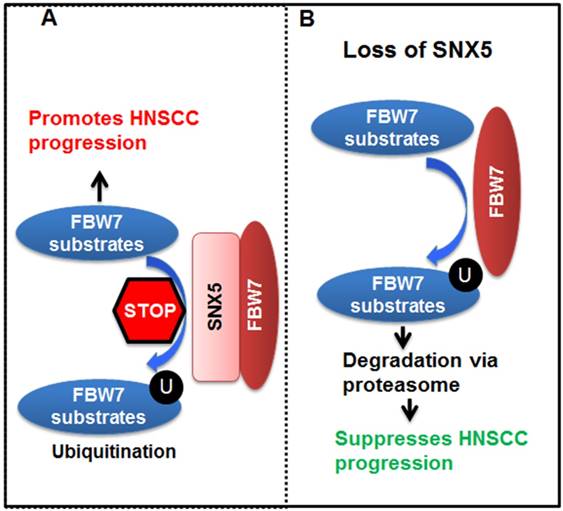
Altogether, our data lead to the model (Fig. 7) that SNX5 binds to FBW7 and blocks FBW7-mediated ubiquitination of oncoproteins. Therefore, SNX5 protects the expression of oncoproteins such as cleaved NOTCH1, c-Myc, and Cyclin E1. In this way, expression of SNX5 promotes HNSCC progression. On the contrary, loss of SNX5 increases the ubiquitination and proteasomal degradation of these oncoproteins, which suppresses HNSCC progression.
Model for SNX5 regulation of HNSCC progression. By interacting with FBW7, SNX5 blocks FBW7-mediated ubiquitination and degradation of oncoproteins such as cleaved NOTCH1, c-Myc, and Cyclin E1. Therefore, expression of SNX5 promotes HNSCC progression. On the contrary, loss of SNX5 enhances the degradation of these oncoproteins and suppresses HNSCC progression.
Discussion
It is well established that SNX5 is a critical regulator of subcellular trafficking events such as macropinocytosis and endosomal sorting. As an important component of the retromer complex, SNX5 is required for mediating endosome-to-trans-Golgi-network retrograde transport of the cation-independent mannose-6-phosphate receptor [45]. SNX5 also controls macrophage macropinocytosis. Loss of SNX5 dramatically decreases macropinocytosis activity of bone-marrow-derived macrophages [46]. Our current research suggests a novel function of SNX5. By interacting with FBW7, SNX5 modulates oncoproteins ubiquitination and degradation. This further expands the knowledge of SNX5 function.
The role of SNX5 in cancer was first identified in thyroid cancers. Expression of SNX5 was observed in papillary thyroid carcinoma (PTC) and well-differentiated thyroid tumors (WDTC), whereas SNX5 was undetectable in poorly differentiated thyroid carcinoma (PDTC) [8]. This suggests that the SNX5 expression is dramatically decreased during the progression of WDTC into PDTC. Loss of SNX5 enhances the tumorigenic signaling driven by thyroid-stimulating hormone (TSH) to promote thyroid cancer progression. The SNX5-/- thyroid tumors showed expanded malignant potential and the pulmonary metastasis of SNX5-/- thyroid tumors was more frequent than that of wild-type tumors [9]. These reports support a potential tumor suppressor role of SNX5 in thyroid cancers. While, our current research indicates that SNX5 expression in HNSCC correlates with worse prognosis of patients (Fig. 1). Loss of SNX5 decreases HNSCC colony formation and tumor growth in xenograft mouse models (Fig. 2, 3 and 4). These results support an oncogenic function of SNX5 in HNSCC. These findings suggest that SNX5 may play distinct function in the progression of thyroid cancer and HNSCC.
Loss of SNX5 significantly decreases cleaved NOTCH1 levels to down-regulate NOTCH1 activation (Fig. 5). Similar findings were also reported in zebrafish that knocking down of SNX5 caused a decrease in Notch activation and overexpression of SNX5 resulted in increased Notch activation [47]. All these results support the function of SNX5 as a regulator of NOTCH signaling. The role of SNX5 in modulating NOTCH1 signaling may not just dependent on FBW7. SNX5 may also regulate NOTCH1 signaling by binding with Mind bomb, an E3 ubiquitin ligase [48]. Mind bomb interacts with the intracellular domain of NOTCH1 ligand Delta to promote its ubiquitination and internalization, which is a critical step for NOTCH1 activation [49]. SNX5 may modulate the NOTCH1 ligand Delta ubiquitination and internalization by interacting with Mind bomb. Therefore, SNX5 may play multiple roles in regulating NOTCH1 signaling by interacting with FBW7 and Mind bomb.
SNX5 controls the expression levels of anti-apoptotic protein Mcl-1 (Fig. 5). Mcl-1 blocks the progression of apoptosis by binding and sequestering the pro-apoptotic proteins Bcl-2 homologous antagonist killer (Bak) and Bcl-2-associated protein X (Bax), which contributes to HNSCC resistance to anti-cancer therapies [17]. It is possible that SNX5 can regulate HNSCC response to anti-cancer therapies by modulating Mcl-1 expression and cell apoptosis.
Acknowledgements
This study has been supported by the grant R03DE025893 from the National Institute of Dental & Craniofacial Research of the National Institutes of Health, and Research Scholar Grant RSG-17-064-01-TBE from the American Cancer Society. This study is also supported by National Natural Science Foundation of China (grant 81572645). Services and products in support of the research project were generated by the VCU Massey Cancer Center Cancer Mouse Model Shared Resource and Tissue and Data Acquisition and Analysis Core, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7-30
2. Solomon B, Young RJ, Rischin D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Seminars in cancer biology. 2018
3. Teasdale RD, Loci D, Houghton F, Karlsson L, Gleeson PA. A large family of endosome-localized proteins related to sorting nexin 1. Biochem J. 2001;358:7-16
4. Carlton J, Bujny M, Rutherford A, Cullen P. Sorting nexins-unifying trends and new perspectives. Traffic. 2005;6:75-82
5. Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919-31
6. van Weering JR, Verkade P, Cullen PJ. SNX-BAR proteins in phosphoinositide-mediated, tubular-based endosomal sorting. Semin Cell Dev Biol. 2010;21:371-80
7. Nguyen LN, Holdren MS, Nguyen AP, Furuya MH, Bianchini M, Levy E. et al. Sorting nexin 1 down-regulation promotes colon tumorigenesis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:6952-9
8. Ara S, Kikuchi T, Matsumiya H, Kojima T, Kubo T, Ye RC. et al. Sorting nexin 5 of a new diagnostic marker of papillary thyroid carcinoma regulates Caspase-2. Cancer science. 2012;103:1356-62
9. Jitsukawa S, Kamekura R, Kawata K, Ito F, Sato A, Matsumiya H. et al. Loss of sorting nexin 5 stabilizes internalized growth factor receptors to promote thyroid cancer progression. The Journal of pathology. 2017;243:342-53
10. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer cell. 2014;26:455-64
11. Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M. et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. The Journal of cell biology. 2008;181:913-20
12. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H. et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004;23:2116-25
13. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C. et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. The Journal of experimental medicine. 2007;204:1813-24
14. Minella AC, Grim JE, Welcker M, Clurman BE. p53 and SCFFbw7 cooperatively restrain cyclin E-associated genome instability. Oncogene. 2007;26:6948-53
15. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374-8
16. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS. et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104-9
17. He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ. et al. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer discovery. 2013;3:324-37
18. Bolesta E, Pfannenstiel LW, Demelash A, Lesniewski ML, Tobin M, Schlanger SE. et al. Inhibition of Mcl-1 promotes senescence in cancer cells: implications for preventing tumor growth and chemotherapy resistance. Molecular and cellular biology. 2012;32:1879-92
19. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE. et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell metabolism. 2017;26:633-47 e7
20. Yu X, Li W, Xia Z, Xie L, Ma X, Liang Q. et al. Targeting MCL-1 sensitizes human esophageal squamous cell carcinoma cells to cisplatin-induced apoptosis. BMC cancer. 2017;17:449
21. Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E. et al. hCDC4 gene mutations in endometrial cancer. Cancer research. 2002;62:4535-9
22. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316-22
23. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer cell. 2013;23:347-61
24. Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N. et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in bioinformatics. 2013;14:671-83
25. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511-5
26. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods. 2014;11:783-4
27. Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8:293-8
28. Ling K, Doughman RL, Iyer VV, Firestone AJ, Bairstow SF, Mosher DF. et al. Tyrosine phosphorylation of type Igamma phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J Cell Biol. 2003;163:1339-49
29. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281-308
30. Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L. et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature methods. 2014;11:399-402
31. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nature reviews Cancer. 2011;11:338-51
32. Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Developmental cell. 2009;16:633-47
33. Sun W, Gaykalova DA, Ochs MF, Mambo E, Arnaoutakis D, Liu Y. et al. Activation of the NOTCH pathway in head and neck cancer. Cancer research. 2014;74:1091-104
34. Lee SH, Do SI, Lee HJ, Kang HJ, Koo BS, Lim YC. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Laboratory investigation; a journal of technical methods and pathology. 2016;96:508-16
35. Henriksson M, Luscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Advances in cancer research. 1996;68:109-82
36. Levens DL. Reconstructing MYC. Genes & development. 2003;17:1071-7
37. Bhattacharya N, Roy A, Roy B, Roychoudhury S, Panda CK. MYC gene amplification reveals clinical association with head and neck squamous cell carcinoma in Indian patients. Journal of oral pathology & medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2009;38:759-63
38. Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. The Journal of biological chemistry. 2001;276:35847-53
39. Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A. et al. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Molecular and cellular biology. 2001;21:7403-15
40. Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S. et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689-94
41. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nature reviews Molecular cell biology. 2002;3:11-20
42. Nakayama N, Nakayama K, Shamima Y, Ishikawa M, Katagiri A, Iida K. et al. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer. 2010;116:2621-34
43. Luhtala S, Staff S, Tanner M, Isola J. Cyclin E amplification, over-expression, and relapse-free survival in HER-2-positive primary breast cancer. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:9813-23
44. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW. et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173-7
45. Wassmer T, Attar N, Bujny MV, Oakley J, Traer CJ, Cullen PJ. A loss-of-function screen reveals SNX5 and SNX6 as potential components of the mammalian retromer. Journal of cell science. 2007;120:45-54
46. Lim JP, Gosavi P, Mintern JD, Ross EM, Gleeson PA. Sorting nexin 5 selectively regulates dorsal-ruffle-mediated macropinocytosis in primary macrophages. Journal of cell science. 2015;128:4407-19
47. Olena AF, Rao MB, Thatcher EJ, Wu SY, Patton JG. miR-216a regulates snx5, a novel notch signaling pathway component, during zebrafish retinal development. Developmental biology. 2015;400:72-81
48. Yoo KW, Kim EH, Jung SH, Rhee M, Koo BK, Yoon KJ. et al. Snx5, as a Mind bomb-binding protein, is expressed in hematopoietic and endothelial precursor cells in zebrafish. FEBS letters. 2006;580:4409-16
49. Itoh M, Kim CH, Palardy G, Oda T, Jiang YJ, Maust D. et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Developmental cell. 2003;4:67-82
Author contact
![]() Corresponding author: Dr. Yue Sun, Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, 521 N 11th Street, Richmond, VA 23298, United States. Fax: 804-828-0150. E-mail: ysun4edu.
Corresponding author: Dr. Yue Sun, Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, 521 N 11th Street, Richmond, VA 23298, United States. Fax: 804-828-0150. E-mail: ysun4edu.