Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(13):2874-2884. doi:10.7150/jca.31329 This issue Cite
Research Paper
CDC25A Facilitates Chemo-resistance in Ovarian Cancer Multicellular Spheroids by Promoting E-cadherin Expression and Arresting Cell Cycles
Yiting Sun1,*, Song Li1,*, Lu Yang2, Di Zhang1, Zeyi Zhao1, Jing Gao1, Lian Liu1, ![]()
1. Department of Medical Oncology, Cancer Center, Qilu Hospital of Shandong University, Jinan, Shandong, 250012, China
2. Key Laboratory of Carcinogenesis and Translational Research, Department of Renal Cancer and Melanoma, Peking University Cancer Hospital and Institute, Beijing, 100142, China
* These authors contributed equally to this work.
Received 2018-11-8; Accepted 2019-4-20; Published 2019-6-2
Abstract
Peritoneal metastasis is the most common pathway for the spread of ovarian cancer and one of the major causes of cancer death. Ovarian cancer cells in ascites prefer to aggregate into the multicellular spheroids (MCS) with an inadequate response to chemotherapy. In this study, gene expression analysis implicated that ovarian cancer MCS had its unique expression pattern and the cell cycle-related pathways were prominently altered in MCS cells compared to the monolayer adherent cells. Flow cytometry and western blots confirmed the cell cycle stagnancy in MCS. Among the cell cycle-related proteins, we found that expression of CDC25A was upregulated in MCS and displayed a time-dependent decrease during the transition from MCS to monolayer adherent cells. Loss-of-function studies showed that CDC25A promoted cisplatin-resistance and paclitaxel-resistance and inhibited the drug-induced apoptosis in ovarian cancer MCS. Mechanically, CDC25A impeded cell cycle progression in MCS cells, enhanced their structure integrity, and maintained upregulation of E-cadherin in MCS cells. Accordingly, addition of NSC95397, a small molecular inhibitor of CDC25A, sensitized the ovarian cancer MCS to chemotherapeutic agents. This provides us a novel strategy for the treatment of ovarian cancer peritoneal metastasis and may help improve the overall survival of ovarian cancer patients.
Keywords: Ovarian Neoplasms, Multicellular Spheroids, CDC25A, Drug Resistance, Cell Cycle, and Molecular Targeted Therapy
Introduction
Ovarian cancer is one of the three major malignancies in the female reproductive system. The incidence of ovarian cancer has been rising in recent years, and the mortality rate ranks first among the gynecologic malignancies [1]. Due to the absence of early symptoms, 60-70% of the patients with ovarian cancer are diagnosed at the late stage, with metastatic lesions beyond the ovaries [2]. Unlike other malignancies, ovarian cancer predominantly disseminates through the intra-abdominal routes [3, 4]. At the first diagnosis of ovarian cancer, approximately two-thirds of the patients developed peritoneal metastasis, and one-third of the patients developed ascites [5]. In addition, 30-50% of the patients with ovarian cancers suffered peritoneal recurrences after radical surgeries [6-8]. Ovarian cancer peritoneal metastasis and ascites production are closely associated with the inferior prognosis as well as deterioration of the life quality [8, 9]. So far, the incomplete understanding of the molecular mechanisms of ovarian cancer peritoneal metastasis hinders the development of effective treatments for it.
The current model of ovarian cancer peritoneal metastasis is a complex process with multiple steps, including cell shedding from the primary tumor, survival in ascites, peritoneal colonization, and so on [10]. When disseminating into the peritoneal cavity, the ovarian cancer cells aggregate into multicellular spheroids (MCS) with advantages for survival [3]. Compared to single exfoliated cells, MCS cells are tolerant to anoikis and able to survive in ascites and peritoneal cavities, thus representing an important survival mechanism for secondary colonization. In addition, MCS possesses a variety of mechanisms for drug-resistance, including the metabolite density gradient which inhibits the access of chemotherapy agents [9], the quiescent cell cycle which is insensitive to the therapies targeting proliferating cells, and the slow metabolism which reduces drug intakes [11]. The MCS is able to establish metastatic lesions at a comparable rate to monolayer cells upon intraperitoneal injection in mice [12]. The presence of MCS was also observed in ascites and the invasion ability of MCS is influenced by ascites derived from patients with ovarian cancer [13, 14]. In order to study the mechanism of ovarian cancer peritoneal metastasis, we previously established an E-cadherin-dependent MCS model of ovarian cancer in vitro to study the biological significance of MCS [15]. We found that the MCS had a stagnant proliferation, prolonged survival time, and drug-resistance to cisplatin in comparison with the monolayer adherent cells [15]. Besides, when re-transformed into monolayer cells, MCS cells obtained even higher abilities to invade and migrate than monolayer adherent cells [16].
Cell division cycle 25 A (CDC25A) is a member of the cell division cycle 25 families [17]. It is a dual-specificity protein phosphatase that removes the inhibitory phosphorylation in cyclin-dependent kinases (CDKs), including CDK4, CDK6, and CDK2, and positively regulates the cell cycle progression by helping pass the G1/S and G2/M checkpoints [17]. Overexpression of CDC25A has been reported in multiple cancers, such as ovarian cancer [18] and hepatocellular carcinomas [19], and correlated to a poor prognosis in patients [19, 20]. The onco-promoting mechanism of CDC25A was considered to be a result of its regulatory role in cell cycle transition [19, 20]. Besides, CDC25A also played critical roles in some other biological processes such as apoptosis [17, 21].
In the present study, we further investigated the differences in the biological behaviors and the underlying mechanisms between MCS and adherent cells and found CDC25A played an important role in the formation and maintenance of MCS as well as the chemo-resistance by arresting cell cycle progression.
Materials and Methods
Cell culture
The SK-H (SKOV-3 expressing high levels of E-cadherin) cell line was obtained from the Cancer Center Lab, Chinese Academy of Medical Sciences (Shanghai, China). Cells were cultured in RPMI-1640 (Gibco, Suzhou, China) with 10% fetal bovine serum (FBS) (Sciencell, Carlsbad, CA, USA), and maintained in a 37oC incubator with a relative humidity of 90% and 5% CO2. Cells were passaged when the confluences reached about 90%.
Establishment of the MCS models
Establishment of MCS was reported in our previous publications [15]. Firstly, 24-well plates were coated by 500 μl poly 2-hydroxyethyl methacrylate (Poly-HEMA) gel (Sigma, St. Louis, MO, USA) per well in the dilution of 12 mg/mL. Then the plates were air-dried in a laminar flow cabinet and washed with PBS three times consequently. A total of 5 x 104 cells were cultured in wells coated with (for MCS suspension) or without (for adherent cells) Poly-HEMA. Cells were not used for the subsequent experiments until the successful formation of MCS under microscopes.
Gene expression profiles
The MCS and monolayer adherent cells were harvested, and the total RNA was extracted using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Two MCS-derived and two monolayer adherent cell-derived RNA samples were applied to Phalanx Human OneArray chips for gene expression profile measurements. A detailed description of Phalanx Biotech company microarray procedure can be found at http://www.OneArray.com.cn. The selection criteria to identify differentially expressed genes are as follows: |Fold change| ≥ 2 and p < 0.05. GO and KEGG enrichment analysis was performed by DAVID gene ontology website.
Cell cycle analysis
MCS cells, monolayer adherent cells, and MCS cells that were dispersed and reattached to the petri dishes for 12h, 24h, and 48h were harvested by trypsinization. These cells were washed with pre-cooled PBS, centrifuged at 400g for 5 min at 4oC, and fixed with 70% pre-cooled ethanol at 4oC overnight. After filtered through 400-mesh filter traps, cells were stained with 5 μg/mL of propidium iodide (PI) in darkness for 30 min. The stained cells were measured on FACS Canto II (BD Biosciences, San Jose, CA), and the data were analyzed using the software Flowjo. To explore the effects of CDC25A on cell cycle, cells that were treated with CDC25A siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or NSC95397 (Millipore, Darmstadt, Germany) were stained and analyzed as described above.
Western blotting
The Bradford protein assay kit (Beyotime, Shanghai, China) was used for protein concentration measurement. The anti-β-actin and anti-GAPDH antibodies were purchased from Abcam. The anti-E-cadherin, anti-N-cadherin, anti-Vimentin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti-Rb, anti-pRb, anti-CDC25C, anti-CDC25A, anti-p53, anti-p27, anti-CDK2, anti-CDK4, anti-CDK6, anti-cyclinD1 and anti-cyclinE1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).The goat anti-rabbit or anti-mouse secondary antibodies were bought from Affinity Biosciences (Cincinnati, OH, USA). The immunoreactive bands were visualized using the MiniBIS Pro gel imaging system (DNR, Israel).
SiRNA transfection
CDC25A siRNA and scramble siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells were transfected with siRNA (100 pmol per 106 cells) before and after the formation of MCS, using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Cell viability assay
Cell viability was measured using Cell Counting Kit-8 (CCK-8) cell assay kits (Bestbio, Shanghai, China) according to the manufacturer's protocol. When MCS was established, cells were cultured in fresh media with DDP (in the final concentration of 30 nM or 90 nM) (Qilu Pharmaceutical, Jinan, Shandong, China) or paclitaxel (in the final concentration of 10 nM or 20 nM) (Qilu Pharmaceutical, Jinan, Shandong, China) for 48h. Cell viabilities were calculated based upon the OD values at 450nm. To measure the effect of NSC95397 (Millipore, Darmstadt, Germany) on chemo-resistance, we pre-treated cells with 32 nM NSC95397 for 48h before cultured in media with DDP or paclitaxel.
Apoptosis analysis
After transfection with CDC25A siRNA or scramble siRNA, or treatment with NSC95397 for 48h cultured, cells were treated with DDP or paclitaxel for 48 hours. Cells were harvested with 0.25% trypsin and washed twice with cold PBS. Then cells were resuspended and incubated with PI and fluorescein isothiocyanate (FITC)-Annexin V (BD Bioscience, Bedford, MA, USA) for 15 minutes in the dark, according to the manufacturer's instructions. Analysis of apoptotic cells was performed on a FACScan flow cytometer, and data were analyzed using the software Flowjo 10.
Graphs and Statistics
Flow cytometry data were graphed by Flowjo 10. All the other data and statistics were performed using the software Graphpad Prism 6. Cell cycle distributions, frequencies of dead cells, and cell viability were compared by two-tailed, unpaired Student's t-test. A p-value of 0.05 was considered to indicate statistical significance.
Results
Ovarian cancer MCS and adherent cells revealed different gene expression profiles
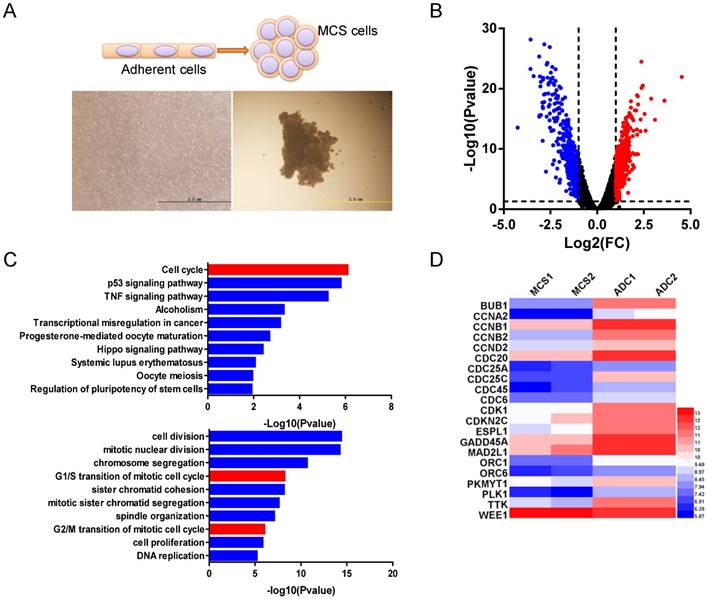
To identify the genes involved in the acquisition of drug-resistance potentials in ovarian cancer MCS, we compared gene expression profiles of MCS and adherent cells by microarray (Figure 1A). Among the 18273 genes analyzed, 980 were differentially expressed between the two clones (fold-change ≥ 2 and Student's t-test p-value < 0.05), with 509 genes upregulated and 471 downregulated in MCS cells (Figure 1B). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that the differentially expressed genes were enriched in the “cell cycle” pathway with the most significance (Figure 1C). Other significantly involved pathways included p53 signaling pathway, TNF signaling pathway, and Hippo signaling pathway (Figure 1C). In line with this, cell cycle-related signaling pathways, such as “G1/S” and “G2/M transition of mitotic cell cycle”, also emerged in the most relevant biological processes by Gene Ontology (GO) analysis (Figure 1C). Within the KEGG term “cell cycle”, a total of 21 genes were differentially expressed between MCS and adherent cells (Figure 1D). All of these genes, except WEE1, were downregulated in MCS cells compared to those in adherent cells (Figure 1D). These data implicated that MCS had its unique expression pattern and the cell cycle pathway was prominently altered.
Different gene expression profiles between ovarian cancer MCS and adherent cells. A. Morphologies of MCS and adherent cells were demonstrated under light microscopes. B. The volcano plot shows the distribution of the differentially expressed genes (MCS vs. ADC). The dotted lines represent the cut-offs. Blue dots represent the lowly expressed genes, and red dots represent the highly expressed genes in MCS (|Fold change| ≥ 2 and p < 0.05). The data were derived from MCS and adherent cell samples in duplicates. C. Top 10 KEGG terms (above) and GO terms (below) from gene set enrichment analysis. The red bars indicate the terms related to cell cycles. D. The differentially expressed genes involved in the KEGG term “cell cycle” are shown in the heatmap. MCS: multicellular spheroid; ADC: adherent cells.
The cell cycle in MCS was arrested at the G1/S checkpoint and dynamically changed when reattached
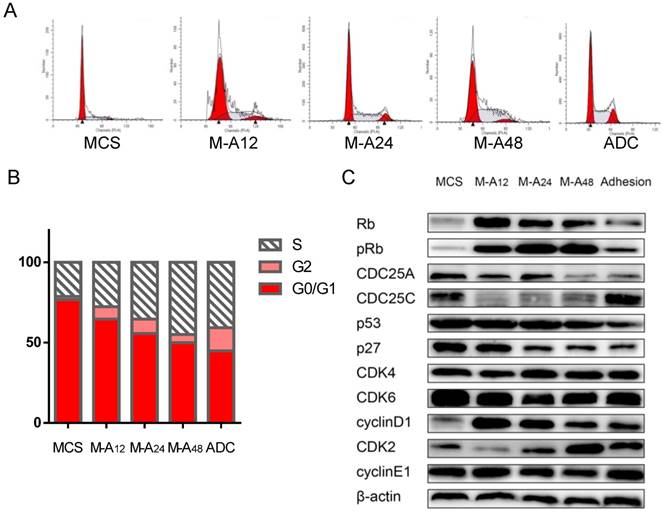
To verify the results of the gene expression profile in MCS, we examined the cell cycle status and several key genes involved in cell cycle regulation at the translational level in MCS and adherent cells. We also dispersed the MCS cells, reattached them to Petri dishes, and evaluated these cells at the time points of the 12th (M-A12), 24th (M-A24), and 48th hours (M-A48) after the attachment. Flow cytometry verified that the cell cycle was hindered at the G1/S checkpoint in MCS cells, which was in accordance with our previous findings. The proportions of G1 phase cells declined consecutively with the time of reattachment; the G1 phase cells contributed to 76.80%, 64.75%, 55.71%, 49.93%, and 44.81% of the MCS, M-A12, M-A24, M-A48, and adherent cells, respectively (Figure 2A and 2B). The probed proteins showed diverse expression patterns (Figure 2C). The molecules CDC25A, p53, and p27 displayed a time-dependent decrease after the transition from MCS to adherent cells (Figure 2C). Expressions of CDC25C and CDK2 were downregulated in MCS and showed a continuous augmentation when reattached (Figure 2C). The Rb and phosphorylated Rb (pRb) also showed lower expressions in MCS than those in adherent cells, but higher expression levels were found in the reattached cells than originally adherent cells (Figure 2C). Among these genes, the CDC25A intrigued us due to its onco-promoting function and the fact that there was no publication reporting its role in MCS so far.
Arrested cell cycle in ovarian cancer MCS. A. The flow cytometry results showed the cell cycle phase distributions in MCS, adhesive cells, and the MCS cells that were dispersed and reattached to the Petri dishes for 12h (M-A12), 24h (M-A24), and 48h (M-A48). B. Distributions of G0/G1, S, and G2 phases of the above data were quantified. C. The cell lysates were probed with the indicated antibodies. MCS: multicellular spheroid; ADC: adherent cells.
CDC25A promoted the G1 stagnancy of ovarian cancer MCS
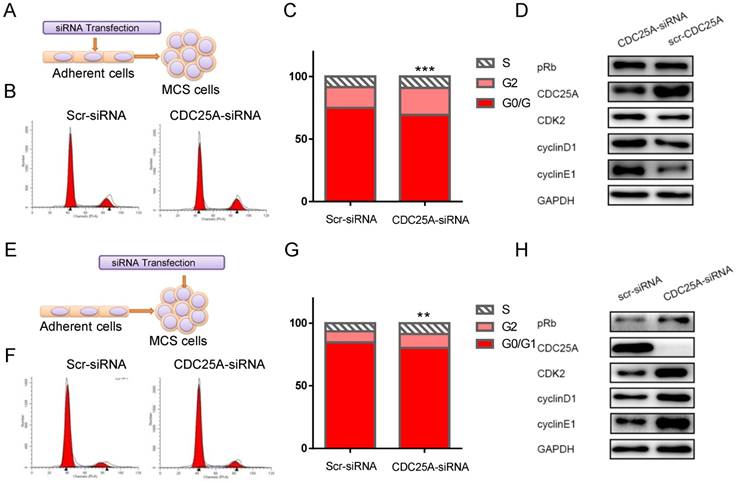
To investigate the biological function of upregulated CDC25A in ovarian cancer MCS, we first manipulated the CDC25A expression in adherent cells and then cultured them in suspension to form MCS (Figure 3A). Knockdown of CDC25A in this model reduced the proportion of cells at G1 phase from 74.97% to 69.31% (p = 0.0001) (Figure 3B and 3C). The expression levels of the G1/S transition-promoting molecules, including CDK2, cyclin D1, cyclin E1, and pRb were all upregulated upon CDC25A suppression (Figure 3D). We also accessed the function of CDC25A in another loss-of-function model by siRNA transfection directly into MCS (Figure 3E). This led to similar effects as in the first model. CDC25A silencing reduced the proportion of cells at G1 phase from 84.72% to 80.30% (p = 0.0017) (Figure 3F and 3G) and increased the expression level of CDK2, cyclin D1, cyclin E1, and pRb (Figure 3H). Taken together, these data indicated that the CDC25A inhibited, instead of promoted, cell cycle progression in ovarian cancer MCS.
Maintenance of the G1 stagnancy by CDC25A in ovarian cancer MCS. A. Adherent cells were transfected with siRNA and then cultured in suspension to form MCS in the first model. B-C. Distributions of G0/G1, S, and G2 phases of the MCS cells were shown by flow cytometry (B) and quantified (C) in the first model. D. The cell lysates in the first model were probed with the indicated antibodies. E. MCS was established first and then was transfected with siRNA in the second model. F-G. Distributions of G0/G1, S, and G2 phases of the MCS cells were shown by flow cytometry (F) and quantified (G) in the second model. H. The cell lysates in the second model were probed with the indicated antibodies. The quantitative data were derived from three independent experiments and expressed as means. MCS: multicellular spheroid; Scr-siRNA: scramble siRNA; CDC25A-siRNA: siRNA targeting CDC25A; **: p < 0.01; ***: p < 0.001.
CDC25A maintained the spheroid structure and upregulated E-cadherin expression in ovarian cancer MCS
Spheroid integrity was observed by microscopy to investigate the role of CDC25A in maintaining the spheroid structure. Treatment of MCS with siRNA before the spheroid formation significantly hindered the development of MCS (Figure 4A). The cell masses with CDC25A silencing displayed a looser morphology with much weaker integrity than the control one (Figure 4B). When directly treated with siRNA, MCS lost partial cell connections and dissociated into fragmentary masses (Figure 4D and 4E). These results indicated that the CDC25A played a critical role in maintaining the organization of MCS.
Maintenance of the spheroid structure and the epithelial property by CDC25A in ovarian cancer MCS. A. Adherent cells were transfected with scr- or CDC25A-siRNA, and then cultured in suspension to form MCS in the first model. B. The morphologies of MCS in the first model were demonstrated under light microscopes. C. The cell lysates in the first model were probed with the indicated antibodies. D. Cells were cultured in suspension to form MCS, and then were transfected with scr- or CDC25A-siRNA in the second model. E. The morphologies of MCS in the second model were demonstrated under light microscopes. F. The cell lysates in the second model were probed with the indicated antibodies. Scr-siRNA: scramble siRNA; CDC25A-siRNA: siRNA targeting CDC25A.
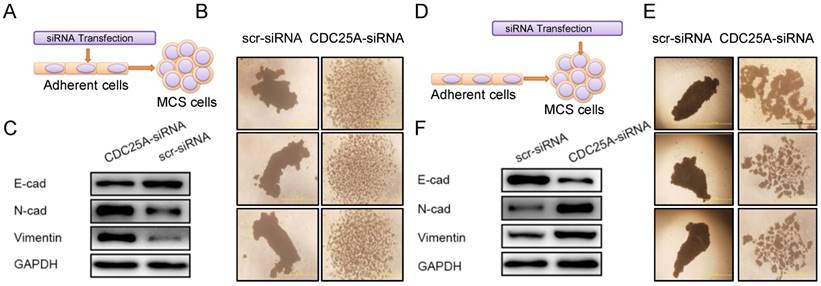
Our previous study revealed that E-cadherin played a key role in maintaining the structure of MCS [15]. Thus, we evaluated the changes of E-cadherin, N-cadherin, and vimentin in ovarian cancer MCS when CDC25A was altered. As shown in Figure 4C and 4F, the epithelial marker E-cadherin was diminished no matter whether the siRNA transfection was applied before (Figure 4C) or after the formation of MCS (Figure 4F). Correspondingly, mesenchymal markers, N-cadherin and vimentin, were augmented upon CDC25A suppression (Figure 4C and 4F). Thus, CDC25A upregulated the expression of E-cadherin and maintained the stable structure of MCS.
CDC25A increased the chemo-resistance of ovarian cancer MCS
The quiescent proliferation state and the condensed structure of ovarian cancer MCS may both contribute to chemo-resistance. Therefore, we next evaluated the role of CDC25A in regulating drug-resistances against cisplatin or paclitaxel. In the first model in which siRNA was transfected prior to the formation of MCS (Figure 5A), CDC25A knockdown (0.6810 vs. 0.7719, p = 0.0576) or 30 nM cisplatin (0.7257 vs. 0.7719, p = 0.3260) alone could not decrease the viability of MCS cells, while the combination of them achieved a significant cell-killing effect (0.4174 vs. 0.7719, p < 0.0001) (Figure 5B). When the concentration was elevated to 90 nM, cisplatin significantly reduced the cell viability (0.5801 vs. 0.7719, p = 0.0040). Addition of CDC25A-siRNA sensitized MCS cells and resulted in much lower cell viability than cisplatin alone (0.3171 vs. 0.5801, p < 0.0001) (Figure 5B). CDC25A interference also sensitized MCS cells to paclitaxel. While CDC25A knockdown (1.1940 vs. 1.1330, p = 0.3126) or 10 nM paclitaxel alone (1.0700 vs. 1.1330, p = 0.3255) could not reach a significance in inhibiting MCS cells, combination of them effectively reduced the cell viability of MCS cells (0.6630 vs. 1.1330, p < 0.0001) (Figure 5C). A concentration of 20 nM paclitaxel alone significantly reduced the cell viability of MCS cells (0.9309 vs. 1.133, p = 0.0006), and addition of CDC25A-siRNA further enhanced the cells' sensitivity to paclitaxel (0.4937 vs. 0.9309, p < 0.0001).
Contribution of CDC25A to the chemo-resistance of ovarian cancer MCS. A. Adherent cells were transfected with scr- or CDC25A-siRNA, and then cultured in suspension to form MCS in the first model. B-C. Cells of the first model were treated with cisplatin (30 nM or 90 nM) (B) or paclitaxel (10 nM or 20 nM) (C) for 48 h and then measured by cell viability assays. D-E. Drug-induced apoptosis in the first model was shown. Cells of the first model were treated with cisplatin (30 nM) (D) or paclitaxel (10 nM) (E), stained with 7AAD and Annexin-V, and measured by flow cytometry (left). The frequencies of cells in each quadrant were quantified on the right. F. Cells were cultured in suspension to form MCS, and then were transfected with scr- or CDC25A-siRNA in the second model. G-H. Drug-resistances of the cells in the second model were shown. Cells of the second model were treated with cisplatin (30 nM or 90 nM) (G) or paclitaxel (10 nM or 20 nM) (H) for 48 h and then measured by cell viability assays. I-J. Drug-induced apoptosis in the second model was shown. Cells of the second model were treated with cisplatin (30 nM) (I) or paclitaxel (10 nM) (J), stained with 7AAD and Annexin-V, and measured by flow cytometry (left). The frequencies of cells in each quadrant were quantified on the right. Treatment conditions were described below each panel. All quantitative data were derived from more than three independent experiments. Cell viability data were expressed as the mean ± SEM, and flow cytometry data were expressed as the mean percentages. Scr-siRNA, scramble siRNA; CDC25A-siRNA, siRNA targeting CDC25A; ns: no significance; *: p < 0.05: **; p < 0.01; ***: p < 0.001; ****: p < 0.0001.
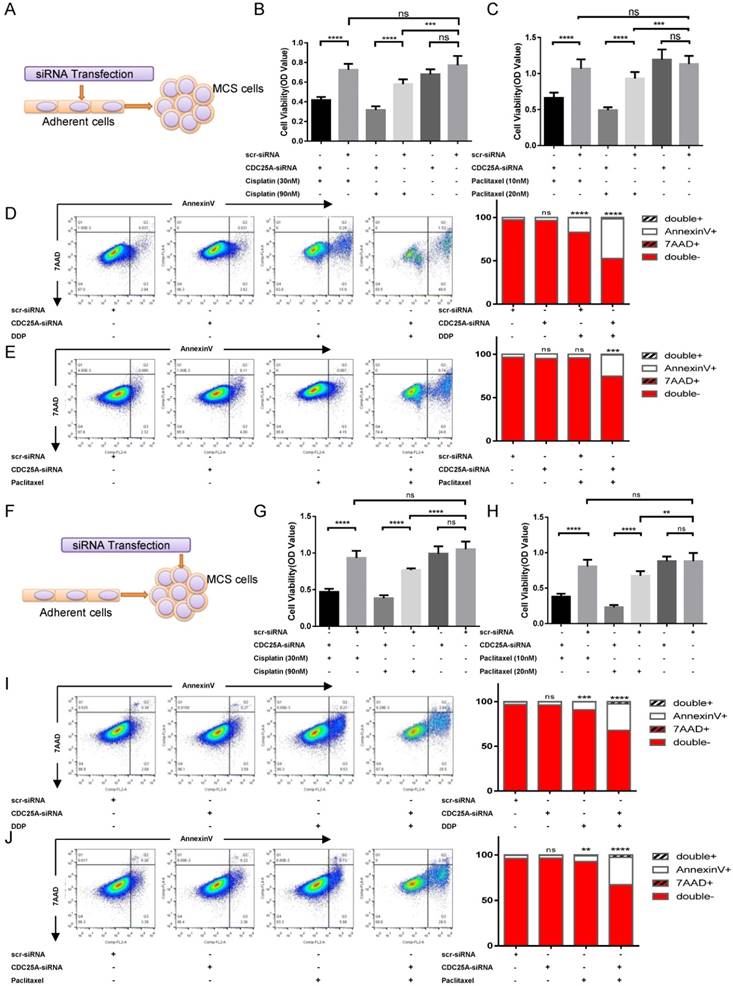
We also assessed the frequencies of drug-induced apoptosis by flow cytometry. The CDC25A-siRNA alone did not alter the frequency of apoptotic cells (AnnexinV+) (2.75% vs. 3.54%, p = 0.0974), while cisplatin (30 nM) alone significantly increased the frequency of AnnexinV+ cells (17.22% vs. 2.75%, p < 0.0001) (Figure 5D). In contrast, combination of them resulted in a much higher frequency of AnnexinV+ cells than cisplatin alone (47.41% vs. 2.75%, p < 0.0001) (Figure 5D). Similarly, the proportion of AnnexinV+ cells was not altered by paclitaxel (10 nM) alone (4.27% vs. 2.79 %, p = 0.0897), but it was significantly increased by addition of CDC25A-siRNA (10 nM) (25.30% vs. 2.79%, p = 0.0003) (Figure 5E).
In the second model in which siRNA was transfected after the formation of MCS (Figure 5F), CDC25A knockdown (0.9932 vs. 1.052, p = 0.3362) or 30 nM cisplatin (0.9349 vs. 1.052, p = 0.0578) alone did not alter the cell viability of MCS, while 90 nM cisplatin (0.7711 vs. 1.052, p < 0.0001) alone moderately lowered the cell viability (Figure 5G). Addition of CDC25A-siRNA significantly enhanced the sensitivity to cisplatin compared to cisplatin alone (0.4691 vs. 0.9349, p < 0.0001 for 30 nM and 0.3843 vs. 0.7711, p < 0.0001 for 90 nM) (Figure 5G).
CDC25A also influenced the paclitaxel-resistance of MCS in a similar pattern. CDC25A knockdown (0.8819 vs. 0.8816, p = 0.9955) and paclitaxel in 10 nM (0.8063 vs. 0.8816, p = 0.2056) did not alter the cell viability of MCS, while paclitaxel in 20 nM (0.6753 vs. 0.8816, p = 0.0014) alone moderately reduced the cell viability (Figure 5H). Combination of CDC25A siRNA and paclitaxel produced a synergetic effect in killing MCS cells compared to paclitaxel alone (0.3791 vs. 0.8063, p < 0.0001 for 10 nM and 0.2307 vs. 0.6753, p < 0.0001 for 20 nM) (Figure 5H).
Flow cytometry showed that cisplatin (9.09% vs. 3.15%, p = 0.0008) (Figure 5I) or paclitaxel (7.23% vs. 3.85%, p = 0.0070) (Figure 5J) alone slightly elevated the frequency of AnnexinV+ cells, while CDC25A siRNA could not promote apoptosis of MCS cells. Addition of CDC25A siRNA significantly elevated the frequency of apoptotic cells induced by cisplatin (32.13% vs. 3.15%, p < 0.0001) (Figure 5I) and paclitaxel (32.70% vs. 3.85%, p < 0.0001) (Figure 5J).
The proteinase inhibitor targeting CDC25A reversed the chemo-resistance of ovarian cancer MCS
Based on the above results, we speculated that the CDC25A inhibitors could be used as a chemosensitizer for treatment of ovarian cancer MCS. We utilized NSC95397 (Millipore, CAS 93718-83-3), a specific targeting inhibitor of CDC25A, to block the effect of CDC25A in MCS. Cell cycle assays indicated that the NSC95397 partially relieved the G1 phase blockage in MCS; the proportion of G1 phase cells in MCS dropped from 80.25% to 70.52% when treated with NSC95397 (p = 0.0024) (Figure 6A). The MCS structures were loosened by NSC95397 and dissociated into fragmentary masses (Figure 6B). Treatment with NSC95397 did not alter the expression of CDC25A, but significantly increased the expression of CDK2, cyclin D1, cyclin E, and pRb (Figure 6C). Meanwhile, the expression level of E-cadherin was decreased, and expression levels of N-cadherin and vimentin were elevated when treated with NSC95397 (Figure 6C), which was consistent with CDC25A downregulation.
Sensitization of ovarian cancer MCS to chemotherapeutic agents by targeting CDC25A. A. Distributions of G0/G1, S, and G2 phases of the MCS cells were shown by flow cytometry (left) and quantified (right). MCS cells were treated with or without NSC95387 for 48h before the flow cytometry assay. B. The morphologies of MCS were observed under light microscopes after treatment with or without NSC95387 for 48h. C. The cell lysates of MCS treated with or without NSC95387 were probed with the indicated antibodies. D-E. Chemo-resistances of the cells treated with or without NSC95387 were shown. Cells were treated with PBS or NSC95387 for 48h, exposed to cisplatin (30 nM or 90 nM) (D) or paclitaxel (10 nM or 20 nM) (E) for 48 h, and then measured by cell viability assays. F-G. The drug-induced apoptosis treated with or without NSC95387 was shown. MCS cells were treated with PBS or NSC95387 for 48 h, exposed to cisplatin (30 nM) (F) or paclitaxel (10 nM) (G), stained with 7AAD and Annexin-V, and measured by flow cytometry (left). The frequencies of cells in each quadrant were quantified on the right. Treatment conditions were described below each panel. All quantitative data were derived from more than three independent experiments. Cell viability data were expressed as the mean ± SEM, and flow cytometry data were expressed as the means. ns: p > 0.05; *: p < 0.05: **; p < 0.01; ***: p < 0.001; ****: p < 0.0001.
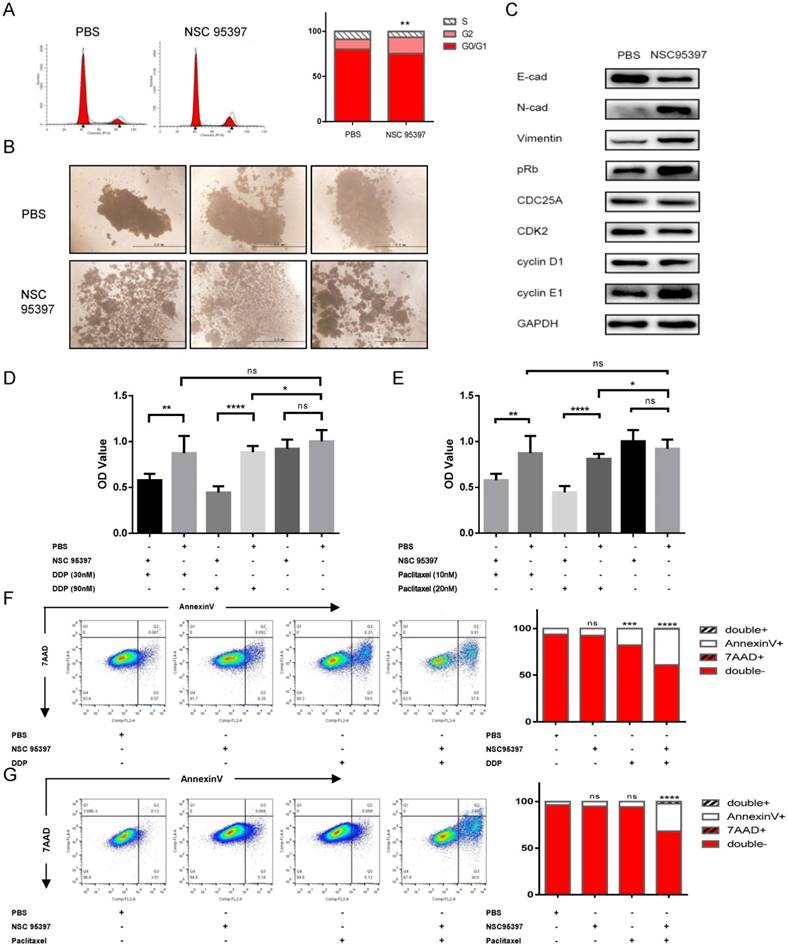
Cell viability assay showed that NSC95397 (0.9217 vs. 1.004, p = 0.1959) and 30 nM cisplatin (0.8746 vs.1.004, p = 0.1534) alone had no effect on MCS cells (Figure 6D). Instead, a combination of them significantly suppressed the cell viability (0.5784 vs. 0.8746, p = 0.0021) (Figure 6D). Cisplatin in 90 nM had a moderate cell-killing effect on MCS cells (0.8839 vs. 1.004, p = 0.0421), and addition of NSC95397 considerably elevated the sensitivity to cisplatin (90 nM) (0.4459 vs. 0.8839, p < 0.0001) (Figure 6D).
Paclitaxel in concentration of 10 nM did not alter the cell viability (0.8746 vs. 0.9217, p = 0.5692) and addition of NSC95397 significantly suppressed the cell viability in MCS (0.5784 vs. 0.8746, p = 0.0021) (Figure 6E). Paclitaxel in 20 nM had a moderate effect on MCS cells (0.8124 vs. 0.9217, p = 0.0256) and the addition of NSC95397 also remarkably sensitized the MCS cells to paclitaxel (0.4459 vs. 0.8124, p < 0.0001) (Figure 6E).
Apoptosis assays indicated that NSC95397 significantly enhanced the frequencies of the drug-induced apoptosis by both cisplatin (39.16% vs. 6.26%, p < 0.0001) (Figure 6F) and paclitaxel (31.87% vs. 3.52%, p < 0.0001) in MCS (Figure 6G). In contrast, NSC95397 or paclitaxel (5.66% vs. 3.52%, p = 0.0544) alone did not induce apoptosis, while cisplatin alone moderately enhanced apoptosis (17.96% vs. 6.26%, p = 0.0003) (Figure 6F and 6G).
Discussion
Peritoneal metastasis is an important cause of morbidity and mortality in patients with ovarian cancer. Formation of MCS contributes to the chemo-resistance in the process of peritoneal metastasis and agents that are able to overcome this drug-resistance can achieve a therapeutically synergistic effect.
In this study, the expression profile analysis indicated that the differentially expressed genes between MCS and monolayer adherent cells were remarkably enriched in the KEGG pathway “cell cycle”. Flow cytometry analysis confirmed that the cell cycle in ovarian cancer MCS was indeed arrested at the G1/S checkpoint and this cell cycle blockage was gradually released during the cell reattachment. Among the cell cycle-related proteins, we found that expression of CDC25A was upregulated in MCS and displayed a time-dependent decrease after the transition from MCS to monolayer adherent cells. Inhibition of CDC25A expression partially relieved the G1/S stagnancy, loosened the spheroid structure, and downregulated E-cadherin expression of MCS cells. Accordingly, targeting CDC25A by siRNA or small molecule inhibitors reduced the drug-resistance to cisplatin or paclitaxel.
In the process of peritoneal metastasis, the disseminated tumor cells aggregate into MCS to obtain multiple advantages in drug-resistance [10, 11]. First, the unvascularized structure generates a metabolite density gradient that inhibits access of chemotherapy agents to the internal cells [9, 10]. Second, the MCS lives in a quiescent state that is resistant to therapies targeting proliferating cells [21]. Third, MCS usually contain cancer-initiating progenitor-like cells which conferred drug-resistance [10, 11]. Our previous study showed that E-cadherin played a crucial role in the formation and maintenance of MCS and downregulation of E-cadherin abolished the structure of MCS [15]. In the present study, inhibition of CDC25A reduced the expression of E-cadherin and promoted the expression of mesenchymal markers, N-cadherin and vimentin. Thus, we speculate that CDC25A may maintain the intact structure of MCS by promoting the expression of E-cadherin. However, the underlying mechanism in detail requires further study. Interfering CDC25A blocked the formation of MCS and loosened their condensed structure, which facilitated the penetration of chemotherapeutic drugs into the interior of the structure and, consequently, reduced the chemo-resistance of the MCS cells. In addition, E-cadherin hinders G1/S progression by induction of overexpression of p27 during contact inhibition [22]. This helps explain the phenomenon in this study that knockdown of CDC25A expression or inhibition of its function partially abolished the cell cycle arrest in MCS. The accelerated G1/S progression would expand the cells in M phase and increase the cytotoxicity of the M phase-specific drug paclitaxel, thus enhancing the sensitivity of MCS to the chemotherapeutics.
Alternative mechanisms of the chemotherapy-sensitizing effects of CDC25A inhibition might exist in ovarian cancer MCS. The commonly used chemotherapeutic drugs in ovarian cancers, cisplatin, and paclitaxel, can both induce apoptosis of cancer cells. Cisplatin may induce cell apoptosis by activation of NF-κB [23], bax [23], and p53 [24], while paclitaxel may activate caspases [25], p53 [24], and bax [25, 26] for cell apoptosis. It has been reported that the cytoplasmic CDC25A increases cells' resistance to oxidative stress-mediated apoptosis by inhibiting ASK1 activity [19]. The ASK1 further asserts its pro-apoptotic effects through the downstream JNK pathway [27-29] or p38 pathway [29, 30]. Therefore, although inhibiting CDC25A alone did not significantly increase the frequencies of apoptotic cells in MCS, these might act synergistically with paclitaxel or cisplatin to promote tumor cell apoptosis.
There are several issues yet to be elusive. First, we noticed contrary CDC25A expression results from microarray and western blot data. This may result from that the transcription of CDC25A mainly occurs in S phase [31], while the proportion of S phase in MCS is much less than that in adherent cells. As a consequence, the mRNA generated from MCS is relatively limited. Another possible explanation is negative feedback of the transcription systems; high level of CDC25A protein reduces the mRNA transcription level through this mechanism [32]. Second, inhibition of CDC25A by siRNA or NSC95397 partially relieved the cell cycle stagnancy in ovarian cancer MCS. Though CDC25A is recognized as a key player in promoting cell cycle G1/S transition [17], it may inhibit cell cycle through alternative pathways [22]. It is reasonable that the dissociated MCS cells under CDC25A inhibition lost part of their contact inhibition and thus entered a growing cell cycle, in which the E-cadherin/p27 might be a responsible pathway [22]. The outcome came from an antagonistic effect of the above pathways.
Conclusions
In summary, CDC25A is overexpressed in ovarian cancer MCS and interfering this molecular promotes G1/S transition, downregulates E-cadherin expression, loosens the structure, and consequently reverses the chemo-resistance to cisplatin and paclitaxel in MCS. This provides us a feasible strategy to overcome the drug-resistance of advanced ovarian cancer with peritoneal metastasis and refractory ascites and may help improve the prognosis of these patients.
Abbreviations
MCS: Multicellular spheroids; CDC25A: Cell division cycle 25 A; CDK: Cyclin-dependent kinases; KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; SK-H: SKOV-3 expressing high levels of E-cadherin; FBS: Fetal bovine serum; Poly-HEMA: Poly 2-hydroxylethyl methacrylate; PI: Propidium iodide.
Acknowledgements
This research was funded by National Natural Science Foundation of China (81172487 to Lian Liu and 81500092 to Song Li) and Natural Science Foundation of Shandong Province, China (ZR201702180008 to Lian Liu).
Author Contributions
Conceptualization: Lian Liu.
Investigation: Yiting Sun, Lu Yang, Di Zhang, Zeyi Zhao and Jing Gao.
Visualization: Yiting Sun and Song Li.
Writing: Yiting Sun and Song Li.
Manuscript review and editing: Song Li and Lian Liu.
Supervision: Lian Liu.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7-30
2. Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nature reviews Cancer. 2005;5:355-66
3. Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. The Lancet Oncology. 2006;7:925-34
4. Ayantunde AA, Parsons SL. Pattern and prognostic factors in patients with malignant ascites: a retrospective study. Annals of oncology: official journal of the European Society for Medical Oncology. 2007;18:945-9
5. Ghaemmaghami F, Hassanzadeh M, Karimi-Zarchi M, Modari-Gilani M, Behtash A, Mousavi N. Centralization of ovarian cancer surgery: do patients benefit? European journal of gynaecological oncology. 2010;31:429-33
6. Ghaemmaghami F, Karimi-Zarchi M, Modares-Gilani M, Mousavi A, Behtash N. Clinical outcome of Iranian patients with advanced ovarian cancer with neoadjuvant chemotherapy versus primary debulking surgery. Asian Pacific journal of cancer prevention: APJCP. 2008;9:719-24
7. Behtash N, Karimi Zarchi M, Ashraf-Ganjoei T. Uterine involvement in advanced epithelial ovarian cancer. European journal of gynaecological oncology. 2010;31:99-101
8. Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177:1053-64
9. Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Frontiers in oncology. 2013;3:256
10. Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecologic oncology. 2009;113:143-8
11. Peart T, Ramos Valdes Y, Correa RJ, Fazio E, Bertrand M, McGee J. et al. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget. 2015;6:22424-38
12. Puiffe ML, Le Page C, Filali-Mouhim A, Zietarska M, Ouellet V, Tonin PN. et al. Characterization of ovarian cancer ascites on cell invasion, proliferation, spheroid formation, and gene expression in an in vitro model of epithelial ovarian cancer. Neoplasia. 2007;9:820-9
13. Allen HJ, Porter C, Gamarra M, Piver MS, Johnson EA. Isolation and morphologic characterization of human ovarian carcinoma cell clusters present in effusions. Experimental cell biology. 1987;55:194-208
14. Zietarska M, Maugard CM, Filali-Mouhim A, Alam-Fahmy M, Tonin PN, Provencher DM. et al. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Molecular carcinogenesis. 2007;46:872-85
15. Xu S, Yang Y, Dong L, Qiu W, Yang L, Wang X. et al. Construction and characteristics of an E-cadherin-related three-dimensional suspension growth model of ovarian cancer. Scientific reports. 2014;4:5646
16. Yang Y, Li S, Sun Y, Zhang D, Zhao Z, Liu L. Reversing platinum resistance in ovarian cancer multicellular spheroids by targeting Bcl-2. OncoTargets and therapy. 2019;12:897-906
17. Shen T, Huang S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anti-cancer agents in medicinal chemistry. 2012;12:631-9
18. Broggini M, Buraggi G, Brenna A, Riva L, Codegoni AM, Torri V. et al. Cell cycle-related phosphatases CDC25A and B expression correlates with survival in ovarian cancer patients. Anticancer research. 2000;20:4835-40
19. Xu X, Yamamoto H, Sakon M, Yasui M, Ngan CY, Fukunaga H. et al. Overexpression of CDC25A phosphatase is associated with hypergrowth activity and poor prognosis of human hepatocellular carcinomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:1764-72
20. Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nature reviews Cancer. 2007;7:495-507
21. Zou X, Tsutsui T, Ray D, Blomquist JF, Ichijo H, Ucker DS. et al. The cell cycle-regulatory CDC25A phosphatase inhibits apoptosis signal-regulating kinase 1. Molecular and cellular biology. 2001;21:4818-28
22. Motti ML, Califano D, Baldassarre G, Celetti A, Merolla F, Forzati F. et al. Reduced E-cadherin expression contributes to the loss of p27kip1-mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis. 2005;26:1021-34
23. Yu S, Gong LS, Li NF, Pan YF, Zhang L. Galangin (GG) combined with cisplatin (DDP) to suppress human lung cancer by inhibition of STAT3-regulated NF-kappaB and Bcl-2/Bax signaling pathways. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;97:213-24
24. Gu J, Tang Y, Liu Y, Guo H, Wang Y, Cai L. et al. Murine double minute 2 siRNA and wild-type p53 gene therapy enhances sensitivity of the SKOV3/DDP ovarian cancer cell line to cisplatin chemotherapy in vitro and in vivo. Cancer letters. 2014;343:200-9
25. Quispe-Soto ET, Calaf GM. Effect of curcumin and paclitaxel on breast carcinogenesis. International journal of oncology. 2016;49:2569-77
26. Salah-Eldin AE, Inoue S, Tsukamoto S, Aoi H, Tsuda M. An association of Bcl-2 phosphorylation and Bax localization with their functions after hyperthermia and paclitaxel treatment. International journal of cancer. 2003;103:53-60
27. Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nature genetics. 1999;21:326-9
28. Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158-62
29. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326-31
30. Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. The Journal of biological chemistry. 2006;281:25215-22
31. Fernandez-Vidal A, Mazars A, Manenti S. CDC25A: a rebel within the CDC25 phosphatases family? Anti-cancer agents in medicinal chemistry. 2008;8:825-31
32. Slobodin B, Han R, Calderone V, Vrielink J, Loayza-Puch F, Elkon R. et al. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell. 2017;169:326-37 e12
Author contact
![]() Corresponding author: Lian Liu, Department of Medical Oncology, Cancer Center, Qilu Hospital of Shandong University, 107 Wenhuaxi road, Jinan, Shandong, 250012, China; Tel: +86 53182169851; Fax: +86 53186927544; Email: lianliusubcom
Corresponding author: Lian Liu, Department of Medical Oncology, Cancer Center, Qilu Hospital of Shandong University, 107 Wenhuaxi road, Jinan, Shandong, 250012, China; Tel: +86 53182169851; Fax: +86 53186927544; Email: lianliusubcom