Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(13):2927-2934. doi:10.7150/jca.31132 This issue Cite
Research Paper
Development and validation of lncRNAs-based nomogram for prediction of biochemical recurrence in prostate cancer by bioinformatics analysis
Ning Shao1,2,#, Hong Tang3,#, Yuanyuan Qu1,2, Fangning Wan1,2, ![]() , Dingwei Ye1,2,
, Dingwei Ye1,2, ![]()
1. Department of Urology, Fudan University Shanghai Cancer Center, Shanghai, 200032, China
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, China
3. Department of Pathology, The Affiliated WuXi No.2 People's Hospital of Nanjing Medical, Wuxi, 214002, China
# These authors contributed equally to the present work and each is considered first author.
Received 2018-10-31; Accepted 2019-4-13; Published 2019-6-2
Abstract
Background: Early biochemical recurrence (BCR) was considered as a sign for clinical recurrence and metastasis of prostate cancer (PCa). The purpose of the present study was to identify a lncRNA-based nomogram that can predict BCR of PCa accurately.
Materials and methods: Bioinformatics analysis, such as propensity score matching (PSM) and differentially expressed genes (DEGs) analyses were used to identify candidate lncRNAs for further bioinformatics analysis. LASSO Cox regression model was used to select the most significant prognostic lncRNAs and construct the lncRNAs signature for predicting BCR in discovery set. Additionally, a nomogram based on our lncRNAs signature was also formulated. Both lncRNAs signature and nomogram were validated in test set. GSEA was carried out to identify various gene sets which share a common biological function, chromosomal location, or regulation.
Results: A total of 457 patients with sufficient BCR information were included in our analysis. Finally, a five lncRNAs signature significantly associated with BCR was identified in discovery set (HR=0.44, 95%CI: 0.27-0.72, C-index = 0.63) and validated in test set (HR=0.22, 95%CI: 0.09-0.56, C-index = 0.65). Additionally, the lncRNAs-based nomogram showed significant performance for predicting BCR in both discovery set (C-index = 0.74) and test set (C-index = 0.78).
Conclusion: In conclusion, our lncRNAs-based nomogram is a reliable prognostic tool for BCR in PCa patients. In addition, the present study put forward the direction for the further investigation on the mechanism of PCa progression.
Keywords: prostate cancer, biochemical recurrence, lncRNA, nomogram
Introduction
Although radical surgery or radiation is demonstrated to be an effective treatment for patients with localized prostate cancer (PCa), approximately 20% of these patients will develop biochemical recurrence (BCR).[1] BCR was defined as two or more consecutive PSA values of >0.20 ng/mL. Previous studies suggested that BCR is a significant predictor for cancer progression, even cancer-specific mortality.[2, 3] Therefore, it is critical to identify the patients with high-risk of BCR after radical prostatectomy (RP).
Many clinical factors like Gleason score, TNM stage and margin status have served in previous models for prediction of BCR.[4, 5] Among these factors, Gleason score is a dominant prognostic factor. However, sampling error and subjectivity in assessing Gleason score is notable confounding factors. Recently, gene expression signatures turn out to have prognostic value in breast cancer and changed clinical care. Hence, previous studies tried to develop gene molecular signature to enhance the predictive power of clinical factors.[2, 6] However, few studies investigated the potential role of long noncoding RNAs (lncRNAs) as novel signatures for predicting BCR. LncRNAs, defined as transcripts containing ≥ 200 nucleotides without coding function, were once considered as transcriptional “noise”.[7, 8] Recently, more and more studies lncRNAs have revealed that lncRNAs play important roles in various biologic processes such as gene regulation, cell proliferation, migration and apoptosis.[9] LncRNAs could act as proto-oncogene or anti-oncogene. Apart from their role in tumor initiation and progression, lncRNAs also have turned out to be promising biomarkers.[10-12]
Up to now, bioinformatics analysis has been extensively applied in molecular experiments and clinical practice. Hence, the aim of our study was to identify significant lncRNAs associated with early BCR by bioinformatics analysis of lncRNAs sequencing data downloaded from The Cancer Genome Atlas Project (TCGA) database. Using Cox regression analysis, a five lncRNAs signature and a nomogram based on the lncRNAs signature that could predict early BCR were constructed. This nomogram may help identify patients with PCa at high-risk of early BCR. Extensive surveillance and aggressive treatment may be needed for these patients. In addition, analysis of the pathway and function of the five lncRNAs can bring new insights into the underlying molecular mechanism of PCa.
Materials and Methods
Data preparation and procedures
The raw lncRNAs sequencing data (Fragments Per Kilobase Of Exon Per Million Fragments Mapped, FPKM) from PCa samples were obtained from TCGA data portal. After that, lncRNAs expression levels were summarized into transcripts per kilobyte million (TPM) values.
A total of 457 patients with sufficient BCR information were included in our analysis. The main endpoint in our study was early BCR, defined as BCR occurring ≤2 years after radical prostatectomy (RP) as previous studies.[13] There were 52 patients with early BCR in TCGA set. Patients of discovery and test series were selected from TCGA set under a stratified-random sampling method based on early BCR, at a ratio of 1/3. That mean 39 (75%) patients with early BCR were assigned to the discovery series. 13 (25%) patients with early BCR were selected for the test series. Finally, the discovery series had 343 patients and the test series had 114 patients.
Patients from discovery set were selected and divided into early BCR group and long-term BCR survival group (no BCR after a minimum of 5 years follow-up). To reduce the bias due to confounding factors that may be involved in early BCR significantly, Propensity score matching (PSM) analysis was performed between the two groups. Age, lymph node status, T stage, and Gleason score were well adjusted. Patients with early BCR and patients with long-term BCR survival were matched 1: 1. Eventually, 39 paired patients were identified. To find different lncRNAs expression between early BCR group and long-term BCR survival groups, differentially expressed genes (DEGs) analysis was conducted using linear models for microarray data (LIMMA) method. Only lncRNAs with P < 0.005 were defined as significantly expressed lncRNAs and considered as candidate lncRNAs for further bioinformatics analysis.
Least absolute shrinkage and selection operator (LASSO) Cox regression model was used to select the most significant prognostic BCR-associated lncRNAs and construct a lncRNAs signature for predicting BCR in discovery set.[14] LncRNAs signature score for each patient based on lncRNAs expression was calculated by their LASSO Cox regression coefficients. According to lncRNAs signature, PCa patients were divided into high or low-risk groups using the mean score. A nomogram based on our lncRNAs signature was also formulated. Age, T/N stage, Gleason score and lncRNAs signature score were included. The prognostic or predictive accuracy of lncRNAs signature was examined by time-dependent receiver operating characteristic (ROC) analysis. The area under the curve (AUC) at different cutoff time was used to measure prognostic or predictive accuracy. Calibration curves were produced by plotting the observed rates against the nomogram predicted probabilities.
Identification of lncRNAs signature associated biological signaling pathway
To identify various lncRNAs signatures associated gene sets which share a common biological function, chromosomal location, or regulation, Gene Set Enrichment Analysis (GSEA) was performed. Therefore, biological pathways of our lncRNAs signature, including Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were also obtained. The FDR < 0.05 and p-value < 0.001 were set as the cut-off criteria.
Statistical analysis
The relationship between lncRNAs and clinical features was assessed by t test for continuous variables and χ2 test for categorical variables. To assess associations between lncRNAs and BCR, Kaplan-Meier curves and log-rank tests were used. To do the multivariable survival analysis, Cox regression model was used. The concordance index (C-index) was performed to evaluate the discriminatory power of the signature. Calibration plots were generated to assess whether actual outcomes approximate predicted outcomes for nomogram. The x-axis represents the prediction calculated use of the nomogram, and the y-axis represents the actual freedom from BCR. All statistical analyses were performed using R (version 3.4.2, www.r-project.org). All statistical tests were 2-sided, and p-value < 0.05 was considered statistically significant.
Results
Patients
A total of 457 patients with sufficient BCR information were included in our study. The average age was 60.8 years. In addition, 181 patients of them had high Gleason scores (≥8) PCa and 43 patients had low Gleason scores (≤6) PCa. Finally, 85 patients had BCR including 52 patients with early BCR.
Development of lncRNAs signature and lncRNAs-based nomogram for prediction of BCR from discovery set
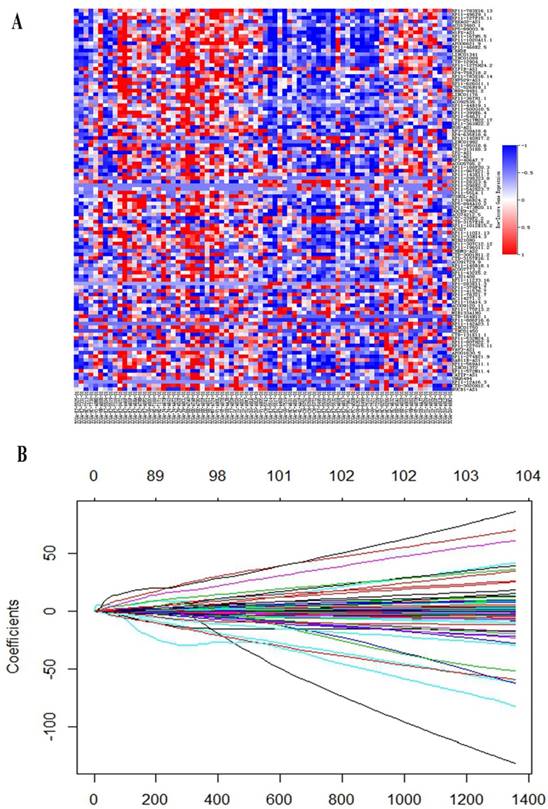
In discovery set, a total of 343 patients were enrolled, including 39 patients with early BCR and 283 patients with long-term BCR survival. After PSM analysis, Patients with early BCR and patients with long-term BCR survival were matched 1: 1. Finally, 39 paired patients were identified. Clinical features before and after PSM analysis are described in Table 1. Before PSM analysis, proportion of patients with high Gleason (> 7) PCa and high tumor stage (> T2) in early BCR group were significantly higher than that in long-term BCR survival group. After PSM analysis, there was no significant difference in age, lymph node status (N stage), tumor stage and Gleason score between the two groups (Supplementary Figure S1). DEGs analysis was conducted to find differently expressed lncRNAs between early BCR and long-term BCR survival groups. Finally, a total of 105 lncRNAs (P < 0.005) were selected for development of lncRNAs signature. (Figure. 1A)
Clinical-pathological features of PCa patients in early BCR and long-term BCR survival groups before and after PSM.
| Before PSM | After PSM | |||||
|---|---|---|---|---|---|---|
| Variable | early BCR | Long-term BCR survival | P | early BCR | Long-term BCR survival | P |
| age | 0.26 | 1.00 | ||||
| < 65 | 12(30.8) | 118(41.7) | 12(30.8) | 13(33.3) | ||
| ≥ 65 | 27(69.2) | 165(58.3) | 27(69.2) | 26(66.7) | ||
| Gleason score | <0.001 | 1.00 | ||||
| 6 | 1(2.6) | 36(12.7) | 1(2.6) | 1(2.6) | ||
| 7 | 11(28.2) | 154(54.4) | 11(28.2) | 10(25.6) | ||
| 8 | 10(25.6) | 34(12.0) | 10(25.6) | 10(25.6) | ||
| 9 | 17(43.6) | 56(19.8) | 17(43.6) | 18(46.2) | ||
| 10 | 0(0) | 3(1.1) | 0(0) | 0(0) | ||
| T stage | 0.01 | 0.58 | ||||
| Tx | 0(0) | 4(1.4) | 0(0) | 0(0) | ||
| T2 | 6(15.4) | 123(43.5) | 6(15.4) | 7(17.9) | ||
| T3 | 32(82.1) | 151(53.4) | 32(82.1) | 32(82.1) | ||
| T4 | 1(2.6) | 5(1.8) | 1(2.6) | 0(0) | ||
| N stage | 0.10 | 0.71 | ||||
| Nx | 3(7.7) | 44(15.5) | 3(7.7) | 4(10.3) | ||
| N0 | 25(64.1) | 194(68.6) | 25(64.1) | 27(69.2) | ||
| N1 | 11(28.2) | 45(15.9) | 11(28.2) | 8(20.5) | ||
PCa: prostate cancer
BCR: biochemical recurrence
PSM: propensity score matching
LASSO Cox regression analysis was performed to build a prognostic signature, which selected five lncRNAs (RP11-783K16.13, RP11-727F15.11, PRKAG2-AS1, AC013460.1, CRNDE) from the 105 lncRNAs identified before. LASSO coefficient profiles of the 105 lncRNAs are presented in Figure. 1B. A formula to calculate the risk score for their risk of BCR was derived based on their individual five lncRNAs expression levels weighted by their regression coefficients as follows:
risk score = (0.07659 × expression level of RP11-783K16.13) + (0.24432 × expression level of RP11-727F15.11) + (0.00427 × expression level of PRKAG2-AS1) + (2.81174 × expression level of AC013460.1) + (0.01615× expression level of CRNDE)
(A) Heat map showed differentially expressed lncRNAs between early BCR patients and patients with long-term BCR survival. (B) LASSO coefficient profiles of the 105 early BCR associated lncRNAs.
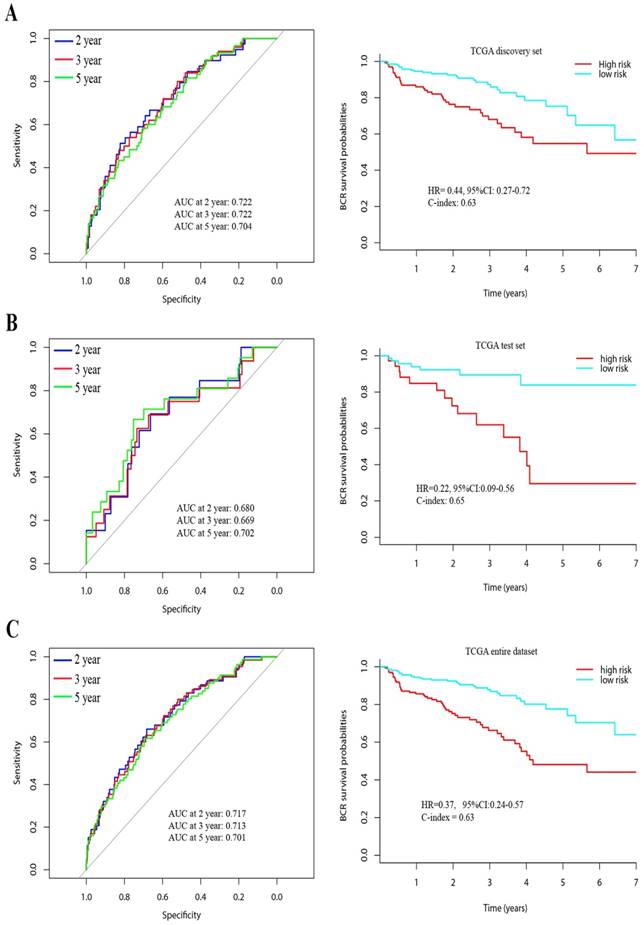
Then, 343 patients were classified into a high-risk group (n = 141) and a low-risk group (n = 202) according to the mean risk score. Time-dependent ROC curves of BCR showed that the AUC at 2, 3 and 5 years was 0.72, 0.72 and 0.70, respectively. (Figure 2A) Using Kaplan Meier method, BCR-free survival analysis suggested that the patients with low-risk scores generally had better BCR-free survival than patients with high-risk scores (HR = 0.44, 95%CI: 0.27-0.72, C-index = 0.63, Figure 2).
Time-dependent ROC curves at 2, 3, and 5 years and Kaplan-Meier survival analysis between patients with low risk scores and high risk scores in discovery set (A), test set (B) and entire dataset (C).
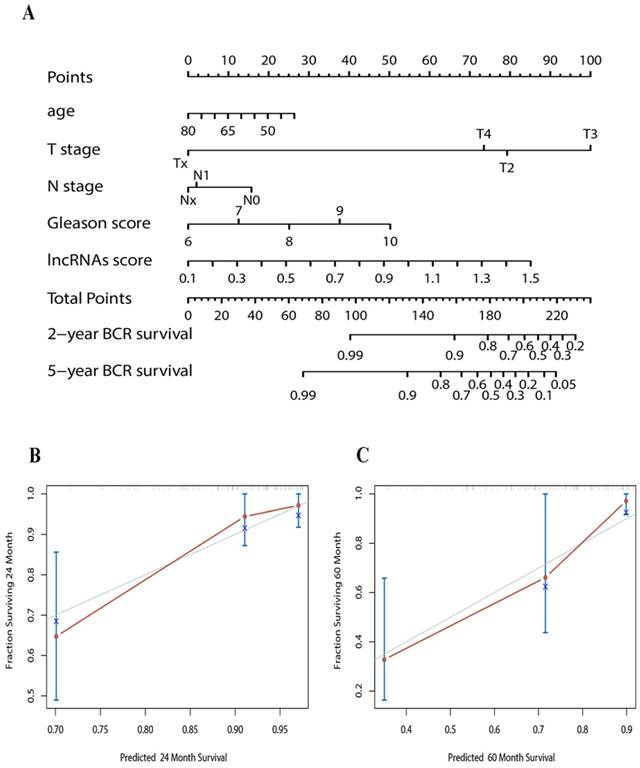
To predict the probability of BCR, we constructed a nomogram that integrated our lncRNAs signature in discovery set. The predictors included age, T/N stage, Gleason score and lncRNAs signature. (Figure 3A) The C-index of the nomogram was 0.74.
(A) Nomogram to predict the risk of BCR in PCa patients (B) Calibration curves of the nomogram to predict BCR at 2 years in test set (C) Calibration curves of the nomogram to predict BCR at 5 years in test set. The actual distant metastasis-free survival is plotted on the y-axis; nomogram predicted probability is plotted on the x-axis.
Validation of lncRNAs signature and lncRNAs-based Nomogram in the test set
In the test set, a total of 114 patients were enrolled, including 13 patients with early BCR and 94 patients with long-term BCR survival. To confirm whether the lncRNAs signature had similar prognostic value in the test set, we did the same analysis. Time-dependent ROC curves of BCR in the test set showed that the AUC at 2, 3 and 5 years was 0.68, 0.67 and 0.70, respectively. (Figure 2B) Using the established formula and cutoff point, 74 patients were classified into low-risk group and 40 patients in high-risk group. Patients with low-risk score also had longer BCR-free survival time than patients with high-risk score (HR = 0.22, 95%CI: 0.09-0.56, C-index = 0.65, Figure 2B). In the entire dataset analysis, the lncRNAs signature yielded similar results. The AUC at 2, 3 and 5 years was 0.72, 0.71 and 0.70, respectively. Patients in low-risk group had longer BCR-free survival time (HR = 0.37, 95%CI: 0.24-0.57, C-index = 0.63, Figure 2C).
Calibration plots showed that the nomograms also did well in the test set. The predictive accuracy of the nomograms is shown in Figure 3B and C. The C-index of the nomogram in the test set was 0.78.
Identification of five lncRNAs signature associated biological signaling pathway
GSEA in TCGA database was carried out to identify the five lncRNA associated biological signaling pathway. Significant gene sets (p < 0.05) were reported as Enrichment Map (Supplementary Figure S2). The risk group system was accompanied with cell cycle process, reproduction, and so on. The target genes or miRNAs of these lncRNAs (Supplementary Table S1) were predicted using lncRNABase, RBPDB, circlncRNAnet and LncRNAMAP online analysis tools. KEGG pathway analyses showed that notable pathway enrichment of the target genes, such as spliceosome, ribosome, RNA degradation, RNA transport, DNA replication and Notch signaling pathway (Supplementary Figure S3). GO term analyses showed that these target genes associated with rRNA processing, RNA binding, nucleic acid binding and so on. (Supplementary Figure S3)
Discussion
Most patients with early BCR will progress to clinical recurrence or metastasis and need immediate intervention. Patients with indolent cancer can be followed without immediate treatment.[3] To avoid unnecessary overtreatment of indolent disease, precise prediction that can stratify patients into high or low-risk groups has a significant clinical value.
Previous studies had reported various gene signatures that could provide prognostic information for PCa BCR.[2] However, few studies focused on the role of lncRNAs as novel signatures for PCa BCR. LncRNAs have shown its important roles in biological processes in various cancers, such as proliferation and angiogenesis of cancer cells.[15] In previous studies, lncRNAs could be used not only as a novel biomarker in the diagnosis of cancers but also as an adjunct to improve the specificity and sensitivity of existing biomarkers.[16] Therefore, lncRNAs were selected as potential parameter in our novel signature for prediction of BCR in PCa. Subsequently, a nomogram based on lncRNAs signature was also constructed in our study.
Five lncRNAs (CRNDE, PRKAG2-AS1, RP11-783K16.13, RP11-727F15.11 and AC013460.1) were identified significantly associated with PCa BCR in this study. Our five lncRNAs signature showed significant discrimination of the BCR both in discovery and test set. Besides, the lncRNAs-based nomogram provided much better discrimination of the BCR in both sets. Despite some biases in the development of lncRNAs signature, the validation in the test set indicated that the bias may not be large in this instance. Early BCR was considered as a sign for clinical recurrence metastasis. Overestimating the risk of patients that more suitable for active surveillance implied a higher disease burden due to overtreatment. Therefore, our nomogram that could identify patients into high or low-risk group of early BCR may help to select appropriate treatments and better clinical management in PCa patients.
In 2018, TA Bismar established a gene molecular signature (HDDA10) for predicting PCa BCR with AUC = 0.65.[6] Nevertheless, our five lncRNAs signature showed better discriminatory power with AUC = 0.72 in discovery set, 0.68 in the test set and 0.72 in the entire dataset. Our lncRNAs-based nomogram could provide new tools and insights on adjuvant therapies in future randomized controlled trials (RCTs). Patients with high-risk of BCR could receive adjuvant or other systemic therapies, while patients with low-risk could be entered into the group with active surveillance. Therefore, evaluation of treatments could be assessed more accurately.
In our five lncRNAs signature, CRNDE was certified that could regulate PI3K/Akt/Wnt/β-catenin axis to exert its oncogenic role in Hepatocellular carcinoma cell proliferation and growth.[17] In addition, CRNDE could promote cervical cancer cell growth, metastasis and progression of bladder cancer.[18, 19] Ding J suggested that CRNDE may promote colorectal cancer cell proliferation via epigenetically silencing DUSP5/CDKN1A expression.[20] Xie H demonstrated that CRNDE may modify susceptibility for various cancers and serve as a new predictive factor for prognosis and diagnosis in various cancers.[21] Hence, these previous studies proved the role of CRNDE in our signature was reasonable and reliable from the other aspect. However, the remaining four lncRNAs had not been investigated before. The future study may focus on these lncRNAs and investigate their function in PCa. Furthermore, these lncRNAs may have potential value in molecular targeted treatments. Although most lncRNAs are not yet functionally annotated in PCa, this study indicated the associated biological signaling pathway of the five lncRNAs through GSEA. The potential molecular function may put forward the direction for the further study on the mechanism of PCa progression.
Conclusion
In a word, a lncRNAs signature that could predict PCa BCR was identified by bioinformatics analysis. Additionally, a lncRNAs-based nomogram was also constructed. Our results showed that the nomogram can effectively classify patients into high or low-risk group of BCR. This nomogram comprising lncRNAs signature might help clinicians selecting personalized clinical management. Additional studies are needed to validate our results and further RCTs can test the role of this nomogram for prediction of the efficacy and safety of adjuvant therapy. In addition, functional studies are also required for better understanding the molecular mechanism in PCa.
Abbreviations
lncRNAs: long noncoding RNAs; BCR: biochemical recurrence; PCa: prostate cancer; PSM: propensity score matching; DEGs: differentially expressed genes; LASSO: least absolute shrinkage and selection operator; GSEA: gene set enrichment analysis.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant 81502192 for Fangning Wan and 81472377, 81672544 for Dingwei Ye).
Author Contributions
Conceptualization, Fangning Wan and Dingwei Ye; Formal analysis, Ning Shao and Yuanyuan Qu; Project administration, Fangning Wan; Supervision, Fangning Wan; Validation, Fangning Wan and Dingwei Ye; Writing - original draft, Ning Shao and Hong Tang; Writing - review & editing, Dingwei Ye.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Luo HW, Chen QB, Wan YP, Chen GX, Zhuo YJ, Cai ZD. et al. Protein regulator of cytokinesis 1 overexpression predicts biochemical recurrence in men with prostate cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;78:116-20
2. Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, Kraft P. et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:2391-6
3. Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N. et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. The Lancet Oncology. 2014;15:1521-32
4. Intasqui P, Bertolla RP, Sadi MV. Prostate cancer proteomics: clinically useful protein biomarkers and future perspectives. Expert review of proteomics. 2018;15:65-79
5. Dall'Era MA, Albertsen PC, Bangma C, Carroll PR, Carter HB, Cooperberg MR. et al. Active surveillance for prostate cancer: a systematic review of the literature. European urology. 2012;62:976-83
6. Abou-Ouf H, Alshalalfa M, Takhar M, Erho N, Donnelly B, Davicioni E. et al. Validation of a 10-gene molecular signature for predicting biochemical recurrence and clinical metastasis in localized prostate cancer. Journal of cancer research and clinical oncology. 2018;144:883-91
7. Sun M, Geng D, Li S, Chen Z, Zhao W. LncRNA PART1 modulates toll-like receptor pathways to influence cell proliferation and apoptosis in prostate cancer cells. Biological chemistry. 2018;399:387-95
8. Zhu H, Yu J, Zhu H, Guo Y, Feng S. Identification of key lncRNAs in colorectal cancer progression based on associated protein-protein interaction analysis. World journal of surgical oncology. 2017;15:153
9. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes & development. 2009;23:1494-504
10. Zhu Y, Li B, Liu Z, Jiang L, Wang G, Lv M. et al. Up-regulation of lncRNA SNHG1 indicates poor prognosis and promotes cell proliferation and metastasis of colorectal cancer by activation of the Wnt/beta-catenin signaling pathway. Oncotarget. 2017;8:111715-27
11. Wang W, Zhao Z, Yang F, Wang H, Wu F, Liang T. et al. An immune-related lncRNA signature for patients with anaplastic gliomas. Journal of neuro-oncology. 2018;136:263-71
12. Liang C, Zhang B, Ge H, Xu Y, Li G, Wu J. Long non-coding RNA CRNDE as a potential prognostic biomarker in solid tumors: A meta-analysis. Clinica chimica acta; international journal of clinical chemistry. 2018;481:99-107
13. Hansen J, Bianchi M, Sun M, Rink M, Castiglione F, Abdollah F. et al. Percentage of high-grade tumour volume does not meaningfully improve prediction of early biochemical recurrence after radical prostatectomy compared with Gleason score. BJU international. 2014;113:399-407
14. Tang XR, Li YQ, Liang SB, Jiang W, Liu F, Ge WX. et al. Development and validation of a gene expression-based signature to predict distant metastasis in locoregionally advanced nasopharyngeal carcinoma: a retrospective, multicentre, cohort study. The Lancet Oncology. 2018;19:382-93
15. Zhang M, Wang W, Li T, Yu X, Zhu Y, Ding F. et al. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;80:73-9
16. Malik B, Feng FY. Long noncoding RNAs in prostate cancer: overview and clinical implications. Asian journal of andrology. 2016;18:568-74
17. Tang Q, Zheng X, Zhang J. Long non-coding RNA CRNDE promotes heptaocellular carcinoma cell proliferation by regulating PI3K/Akt /beta-catenin signaling. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;103:1187-93
18. Cheng J, Chen J, Zhang X, Mei H, Wang F, Cai Z. Overexpression of CRNDE promotes the progression of bladder cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;99:638-44
19. Meng Y, Li Q, Li L, Ma R. The long non-coding RNA CRNDE promotes cervical cancer cell growth and metastasis. Biological chemistry. 2017;399:93-100
20. Liu T, Zhang X, Yang YM, Du LT, Wang CX. Increased expression of the long noncoding RNA CRNDE-h indicates a poor prognosis in colorectal cancer, and is positively correlated with IRX5 mRNA expression. OncoTargets and therapy. 2016;9:1437-48
21. Ellis BC, Molloy PL, Graham LD. CRNDE: A Long Non-Coding RNA Involved in CanceR, Neurobiology, and DEvelopment. Frontiers in genetics. 2012;3:270
Author contact
![]() Corresponding authors: Dingwei Ye and Fangning Wan: Department of Urology, Fudan University Shanghai Cancer Center and Department of Oncology, Shanghai Medical College, Fudan University, 270 Dongan Road, Shanghai, 200032, China. Email: 17111230009edu.cn and fnwan06edu.cn, Tel.:+86 17317821743
Corresponding authors: Dingwei Ye and Fangning Wan: Department of Urology, Fudan University Shanghai Cancer Center and Department of Oncology, Shanghai Medical College, Fudan University, 270 Dongan Road, Shanghai, 200032, China. Email: 17111230009edu.cn and fnwan06edu.cn, Tel.:+86 17317821743