Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(8):1862-1869. doi:10.7150/jca.28379 This issue Cite
Research Paper
Fine Mapping in Chromosome 3q28 Identified Two Variants Associated with Lung Cancer Risk in Asian Population
Yang Wen1*, Chen Zhu2*, Ni Li3, Zhihua Li1,4, Yang Cheng1, Jing Dong1, Meng Zhu1, Yuzhuo Wang1, Juncheng Dai1,5, Hongxia Ma1,5, Guangfu Jin1,5, Min Dai3, Zhibin Hu1,5, Hongbing Shen1,5 ![]()
1. Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, China.
2. Zhejiang Provincial Office for Cancer Prevention and Control, Zhejiang Cancer Center/Zhejiang Cancer Hospital, Hangzhou 310004, China.
3. National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China.
4. Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China.
5. Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Jiangsu Collaborative Innovation Center of Cancer Personalized Medicine, Nanjing Medical University, Nanjing, 211166, China.
*Y.W. and C.Z. contributed equally to this work.
Received 2018-7-8; Accepted 2019-1-20; Published 2019-4-21
Abstract
Genome-wide association studies (GWASs) have consistently identified chromosome 3q28 as a lung cancer susceptibility region. To further characterize the potential genetic mechanism of the variants in this region, we conducted a fine-mapping study on chromosome 3q28 region. We performed a target resequencing in 200 lung cancer cases and 300 controls in the screening and followed by validation in multi-ethnic lung cancer GWASs with 12,843 cases and 12,639 controls. For our identified novel variants, we conducted expression quantitative trait loci (eQTL) analysis to reveal the potential target genes. Two susceptibility variants were identified (rs4396880: G>A, OR = 0.35, 95%CI: 0.20-0.62, P = 3.01×10-4; and rs3856776: C>T, OR = 2.05, 95%CI: 1.32-3.18, P = 1.49×10-3) and further supported in Asian population (rs4396880: OR = 0.88, P = 7.43×10-6; and rs3856776: OR =1.17, P = 1.64×10-4). The eQTL analysis showed the A allele of rs4396880 was significantly associated with higher mRNA expression of TP63 (P = 1.70×10-4) in lung tissues, while rs3856776 showed significant association with the expression of LEPREL1-AS1 (P = 6.90×10-3), which was the antisense RNA of LEPREL1 and could suppress the translation of LEPREL1. Notably, LEPREL1 was aberrantly downregulated (P = 2.54×10-18) in lung tumor tissues based on TCGA database. In conclusion, this is the first fine-mapping analysis of 3q28 region in Han Chinese, and we found two variants associated with lung cancer susceptibility in Asian population. What's more, rs3856776 was newly identified and might modulate lung cancer susceptibility by suppressing the function of LEPREL1.
Keywords: lung cancer, chromosome 3q28, fine mapping, susceptibility loci, Chinese population
Introduction
Lung cancer is one of the most common malignancies and the leading cause of cancer death worldwide with over one million deaths annually [1, 2]. Tobacco smoking confers 90% lung cancer cases, whereas inherited genetic factors also play an important role in individual predisposition to lung cancer [3]. Genome-wide association studies (GWAS) have significantly improved our understanding of inherited susceptibility to lung cancer [4-7].
According to Amos CI's paper, GWAS has found 45 lung cancer susceptibility loci [8]. Among them, chromosome 3q28 was first reported by Miki et al. in Japanese and Korean descent and two common variants rs10937405 and rs4488809 at 3q28 were identified associated with lung cancer risk [9]. This association was further replicated by our previous study in Chinese and European populations, respectively [5, 10, 11]. Genetic variants within this region were also associated with lung cancer risk, especially with adenocarcinoma in non-smoking Asian women [9, 12]. However, most of these variants reported by GWAS are merely the associated markers and the underlying molecular bases of these associations are unknown. Fine mapping is useful to survey the possible variants for further follow-up studies to understand the biological basis [13-15].
In the past several years, fine mapping studies of lung cancer have been performed in 15q25, 5p15.33 and major histocompatibility complex (MHC) regions, while the chromosome 3q28 remains unclear [16-19]. Therefore, to localize the potentially causal variants at 3q28 associated with lung cancer susceptibility, we conducted a two-stage fine-mapping study, which consisted of a target region resequencing on 200 lung cancer cases and 300 controls (Discovery cohort) and an association analysis in silico with 12,843 lung cancer cases and 12,639 controls (Validation cohort). The Expression quantitative trait loci (eQTL) analysis and gene differential expression analysis were further applied to explore the possible target genes through which genetic variants in 3q28 modified lung cancer susceptibility. This study would provide us a deeper insight into the genetic mechanisms of variants in 3q28 in the development of lung cancer.
Materials and Methods
Study subjects
Discovery cohort
Two hundred newly diagnosed lung cancer patients and 300 age-matched and sex-matched controls were involved in the study. These individuals were also a proportion of our previous GWAS subjects [5, 20]. Lung cancer cases were recruited from the Cancer Hospital of Jiangsu Province and the First Affiliated Hospital of Nanjing Medical University since 2003. Patients were confirmed non-small cell lung cancer (NSCLC) without radiation, chemotherapy or had a history of cancer, metastasized cancer from other organs. Controls were selected from individuals participating in a community-based screening program for non-infectious diseases conducted in Jiangsu province during the same period when the cases were recruited. The controls were frequency matched to the cases on age (±5 years) and gender.
All of the participants were genetically unrelated ethnic Han Chinese individuals, and they each donated approximately 5-ml of venous blood. Smoking information were collected through face-to-face interviewed by trained interviewers and those who had smoked less than an average of one cigarette per day and < 1 year in their lifetime were defined as nonsmokers; while others were considered as smokers. This study was conducted with approval of the institutional review board of Nanjing Medical University and informed consent was obtained from all participants.
Validation cohort
The validation cohort included our previous GWAS subjects (NJMU GWAS), consisting of 2,331 lung cancer cases and 3,077 controls form Nanjing and Beijing; and additional 10,512 lung cancer cases and 9,562 controls from the Female Lung Cancer Consortium in Asia (FLCCA) [21] and the National Cancer Institute (NCI) GWASs which were obtained via the database of Genotypes and Phenotypes (dbGAP) [22]. The FLCCA (dbGaP Study Accession: phs000716.v1.p1) investigated the etiology of lung cancer among never-smoking women in Asia, including 5,510 lung cancer cases and 4,544 controls from 14 studies from mainland China, South Korea, Japan, Singapore, Taiwan and Hong Kong [23] ( http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000716.v1.p1). Among these subjects, 4,922 lung cancer cases and 3,959 controls are available in dbGAP. The NCI GWASs (dbGaP Study Accession: phs000336.v1.p1) were derived from one population-based case-control study and three cohort studies specifically: the Environment and Genetics in Lung Cancer Etiology (EAGLE) (dbGaP Study Accession: phs000093.v2.p2), the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC), the Prostate, Lung, Colon, Ovary Screening Trial (PLCO) and the Cancer Prevention Study II Nutrition Cohort (CPS-II) (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000336.v1.p1). The four studies (EAGLE, ATBC, CPS-II and PLCO) were divided into two data sets because the EAGLE study provided the smoking information and the others didn't offer. We obtained EAGLE study data from dbGaP phs000093.v2.p2, which included 1,945 cases and 1,992 controls (SLD) in Italy between 2002 and 2005. The other three studies included 3,782 cases and 3,840 controls (CADM, dbGap phs000336.v1.p1). Among these three studies, ATBC is a randomized primary prevention trial in Finland between 1985 and 1993. Both PLCO and CPS-II were conducted in U.S. states. The population characteristics of these studies were shown in Table S1.
Quality control and imputation of GWAS data sets
The NJMU GWAS was conducted using Affymetrix Genome-Wide Human SNP Array 6.0 with standard quality control procedures conducted in previous study [5, 20]. Though the data sets from the dbGaP were described to be deposited after the initial quality control, we performed standard quality control on these data sets. We eliminated individuals with low call rates (95%), familial relationships and extreme heterozygosity rates, and excluded SNPs with low call rates (95%), MAF (minor allele frequency) < 0.05 and P < 1×10-6 for the Hardy-Weinberg equilibrium (HWE). After quality control, 4,796 cases and 3,741 controls from FLCCA, 1,937 cases and 1,984 controls from SLD, and 3,779 cases and 3,837 controls from CADM remained. The genotyping data were imputed using the 1000 Genomes Project data (the Phase III integrated variant set release, across 2,504 samples) as a reference. We then phased the haplotypes with Shapeit v2 (http://www.shapeit.fr/, Phasing step) and imputed with IMPUTE2 (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html). Poorly imputed SNPs defined by an information measure < 0.40 with IMPUTE2 were further excluded from the analysis.
Target region sequencing and genotyping
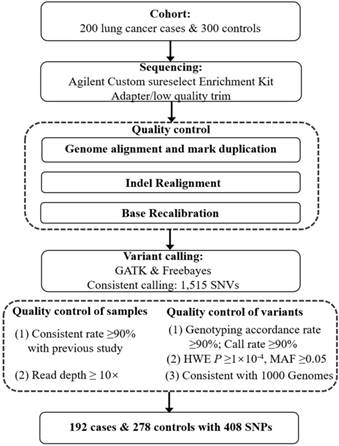
We used the HapMap Project database (phase II + III Feb 09, on NCBI B36 assembly, dbSNP126), to explore the LD structure around the SNP rs4488809, as previously reported by Miki et al. [9]. Finally, an LD block spanning from chr3: 189214000 to chr3: 189428000 (hg19) was included in our study. In total, 248 probes (total size: 131.73 kb; coverage: 61.56%) were designed using Agilent Sure Select software (http://earray.chem.agilent.com/eArray) and captured through Agilent Sure Select protocol (Agilent Technologies, Santa Clara, California, USA). The following sequencing experiment was performed on a Genome Analyzer IIx (Illumina, San Diego, CA) [24]. Sequencing adapters and low-quality reads were filtered using the FASTQ Quality Filter tool. The high-quality reads were aligned to the human genome reference sequence (hg19) using Burrows-Wheeler Aligner (BWA, V.0.5.9) (http://bio-bwa.source-forge.net) [25]. Duplicates were marked using Picard Tools (http://picard.sourceforge.net/), and the base quality scores were recalibrated using Genome Analysis Toolkit (GATK, V.1.0.5974). Variants were called by GATK and Freebayes, and only those variants identified by both tools were taken into account.
We ruled out 8 lung cancer cases and 22 control subjects because they (i) had a low consistent rate (< 90%) as determined by comparing overlapped single nucleotide variants (SNVs) against existing NJMU GWAS data or (ii) yielded a read depth < 10× across samples. A total of 1,515 variants were detected through targeted resequencing. Among them, 1107 variants were excluded from subsequent analyses for the following reasons: (i) call rate of genotype < 90%; (ii) P value for HWE < 1×10 -4 in case, control or all subjects; (iii) MAF < 0.05 (control); (iv) concordance rate < 90% with the previous GWAS for overlapping variants if applied; or (v) not in the 1000 Genomes Project (www.1000genomes.org) (QC details in Figure 1). Finally, 408 common SNVs in 192 cases and 278 controls were retained for subsequent association analysis. Gemini (V.0.18.3) and snpEff (V.3_6) were used to annotate the function of the genetic variants from the targeted resequencing. The associations between each variant and lung cancer risk in the discovery cohort were calculated using logistic regression model with adjustment for age, sex and smoking statue in PLINK (V.1.90). Conditional analysis was performed using an approximated joint regression model in GCTA [26] to select index variants in 3q28 region, and those with nominal evidence of association (P < 0.05) were selected for further validation in multiple GWAS data sets.
Flowchart of this study.
Validation of identified SNVs
We validated the SNVs identified in targeted resequencing using multiple lung cancer GWASs in both Asian and European populations. Association analysis was performed using SNPTEST v2.5 under a probabilistic dosage model with adjustment for age, sex, smoking pack-year and the first principal component (PCA) in NJMU GWAS, age in FLCCA GWAS, age, sex, smoking status and the first PCA in SLD GWAS, and age, sex and the first PCA in CADM GWASs. The effect estimates and P values of candidate SNVs in each GWAS data set were then combined using SNPTEST separately by Asian and European populations. Finally, fixed-effect meta-analysis was conducted to assess pooled genetic effects in the validation stage. The Cochran's Q statistic and I2 were calculated using STATA software (V.8.0, College Station, TX, USA) to test the heterogeneity between studies. General analyses were performed with R software (version 3.1.1; The R Foundation for Statistical Computing).
Functional element analysis and gene expression analysis
These novel SNPs were investigated for the presence of chromatin histone marks and hypersensitive DNAse elements using data from ENCODE included in HaploReg v4.1 (http://www.broadinstitute.org/mammals/haploreg/haploreg.php) and the UCSC genome browser (https://genome.ucsc.edu/cgi-bin/hgGateway). The results and boxplots of the eQTL analysis in the lung tissue (383 samples) from the Genotype-Tissue Expression (GTEx) project were obtained from the version of GTEx Portal.v7 (http://www.gtexportal.org/).
Results
Characteristics of study subjects
In the current study, 192 lung cancer cases and 278 controls were finally included in the discovery cohort after quality control. Among the cases, 63 samples were squamous cell carcinoma (SCC) and 129 cases belonged to lung adenocarcinoma (AD). Male individuals accounted for more than 70% of subjects in discovery cohort. The distributions of gender and age were similar in cases and controls. Notably, cases had a higher proportion of smoking than that in controls (67.2% vs 47.5%). Characteristics of subjects in validation cohort had been described in our previous studies. The detailed information of study subjects in discovery and validation cohorts was summarized in Table S1.
Association results in discovery cohort
Of the 408 variants that were analyzed, 34 were significantly associated with lung cancer risk at P values less than 0.05, including three genetic variants which were identified by previous GWASs (rs4488809, rs10937405 and rs13314271) (Table S2). The directions of effects between the three SNPs and lung cancer risk were consistent with previous reports, and effect sizes were similar. Through approximate conditional analysis with GCTA, we identified six novel SNPs significantly associated with lung cancer risk (Table 1): rs17443378 (OR = 1.96, P = 0.026), rs3856776 (OR = 2.05, P = 1.49×10-3), rs4396880 (OR = 0.35, P = 3.01×10-4), rs4687064 (OR = 1.69, P = 0.034), rs57830317 (OR = 2.55, P = 2.76×10-4) and rs6798700 (OR = 0.29, P = 3.08×10-5).
Multiple distinct signals of association with lung cancer risk at the TP63 in the discovery and validation stages.
| SNP | Ref/Effect Allele | Discovery | Validation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF a | OR(95%CI) b | P b | Asian c | European d | Meta e | |||||||
| Cases | Controls | OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | Phet | ||||
| rs17443378 | C/G | 0.28 | 0.32 | 1.96(1.08-3.54) | 0.026 | 0.90(0.85-0.95) | 1.10×10-4 | 0.98(0.93-1.03) | 0.432 | 0.94(0.91-0.98) | 2.00×10-3 | 0.027 |
| rs3856776 | C/T | 0.13 | 0.08 | 2.05(1.32-3.18) | 1.49×10-3 | 1.17(1.08-1.27) | 1.64×10-4 | 1.01(0.96-1.07) | 0.701 | 1.06(1.01-1.11) | 0.016 | 0.003 |
| rs4396880 | G/A | 0.26 | 0.35 | 0.35(0.20-0.62) | 3.01×10-4 | 0.88(0.84-0.93) | 7.43×10-6 | 0.98(0.92-1.03) | 0.425 | 0.92(0.89-0.96) | 3.68×10-5 | 0.006 |
| rs4687064 | T/A | 0.22 | 0.18 | 1.69(1.04-2.76) | 0.034 | 0.89(0.84-0.94) | 5.66×10-5 | 0.95(0.88-1.03) | 0.202 | 0.91(0.87-0.95) | 5.19×10-5 | 0.186 |
| rs57830317 | A/C | 0.29 | 0.22 | 2.55(1.54-4.23) | 2.76×10-4 | 0.95(0.90-1.01) | 0.086 | 1.00(0.95-1.06) | 0.918 | 0.98(0.94-1.02) | 0.230 | 0.206 |
| rs6798700 | G/A | 0.27 | 0.26 | 0.29(0.16-0.51) | 3.08×10-5 | 0.94(0.89-0.99) | 0.027 | 1.02(0.97-1.08) | 0.480 | 0.98(0.94-1.02) | 0.267 | 0.034 |
a EAF, effect allele frequency; b Derived from the GCTA joint regression model with adjustment for age, sex and smoking status; c Derived from the fixed effect meta-analysis of NJMU and FLCCA GWASs; d Derived from the fixed effect meta-analysis of the NCI studies (the EAGLE, ATBC, CPSII and PLCO studies). e Derived from the fixed-effect meta-analysis of the NJMU, FLCCA, EAGLE, ATBC, CPSII and PLCO studies.
Validation in lung cancer GWASs and evaluation for independence
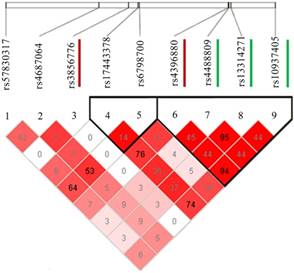
To validate the SNVs identified by targeted resequencing, we performed association analysis in multiple lung cancer GWASs divided by Asian and European populations (Table 1). Although no SNVs reached a genome-wide significance level of association (P < 5.0 × 10-8), two of the six SNVs showed a consistent association with lung cancer risk in Asian population. They are rs3856776 (OR = 1.17, 95% CI: 1.08-1.27, P = 1.64 × 10-4), and rs4396880 (OR = 0.88, 95% CI: 0.84-0.93, P = 7.43 × 10-6). However, no associations were observed for the two SNPs in European population. Based on LD analysis (Figure 2), rs4396880 was in LD with previous reported risk variants rs4488809, rs10937405 and rs13314271 (r2 = 0.44 to 0.95), while rs3856776 could not been tagged by these variants (r2 < 0.05). We applied conditional analysis to test the independence of rs4396880 and rs3856776 and used the predefined variants as covariates. Interestingly, after conditioning on the previously reported SNVs in this region, denoted by rs4488809 and rs10937405, only the association observed for rs4396880 was largely attenuated (P_Conditional=0.136 for rs4396880), while the association for rs3856776 remained essentially unchanged (Table 2). Furthermore, after conditioning on rs4396880 and rs3856776, the GWAS SNVs were no longer significant in the targeted resequencing analysis (all P_Conditional > 0.05, Table S3). We then performed stratification analysis of the two identified variants by age, gender, smoking status and histology and did not find significant effect difference between subgroups (Table 3).
LD plot of the two SNPs (rs4396880 and rs3856776) and other significant SNPs identified in previous studies. LD between the newly found two distinct signals (rs3856776 and rs4396880) and the three previously reported SNPs at this locus are based on our data. The 2 SNPs associated with lung cancer susceptibility in this article are marked with red lines and the 3 SNPs identified previously are marked with green lines.
Independent effect analysis by conditioning on rs4488809 and rs10937405.
| SNP | Ref/Eff Allele | MAF | Step 1 | Step 2 | |||
|---|---|---|---|---|---|---|---|
| case | control | OR (95%CI) a | P a | OR (95%CI) b | P b | ||
| rs4396880 | G/A | 0.26 | 0.35 | 0.64(0.47-0.87) | 3.88×10-3 | 0.33(0.08-1.41) | 0.136 |
| rs3856776 | C/T | 0.13 | 0.08 | 1.84(1.19-2.85) | 6.05×10-3 | 1.80(1.15-2.81) | 9.93×10-3 |
a adjusted for age, sex and smoking status. b adjusted for age, sex, smoking status, rs4488809 and rs10937405.
Stratification analysis of the associations of rs3856776 and rs4396880 with lung cancer risk.
| Characteristics | rs3856776 | rs4396880 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case a | Control a | OR(95%CI) b | P b | P_het c | Case a | Control a | OR(95%CI) b | P b | P_het c | ||
| Age | |||||||||||
| <60 | 82/16/4 | 123/21/0 | 1.65(0.90-3.01) | 0.104 | 0.626 | 52/45/5 | 62/67/15 | 0.73(0.48-1.11) | 0.144 | 0.404 | |
| ≥60 | 67/19/4 | 113/21/0 | 2.06(1.07-3.98) | 0.030 | 54/29/7 | 56/59/19 | 0.56(0.35-0.88) | 0.012 | |||
| Sex | |||||||||||
| Male | 109/24/7 | 165/33/0 | 1.68(1.02-2.79) | 0.043 | 0.511 | 76/55/9 | 77/95/26 | 0.59(0.41-0.85) | 0.004 | 0.454 | |
| Female | 40/11/1 | 71/9/0 | 2.38(0.96-5.90) | 0.062 | 30/19/3 | 41/31/8 | 0.76(0.44-1.33) | 0.344 | |||
| Smoke | |||||||||||
| Never | 48/13/2 | 125/21/0 | 2.24(1.09-4.61) | 0.029 | 0.493 | 37/23/3 | 67/57/22 | 0.64(0.39-1.03) | 0.066 | 1 | |
| Ever | 101/22/6 | 111/21/0 | 1.63(0.94-2.84) | 0.081 | 69/51/9 | 51/69/12 | 0.64(0.43-0.95) | 0.028 | |||
| Histology d | |||||||||||
| SCC | 49/10/4 | 236/42/0 | 1.73(0.91-3.29) | 0.091 | 0.871 | 33/24/6 | 118/126/34 | 0.72(0.44-1.15) | 0.177 | 0.549 | |
| AD | 100/25/4 | 236/42/0 | 1.85(1.13-3.04) | 0.014 | 73/50/6 | 118/126/34 | 0.60(0.42-0.85) | 0.004 | |||
a Sample size of subjects with wild type homozygote/heterozygote/variant homozygote; b Adjusted for age, sex and smoking status where is appropriate; c P for heterogeneity; d SCC, squamous cell carcinoma; AD, adenocarcinoma.
Functional evaluation
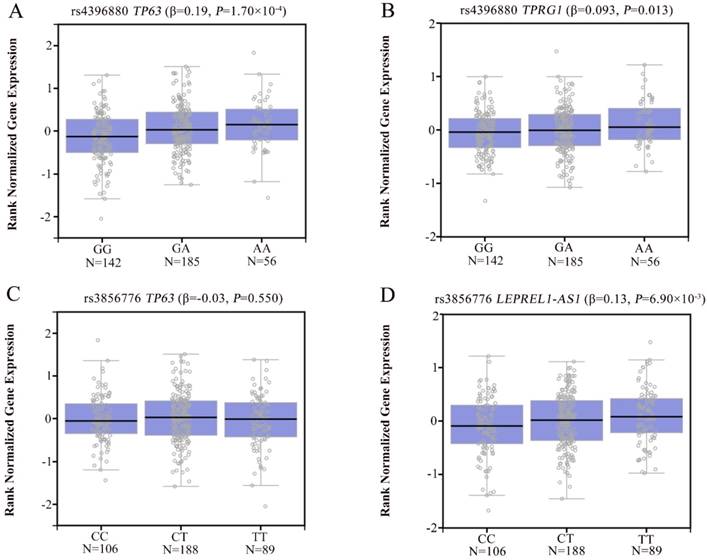
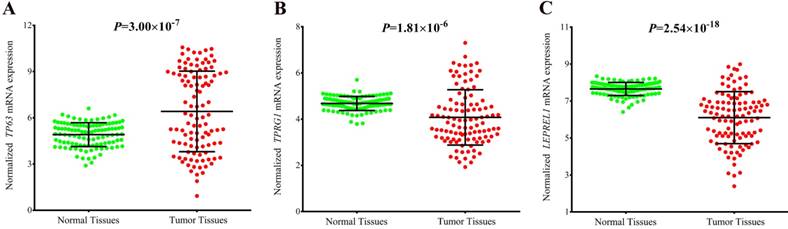
To explore the potential mechanism of the associations, we annotated the two newly identified variants using public databases (ENCODE and Roadmap Epigenomics Project database). We found that rs4396880 was located in DNase I hypersensitivity site (DHS) and regulatory histone modification signals in multiple cell types. Further eQTL annotation with GTEx v7 database suggested that the A allele of rs4396880 was associated with the increased mRNA expression of TP63 in 383 normal lung tissues (β = 0.19, P = 1.70×10-4, Figure 3A). Besides, we also found that rs4396880 showed significant association with tumor protein p63 regulated 1 (TPRG1) expression (β = 0.093, P = 0.013, Figure 3B). TP63 was aberrantly upregulated in lung tumor tissues based on TCGA database (P = 3.00×10-7, Figure 4A), while the expression of TPRG1 was significantly decreased in tumor tissues (P = 1.81×10-6, Figure 4B). For rs3856776, genotypes of this variant showed no significant association with TP63 (β = -0.03, P = 0.550, Figure 3C). Notably, SNP rs3856776 was associated with the expression of antisense LEPREL1-AS1 (β = 0.13, P = 6.90×10-3, Figure 3D). Interestingly, LEPREL1 expression was aberrantly downregulated in lung tumor tissues (P = 2.54×10-18, Figure 4C).
The eQTL analysis of rs4396880 and rs3856776 in 383 normal lung tissues based on GTEx database. Rs4396880 was significantly associated with TP63 (A) and TPRG1 expression (B); Rs3856776 showed no significant association with the expression of TP63 (C), but was significantly associated with LEPREL1-AS1 expression (D).
Expression of TP63, TPRG1 and LEPREL1 in lung tumor tissues and adjacent normal tissues based on TCGA database. TP63 was aberrantly upregulated in tumor tissues (A); The expression of TPRG1 was significantly decreased in tumor tissues (B); LEPREL1 expression in tumor tissues was significantly downregulated compared with that in adjacent normal tissues (C).
Discussion
In this study, we provided a relatively comprehensive genetic landscape at 3q28 in Chinese subjects by sequencing ~214 kb on TP63 region. We newly identified two genetic variants (rs4396880 and rs3856776) that were associated with lung cancer risk and subsequently validated in large-scale GWASs within Asian population. What's more, rs3856776 was a novel independent lung cancer susceptibility variant.
The TP63 gene whose product is the tumor protein P63, transcriptionally regulates genes involved in DNA repair [27, 28] and is important for normal development and differentiation of stratified epithelial tissues as well as for human carcinogenesis [29, 30]. In addition, p63 has been found to play an important role in cell cycle arrest and apoptosis through its interaction with mutant p53 [31, 32]. Altered TP63 expression has been linked to the development and progression of various types of cancers, including lung cancer [31, 33]. The most commonly reported variant variants in the TP63 gene, rs4488809 and rs10937405, were first identified in lung cancer GWAS in Asia [9], and consistently replicated in other Asian populations [5] and Europeans [34]. In our study, the variant rs4396880 was 40bp away from rs4488809 (r2 = 0.46, D' = 1.00) and 27kb from rs10937405 (r2 = 0.95, D' = 0.98). These three SNPs were included in the TP63 isoforms of TAp63 that are transcribed using a promoter upstream of exon 1, but not in other isoforms (△Np63) that are regulated by another promoter in intron 3 [35]. Interestingly, certain biologic hypotheses based on ENCODE are consistent with our statistical evidence. Rs4396880 demonstrates evidence of the DNase I hypersensitivity site (DHS) and histone modification marks H3K27Ac, H3K36M3 and H3K27Me3 consistent with promoter and enhancer activity in several cell lines, suggesting that it may have a functional role in the regulation of TP63 gene expression. Notably, SNP rs4396880 was also associated with the expression of TPRG1, which was significantly downregulated in lung tumor tissues. However, limited study was available about the function of TPRG1. Highlighting these findings, we speculate that rs4396880 A allele may modulate lung cancer risk by increased mRNA expression of TP63.
In the current study, we also identified an intergenic variant rs3856776 in 3q28 (50kb upstream of TP63) that was independently associated with lung cancer risk. The variant was not in LD with the previously reported variants. Interestingly, SNP rs3856776 was not associated with the expression of TP63, suggesting that this variant could modulate lung cancer susceptibility through other mechanisms. Importantly, a significant association between rs3856776 and antisense RNA LEPREL1-AS1 expression was observed according to GTEx database. LEPREL1 (leprecan-like 1, also known as P3H2) encodes a member of the P3H2 dioxygenases and participates in the collagen chain assembly, stability and cross-linking [36, 37]. Mutation in LEPREL1 had been reported associated with the risk of myopia [38, 39]. Previous studies indicated that LEPREL1 was significantly downregulated in breast cancer, lymphoma and hepatocellular carcinoma tissues and could function as a tumor suppressor [40-42]. Notably, in this study, we found that the expression of LEPREL1 was also aberrantly decreased in lung tumor tissues, suggesting that LEPREL1 might also play a tumor suppressor role in lung cancer. The antisense RNA of LEPREL1 (LEPREL1-AS1) could suppress the translation of LEPREL1. Taken together, rs3856776 might contribute to lung cancer risk by regulating the expression of LEPREL1-AS1 through suppressing the translation of tumor suppressor gene LEPREL1.
In summary, we performed the first fine mapping studying focusing on 3q28 region, two variants were identified significantly associated with lung cancer risk in Asian population. What's more, rs3856776 was a novel independent lung cancer risk variant and might influence lung cancer susceptibility through suppressing the function of LEPREL1. However, this study also had some limitations. First, we only analyzed the common variants, rare variants were excluded because of the limited sample size in discovery cohort. Second, for our identified variants, we speculated their potential target genes and possible mechanisms through bioinformatics methods, functional studies are needed to validate our findings.
Abbreviations
GWAS: genome-wide association study; MHC: major histocompatibility complex; eQTL: expression quantitative trait loci; NSCLC: non-small cell lung cancer; SNP: single nucleotide polymorphism; NCI: National Cancer Institute; QC: quality control; MAF: minor allele frequency; HWE: Hardy-Weinberg equilibrium; PCA: principal component; SCC: squamous cell carcinoma; AD: adenocarcinoma; TFBS: transcription factor binding site; DHS: DNase Hypersensitive Site.
Supplementary Material
Supplementary tables.
Acknowledgements
The authors would like to thank the patients and the supporting staff in this study. This work was supported by the Key international (regional) cooperative research project (81820108028), National Natural Science of China (81521004), the Priority Academic Program for the Development of Jiangsu Higher Education Institutions [Public Health and Preventive Medicine] and Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (PPZY2015A067).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015;136:E359-86
3. Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nature reviews Cancer. 2003;3:733-44
4. Zhang H, Cai B. The impact of tobacco on lung health in China. Respirology. 2003;8:17-21
5. Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z. et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nature genetics. 2011;43:792-6
6. Li Y, Sheu CC, Ye Y, de Andrade M, Wang L, Chang SC. et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. The Lancet Oncology. 2010;11:321-30
7. Yoon KA, Park JH, Han J, Park S, Lee GK, Han JY. et al. A genome-wide association study reveals susceptibility variants for non-small cell lung cancer in the Korean population. Human molecular genetics. 2010;19:4948-54
8. Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:363-79
9. Miki D, Kubo M, Takahashi A, Yoon KA, Kim J, Lee GK. et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nature genetics. 2010;42:893-6
10. Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nature genetics. 2014;46:736-41
11. McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126-32
12. Hosgood HD 3rd, Wang WC, Hong YC, Wang JC, Chen K, Chang IS. et al. Genetic variant in TP63 on locus 3q28 is associated with risk of lung adenocarcinoma among never-smoking females in Asia. Human genetics. 2012;131:1197-203
13. Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J. et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48:374-86
14. Choi J, Xu M, Makowski MM, Zhang T, Law MH, Kovacs MA. et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nat Genet. 2017;49:1326-35
15. Painter JN, Kaufmann S, O'Mara TA, Hillman KM, Sivakumaran H, Darabi H. et al. A Common Variant at the 14q32 Endometrial Cancer Risk Locus Activates AKT1 through YY1 Binding. Am J Hum Genet. 2016;98:1159-69
16. Dong J, Cheng Y, Zhu M, Wen Y, Wang C, Wang Y. et al. Fine mapping of chromosome 5p15.33 identifies novel lung cancer susceptibility loci in Han Chinese. Int J Cancer. 2017;141:447-56
17. Kachuri L, Amos CI, McKay JD, Johansson M, Vineis P, Bueno-de-Mesquita HB. et al. Fine mapping of chromosome 5p15.33 based on a targeted deep sequencing and high density genotyping identifies novel lung cancer susceptibility loci. Carcinogenesis. 2016;37:96-105
18. Cheng Y, Wang C, Zhu M, Dai J, Wang Y, Geng L. et al. Targeted sequencing of chromosome 15q25 identified novel variants associated with risk of lung cancer and smoking behavior in Chinese. Carcinogenesis. 2017;38:552-8
19. Qin N, Wang C, Zhu M, Lu Q, Ma Z, Huang M. et al. Fine-mapping the MHC region in Asian populations identified novel variants modifying susceptibility to lung cancer. Lung Cancer. 2017;112:169-75
20. Dong J, Hu Z, Wu C, Guo H, Zhou B, Lv J. et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population. Nature genetics. 2012;44:895-9
21. Hsiung CA, Lan Q, Hong YC, Chen CJ, Hosgood HD, Chang IS. et al. The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS genetics. 2010;6:e1001051
22. Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M. et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. American journal of human genetics. 2009;85:679-91
23. Lan Q, Hsiung CA, Matsuo K, Hong YC, Seow A, Wang Z. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet. 2012;44:1330-5
24. Zuzarte PC, Denroche RE, Fehringer G, Katzov-Eckert H, Hung RJ, McPherson JD. A two-dimensional pooling strategy for rare variant detection on next-generation sequencing platforms. PloS one. 2014;9:e93455
25. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
26. Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nature genetics. 2012;44:369-75 S1-3
27. Flores ER. The roles of p63 in cancer. Cell cycle. 2007;6:300-4
28. Lin YL, Sengupta S, Gurdziel K, Bell GW, Jacks T, Flores ER. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS genetics. 2009;5:e1000680
29. Tomkova K, Tomka M, Zajac V. Contribution of p53, p63, and p73 to the developmental diseases and cancer. Neoplasma. 2008;55:177-81
30. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413-31
31. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell death and differentiation. 2011;18:1487-99
32. Shakya R, Tarulli GA, Sheng L, Lokman NA, Ricciardelli C, Pishas KI. et al. Mutant p53 upregulates alpha-1 antitrypsin expression and promotes invasion in lung cancer. Oncogene. 2017;36:4469-80
33. Graziano V, De Laurenzi V. Role of p63 in cancer development. Biochimica et biophysica acta. 2011;1816:57-66
34. Wang Y, Broderick P, Matakidou A, Vijayakrishnan J, Eisen T, Houlston RS. Variation in TP63 is associated with lung adenocarcinoma in the UK population. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2011;20:1453-62
35. Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Molecular cancer research: MCR. 2004;2:371-86
36. Jarnum S, Kjellman C, Darabi A, Nilsson I, Edvardsen K, Aman P. LEPREL1, a novel ER and Golgi resident member of the Leprecan family. Biochem Biophys Res Commun. 2004;317:342-51
37. Pokidysheva E, Boudko S, Vranka J, Zientek K, Maddox K, Moser M. et al. Biological role of prolyl 3-hydroxylation in type IV collagen. Proc Natl Acad Sci U S A. 2014;111:161-6
38. Mordechai S, Gradstein L, Pasanen A, Ofir R, El Amour K, Levy J. et al. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am J Hum Genet. 2011;89:438-45
39. Guo H, Tong P, Peng Y, Wang T, Liu Y, Chen J. et al. Homozygous loss-of-function mutation of the LEPREL1 gene causes severe non-syndromic high myopia with early-onset cataract. Clin Genet. 2014;86:575-9
40. Shah R, Smith P, Purdie C, Quinlan P, Baker L, Aman P. et al. The prolyl 3-hydroxylases P3H2 and P3H3 are novel targets for epigenetic silencing in breast cancer. Br J Cancer. 2009;100:1687-96
41. Hatzimichael E, Lo Nigro C, Lattanzio L, Syed N, Shah R, Dasoula A. et al. The collagen prolyl hydroxylases are novel transcriptionally silenced genes in lymphoma. Br J Cancer. 2012;107:1423-32
42. Wang J, Xu X, Liu Z, Wei X, Zhuang R, Lu D. et al. LEPREL1 Expression in Human Hepatocellular Carcinoma and Its Suppressor Role on Cell Proliferation. Gastroenterol Res Pract. 2013;2013:109759
Author contact
![]() Corresponding author: Hongbing Shen, Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China, Tel.: 186-25-8686-8439, Fax: 186-25-8686-8499, E-mail: hbshenedu.cn
Corresponding author: Hongbing Shen, Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China, Tel.: 186-25-8686-8439, Fax: 186-25-8686-8499, E-mail: hbshenedu.cn