Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(7):1651-1662. doi:10.7150/jca.28077 This issue Cite
Research Paper
Salivary Microbial Dysbiosis is Associated with Systemic Inflammatory Markers and Predicted Oral Metabolites in Non-Small Cell Lung Cancer Patients
Weiquan Zhang1,2, Junwen Luo1,2, Xiaopeng Dong1, Shukang Zhao1,2, Yingtao Hao1, Chuanliang Peng1, Hubo Shi3, Yong Zhou4, Lei Shan1, Qifeng Sun1, Yuyang Li5, Xiaogang Zhao1 ![]()
1. Department of Thoracic Surgery, The Second Hospital of Shandong University, Jinan, Shandong 250033, China
2. School of Medicine, Shandong University, Jinan, Shandong 250012, China
3. Department of Thoracic Surgery, Shandong Provincial Chest Hospital, Jinan, Shandong 250013, China
4. Cancer Center, The Second Hospital of Shandong University, Jinan, Shandong 250033, China
5. Department of Breast Thyroid Surgery, Shandong Provincial Hospital affiliated to Shandong University, Jinan, Shandong 250021, China.
Received 2018-6-24; Accepted 2019-2-21; Published 2019-3-22
Abstract
An increasing number of studies have suggested the dysbiosis of salivary microbiome has been linked to the advancement of multiple diseases and proved to be helpful for the diagnosis of them. Although epidemiological studies of salivary microbiota in carcinogenesis are mounting, no systemic study exists regarding the oral microbiota of non-small cell lung cancer (NSCLC) patients. In this study, we presented the characteristics of the salivary microbiota in patients from NSCLC and healthy controls by sequencing of the 16S rRNA microbial genes. Our result revealed distinct salivary microbiota composition in patients from NSCLC compared to the healthy controls. As principal co-ordinates analysis (PCoA) showed, saliva samples clearly differed between the two groups, considering the weighted (p = 0.001, R2 = 0.17), and unweighted (p = 0.001, R2 = 0.25) UniFrac distance. Phylum Firmicutes (31.69% vs 24.25%, p < 0.05) and its two genera Veillonella (15.51%% vs 9.35%, p < 0.05) and Streptococcus (9.96% vs 6.83%, p < 0.05) were strongly increased in NSCLC group compared to the controls. Additionally, the relative abundances of Fusobacterium (3.06% vs 4.92%, p = 0.08), Prevotella (1.45% vs 3.52%, p < 0.001), Bacteroides (0.56% vs 2.24%, p < 0.001), and Faecalibacterium (0.21% vs 1.00%, p < 0.001) in NSCLC group were generally decreased. Furthermore, we investigated the correlations between systemic inflammation markers and salivary microbiota. Neutrophil-lymphocyte ratio (NLR) positively correlated with the Veillonella (r =0.350, p = 0.007) and lymphocyte-monocyte ratio (LMR) negatively correlated with Streptococcus (r =-0.340, p = 0.008). Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways inferred by phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) showed that pathways related to xenobiotics biodegradation and metabolism (p < 0.05) and amino acid metabolism (p < 0.05) were enriched in the NSCLC group. Folate biosynthesis (p < 0.05) significantly decreased in NSCLC group. The specific correlations of clinical systemic inflammation markers and predicted KEGG pathways also could pronounce a broad understanding of salivary microbiota in patients with NSCLC. Moreover, our study extended the new sight into salivary microbiota-targeted interventions to clinically improve the therapeutic strategies for salivary dysbiosis in NSCLC patients. Further investigations of the potential mechanism of salivary microbiota in the progression of NSCLC are still in demand.
Introduction
Lung cancer, one of the most prevalent cancers globally, is the leading cause of cancer-related deaths among males, [1] with an increased incidence in recent years in China [2]. Lung cancer is divided into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), with NSCLC accounting for about 80% of lung cancers and SCLC accounting for 20%. NSCLC is often asymptomatic or causes only nonspecific symptoms in its early stages. The five-year survival rate for NSCLC is only 15%, in part because the disease is usually diagnosed at a late stage and is frequently metastatic and incurable [3, 4]. Early detection is crucial for reducing the morbidity and mortality of NSCLC. However, it is difficult to promote current approaches to detect NSCLC, due to the high cost and low positive detection rate at the early stages of the disease. Currently, practical and effective approaches to the early diagnosis of NSCLC are desperately needed.
Tissue and blood samples have been widely used in the diagnosis and research of NSCLC [5]. In addition, human saliva has become an attractive medical diagnostic fluid as its collection is convenient and non-invasive. Saliva-based epidermal growth factor receptor (EGFR) gene mutation detection in patients with NSCLC is a method that has been shown to fulfil the clinical requirements for detection of EGFR mutation in patients with NSCLC, and this method could be reinforced with tissue DNA testing or used as a complement to biopsy [6]. Certain proteins in saliva have also been shown to indicate oral or systematic diseases, suggesting this method could be used for clinical screening and detection of lung cancer [7]. Increasingly, studies [8-11] have also reported an increased risk of certain types of tumourss that are related to the dysbiosis of salivary microbiota. The oral cavity is a large microbiome habitat in the human body and composed of more than 700 species of bacteria, among which more than 50% have not been cultivated [12]. Previous studies have identified the core microbiome related to health [13] and shown that shifts in the core microbiome are associated with the immuno-inflammatory response [14]. A number studies [15-18] have also shown that microbiota markers in saliva are well-known diagnostic and prognostic biomonitors for diverse diseases. In addition, salivary Capnocytophaga and Veillonella have been identified as potential biomarkers for lung cancer detection and classification [19]. Hence, characterizing the salivary microbiota in NSCLC patients could potentially help delineate the pathogenic role of salivary dysbiosis in the progression of NSCLC and direct the management of microbiota-targeted therapies.
To validate the above assumptions and predictions, we directly compared the salivary microbiota composition in NSCLC patients which are newly diagnosed and untreated with the healthy controls, by sequencing the 16S Ribosomal RNA gene in saliva samples. Our study also investigated correlations between salivary microbiota, systemic inflammatory markers, and the metabolites of salivary microbiota.
Materials and Methods
Patient Recruitment
This study complied with the ethical guidelines outlined in the 1975 Declaration of Helsinki and has been approved by the Research Ethics Board of the Second Hospital of Shandong University (Jinan, China) and Shandong Provincial Chest Hospital (Jinan, China). Prior to inclusion in this study, written informed consent was obtained from all of the participants.
Study subjects were newly diagnosed NSCLC patients seen at the Second Hospital of Shandong University and Shandong Provincial Chest Hospital in Jinan, from March 2016 to October 2016. NSCLC patients eligible for inclusion in this study were Han Chinese local inhabitants who had lived in Jinan for at least 5 years prior to the date of sample collection and had a definite postoperative pathological diagnosis of NSCLC stage IIB or IIIA. No patients received chemotherapy, radiation therapy, or surgery before sample collection. Subjects were excluded if they had been diagnosed as any diseases other than NSCLC, taken oral antibiotic or received probiotic therapy less than 6 months from the date of consent [20]. External factors also included smoking and drinking, which are significantly associated with salivary community types [21-23], or were unwilling to sign the informed consent. Control subjects were recruited from the Physical Examination Centre of the Second Hospital of Shandong University. The healthy status of the control subjects was self-reported and the control group was frequently matched to the study subjects by gender, age, and body mass index (BMI). Additionally, the clinical characteristics of all of the subjects are listed in Table 1.
Subject and sample data.
| Parameter | NSCLC (N=39) | Healthy Controls (n=20) | p-value | ||
|---|---|---|---|---|---|
| Age (years; mean ± SD) | 59.49 (8.43) | 59.45(7.33) | 0.987 | ||
| Females/Males, No. (%) | 10/29 (25.64%/74.36%) | 7/13 (35.00%/65.00%) | 0.452 | ||
| BMI (mean ± SD) (range) | 23.49 (2.12) | 23.01 (1.47) | 0.369 | ||
| NLR, mean (SD) | 2.64 (1.24) | 1.86 (0.47) | 0.001** | ||
| PLR, mean (SD) | 137.47 (55.52) | 119.30 (27.71) | 0.099 | ||
| LMR, mean (SD) | 6.36 (11.40) | 7.75 (2.67) | 0.595 | ||
| AMC, mean (SD) | 0.44 (0.17) | 0.30 (0.10) | <0.001*** | ||
| IL-6, mean (SD) | 4.53 (5.05) | 2.79 (1.56) | 0.141 | ||
| IL-1β, mean (SD) | 3.96 (3.04) | 9.64 (20.36) | 0.229 | ||
| Final diagnosis | NA | ||||
| Adenocarcinoma | 21 (53.85%) | ||||
| Squamous cell cancer | 18 (46.15%) | ||||
| Stage I/II/III/IV | 10/12/17/0 (26.83%/29.27%/43.90%/0) | ||||
p Values were based on t-test or χ2 test (two-sided). BMI, body mass index; NLR, neutrophil-lymphocyte ratio; PLR, platelet-lymphocyte ratio; LMR, lymphocyte-monocyte ratio; AMC, absolute monocyte count; IL-6, Interleukin 6.
Enzyme-linked Immunosorbent Assay
All of the blood samples were collected immediately after hospitalization and centrifuged at 1,000 g at 4°C for 15 minutes before being stored at -80°C for further analysis. Aliquots of serum were used to determine interleukin-6 (IL-6), IL-1β, IgA, and IgG concentration. Measurement of each factor was performed using specific ELISA kits obtained from ProteinTech Group (Wuhan, Hubei, China) according to the manufactures' instructions. All of the samples were assayed in duplicate and the average was used in the statistical analyses.
Saliva Sample Collection and Bacterial DNA extraction
Saliva samples for genomic DNA extraction were collected from each participant in the morning before brushing and breakfast, and placed into a 50-mL conical tube. Approximately 3mL of saliva was obtained from each individual, and samples were then transferred to the laboratory immediately in an icebox and stored at -80°C until further processing. Genomic DNA was extracted with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturers' protocols. The concentration and purity of extracted DNA were determined on 1% agarose gels. Then each sample was quantified with a Nano Drop ND-1000 spectrophotometer (Thermo Electron Corporation). All of the DNA was stored at -20°C until further analysis.
16S rRNA PCR Amplification and Sequencing
16S rRNA genes of distinct regions (V1-V2) were amplified with specific primers targeting 27F and 355R of the bacterial 16S gene bacterial barcode (27F: 5′-AGAGTTTGATCMTGGCTCAG-3′ and 355R: 5′-GCTGCCTCCCGTAGGAGT-3′). All of the PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs). Then, the PCR products were separated by 2% agarose gel electrophoresis and purified with the Qiagen Gel Extraction Kit (Qiagen, Germany) for further experiments. Sequencing libraries were generated using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) and sequenced on an IlluminaHiSeq2500 platform.
Bioinformatics Analysis
The paired-end reads were merged using FLASH software (V1.2.7, http://ccb.jhu.edu/software/FLASH/) [24], and filtered using the QIIME software package (Quantitative Insights into Microbial Ecology, QIIME, V1.7.0, http://qiime.org/index.html) [25, 26] to obtain high-quality clean tags. Operational taxonomic units (OTUs) with a 97% similarity cut off were clustered using Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/) and chimeric sequences were detected and removed using UCHIME algorithm (UCHIME algorithm, http://www.drive5.com/usearch/manual/uchime_algo.html). For each representative sequence of OTUs, the GreenGene Database (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi) [27] was used based on the RDP classifier (Ribosomal Database Project, RDP, Version 2.2, http://sourceforge.net/projects/rdp-classifier/) [28] algorithm to annotate the taxonomic information.
OTU abundance information was normalized using a standard of sequence number corresponding to the sample with the least sequences. Subsequent analysis of alpha diversity and beta diversity was performed based on the output of normalized data.
Alpha diversity indices and richness estimators were calculated according to OTU information using QIIME software and displayed with R software (Version 2.15.3, http://www.R-project.org). To determine global differences in microbial composition, weighted and unweighted UniFrac distance for PCoA and non-metric multidimensional scaling (NMDS) were also used to visualize the differences in microbiota between the NSCLC and the healthy controls. The linear discriminant analysis (LDA) effect size (LEfSe) method (http://huttenhower.sph.harvard.edu/lefse/), which emphasizes the statistical significance [29], was used to detect microbial biomarkers and differentiate the NSCLC samples from the healthy controls, with a significance alpha of 0.05 and an LDA score > 4.0. The Analysis of Similarity (ANOSIM) test was carried out to analyse significant differences in microbial communities between NSCLC and healthy controls using the vegan package in R software. We also used canonical correspondence analysis (CCA) to further explore the possible clinical inflammatory markers that correspond to salivary microbiota community structure in R software. CCA is a multivariate method used to illustrate the relationships between biological structures of species and environmental variables, which is widely used in community ecology. The Monte Carlo test method was used to test the variance corresponding to sequencing data with the number of randomized permutations at N = 999 at the significance level of α < 0.05. Metastat analysis was also used to test differences in salivary microbiota between NSCLC and healthy controls.
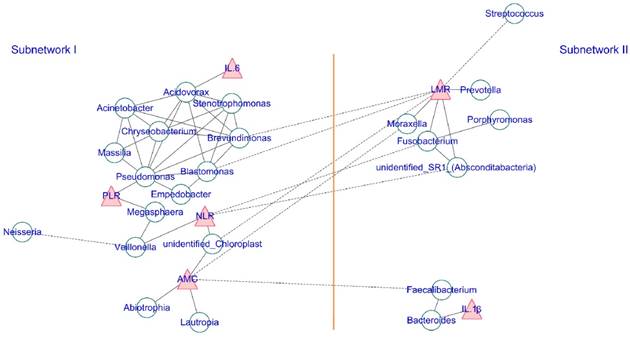
Additionally, the co-occurrence network of the top 35 abundant genera and the clinical inflammatory markers was generated to visualize the correlations between host-microbiota and clinical inflammatory markers according to the Spearman's rank correlation analysis. A strict p-value threshold (p ≤ 0.001, and ¦correlation r¦ > 0.6) was applied to filter the strong correlations among host microbiota and a less strict threshold (p ≤ 0.05, and ¦correlation r¦ > 0.3) was adopted to filter out the significant correlations between the clinical inflammatory markers and genera. The co-occurrence networks were then visualized using Cytoscape (Version 3.6.1) with a force-directed algorithm. Each green round node represents a genus, and each red triangle represents an inflammatory marker. The solid and dashed edges represent a positive and negative correlation, respectively.
Finally, the salivary microbial functional gene contents were predicted using PICRUSt v1.0.0 [30]. In brief, the cleaned amplicon sequences were re-agglomerated into OTUs using QIIME software against the Greengenes database [31] with a similarity cut off value of 97%. After OTU picking, the OTU-table was normalized and then the metagenome functional predictions were predicted via PICRUSt [30] and mapped on KEGG orthologues [32] level-2 and level-3 pathways using HUMAnN [32, 33]. LEfSe was also utilized to evaluate differentially abundant predicted microbiome functions between NSCLC and healthy controls. Then, a Spearman correlation of the KEGG level-3 functional orthologues and the clinical inflammatory markers was calculated in R and significant correlations (p< 0.05, and ¦correlation r¦ > 0.3) were exported for visualization.
Statistical Methods
The results are presented as counts (percentage) for the categorical variables, and mean ± standard deviation for the continuous variables. Chi-squared test was used to compare the differences in the proportions for the categorical variables. Unpaired- sample Student's t-test or Mann Whitney U test were used to compare the differences in the independent samples for the continuous variables with False Discovery Rate (FDR) correction. All of the tests for significance were two-sided, and p < 0.05 was considered statistically significant. Analyses were performed using the SPSS statistical package, version 24.0 (SPSS).
Results
Physiological Characteristics of Study Subjects
A total of 39 NSCLC patients and 20 healthy subjects were enrolled in this study protocol. Groups of enrolled subjects were similar with respect to demographic characteristics (p = 0.987 for age and p = 0.452 for sex), and BMI (p = 0.369). It is important to note that the neutrophil-lymphocyte ratio (NLR) and absolute monocyte count (AMC) were significantly elevated in the NSCLC group (p = 0.001 and p < 0.001 respectively). However, there was no significant difference in other inflammatory indicators between the two groups. The detailed characteristics of the cohort are summarized in Table 1.
Diversity of Salivary Microbiota in NSCLC Patients and Healthy Subjects
A total of 3,207,485 taxon reads were analysed after sequence de-noising, trimming and chimera picking. The reads were clustered into 37,394 OTUs at the 97% similarity threshold level, with 25,270 OTUs in the NSCLC group and 12,124 OTUs in controls. We first checked the sequencing depth by plotting the rarefaction curve for each sample (Figure S1). Most of the samples reached plateau, indicating the adequacy of the sequencing. Moreover, the value of Good's coverage in our study was more than 0.99 in both groups, which suggested that the reads obtained from the two groups were representative of most of the bacteria presented in the study. Next, we compared the microbial diversity and richness of NSCLC patients with the healthy subjects using the ACE, Chao1, Shannon, and Simpson indices. The NSCLC patients tended to have a higher richness and a lower diversity than healthy subjects in our study. As shown in Figure S2A-B, the Chao1' and ACE' richness indices in healthy subjects were slightly lower than those in the NSCLC group, however, this was not statistically significant. Conversely, the Simpson' diversity index was significantly decreased in the NSCLC group (Figure S2D). The summary information is shown in Table 2.
Comparison of phylotype coverage and diversity estimation of the 16S rRNA gene libraries at 97% similarity from the sequencing analysis
| Group | Observed species | Good's coverage (%) | PD-whole tree | Richness estimator | Diversity index | ||||
|---|---|---|---|---|---|---|---|---|---|
| ACE | 95% CI | Chao 1 | 95% CI | Shannon | Simpson | ||||
| NSCLC | 531 | 99.50% | 54.56 | 659.08 | 621.15-697.00 | 652.43 | 614.26-690.61 | 5.50 | 0.92 |
| Healthy Controls | 510 | 99.50% | 53.08 | 633.62 | 614.87-652.36 | 642.99 | 620.09-665.88 | 5.65 | 0.94 |
1 The operational taxonomic units (OTUs) were defined with 97% similarity level. 2 The coverage percentage (Good's) and richness estimators (ACE and Chao1), and diversity indices (Shannon and Simpson) were calculated using the R software. CI, confidence interval.
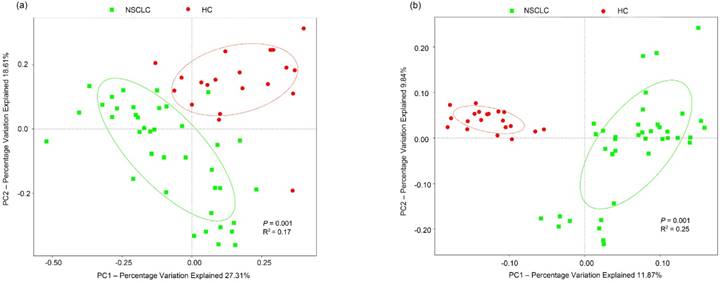
To test whether the complexity of salivary microbiota distinguished the NSCLC group from healthy controls, PCoA was applied to compare the overall structure of microbial communities between the two groups. Although the salivary microbiota showed an interindividual difference, the saliva samples clearly differed between the two study groups when considering the weighted (p = 0.001, R2 = 0.17), and unweighted (p = 0.001, R2 = 0.25) UniFrac distance in the PCoA (Figure 1). However, samples in our study did not cluster by patient's clinical stage, T-stage, node positivity, or pathology. NMDS of the bacterial community also showed clear separation (Figure S3). Additionally, we performed an ANOSIM, and the results suggested that the structure of the salivary microbiota of NSCLC patients was significantly different from that of the healthy controls (ANOSIM, p = 0.012, R = 0.127).
Principal coordinates analysis of the salivary microbiota. Overall salivary microbiota of patients with non-small cell lung cancer is statistically significantly different from that of healthy individuals as represented by the first two principal coordinates analysis of (a) weighted and (b) unweighted UniFrac distances. Each point represents a single sample, with plus sign and ellipses representing the fitted mean and 68% confidence interval of each group respectively. NSCLC represent non-small cell lung cancer group, and HC, healthy controls.
Compositional Analysis of Salivary Microbiota
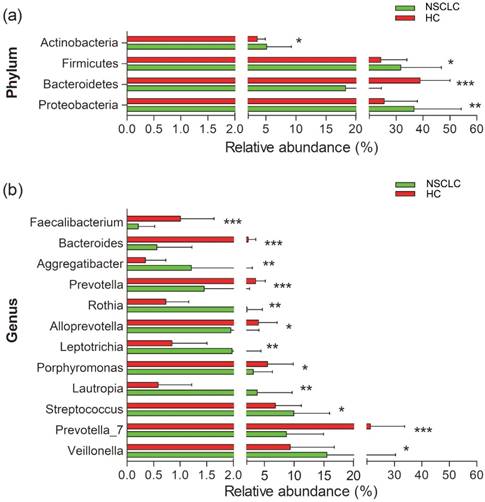
We next compared the relative abundance of microbiota composition of NSCLC patients with healthy controls. More than 200 genera were classified from the salivary microbiota. The predominant bacterial composition (>1% of the total sequences in either group) at the phyla level included Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria, comprising 97.01% and 98.17% of the salivary microbiota in the NSCLC patients and healthy subjects, respectively (Figure S4). The dominant genera between NSCLC and healthy subjects were different. Neisseria was the most abundant genus in the NSCLC group, and the second dominant genus was Veillonella. Conversely, the predominant genus in healthy controls was Prevotella_7, followed by Neisseria (Figure S4). Among all the predominant taxa at genus level, we found 13 statistically significant differences between the two groups. The relative abundance of Veillonella, Streptococcus, Lautropia, Leptotrichia, Rothia, and Aggregatibacter was significantly higher in the NSCLC group compared with the healthy control group. We also found significantly lower levels of Prevotella_7, Fusobacterium, Porphyromonas, Alloprevotella, Prevotella, Bacteroides, and Faecalibacterium in NSCLC patients than in the healthy control group (Figure 2).
Relative abundance of significantly different genera at phylum (a), and genus (b) levels between non-small cell lung cancer patients and healthy controls. *p<0.05, ** p<0.01, ***p<0.001. NSCLC represent non-small cell lung cancer group, and HC, healthy controls.
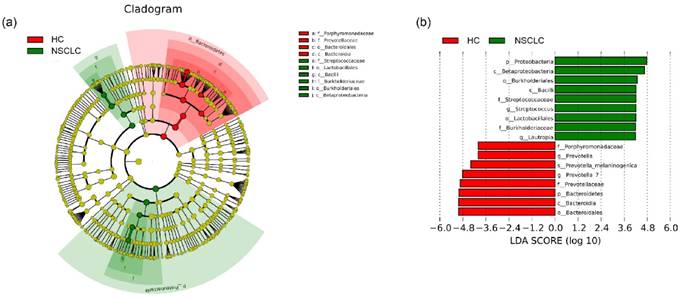
Lastly, LEfSe [29] was used to identify the greatest differences in taxa by comparing the NSCLC patients with healthy subjects (Figure 3). We found that the NSCLC group was associated with a significantly higher amount of Proteobacteria and Firmicutes. Lautropia was the prominent biomarker for the NSCLC group in the genus level, and thus, the NSCLC group might be designated by the Lautropia dominant cluster. Other biomarkers for the NSCLC group included Streptococcus, Burkholderiaceae, Burkholderiales, Lactobacillales, Betaproteobacteria, and Bacilli. For healthy controls, Bacteroidetes was the most abundant phylum, and Prevotella and Prevotella_7 were the most prominent genus level biomarkers. Thus, the healthy control group might be designated as the Prevotella and Prevotella_7 dominant cluster. The other biomarkers of healthy controls included Porphyromonadaceae, Prevotellaceae, Bacteroidia, and Bacteroidales.
Different structures of salivary microbiota between non-small cell lung cancer and healthy control groups. (a) Cladograms of bacterial lineages with significantly different representation in non-small cell lung cancer and the healthy control groups. Taxonomic cladogram obtained from LEfSe analysis of 16S sequences (The diameter of each circle is proportional to taxon abundance). (b) Histogram of the linear discriminant analysis (LDA) scores for differentially abundant bacterial taxa between non-small cell lung cancer patients and healthy controls. Only taxa meeting an LDA significant threshold > 4.0 are shown. Red (HC) indicates the healthy controls, and green (NSCLC), non-small cell lung cancer group
Systemic Inflammatory Markers Correlated with Salivary Microbiota
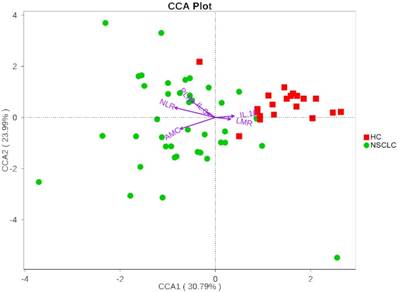
We next investigated whether the salivary microbiota interact with clinical systemic inflammation-related markers including NLR, PLR, LMR, AMC, IL-6, and IL-1β. Here, we used CCA to visualize the relationship between systematic inflammatory markers and salivary microbiota (Figure 4). The inflammatory factors were plotted with purple arrows, and each factor's weight was proportional to its arrow length. The first CCA axis explained 30.79% of the total variance and was negatively correlated with NLR, AMC, PLR, and IL-6, and positively correlated with LMR and IL-1β. The effects of IL-6, IL-1β, and LMR were weaker than those of AMC, NLR, and PLR, as indicated by the lengths of the vectors (Figure 4). The CCA figure shows that the strongest determinant for microbial communities was NLR. Veillonella and Fusobacterium, the two most dominant genera, were significantly correlated with NLR (Spearman r = 0.35, p = 0.007; Spearman r = -0.41, p = 0.001, respectively). CCA axis 2, which explained 23.99% of the total variance, was positively correlated with NLR, PLR, IL-6, and IL-1β and negatively correlated with AMC and LMR. These results suggested that the salivary microbiota correlated with its host systemic inflammatory statues.
Canonical Correspondence Analysis (CCA) illustrating relations between bacteria taxa and systemic inflammatory markers in our study groups. Arrows indicate the direction and magnitude of the systemic inflammatory markers associated with bacterial community structures. The explained variance of the principal axes [Axis 1 (horizontally) and Axis 2 (vertically)] are 30.79% and 23.99%, respectively. NSCLC represent non-small cell lung cancer group, and HC, healthy controls.
Additionally, we also calculated the correlation among abundance of the top 35 genera and screened strong correlations with p < 0.001 and ¦r¦ > 0.6. The correlation between the systemic inflammatory markers and genus was filtered by a loose threshold of p < 0.05 because of the graded input of these factors. The overall result is displayed in Figure 5, and the detailed list is presented in Table S1. Clear visualization showed that the salivary microbiota formed two major mutualistic sub-networks (Sub-network I and Sub-network II in Figure 5). The correlations were all positive within each sub-network, and the correlations among different sub-networks were all negative. The Stenotrophomonas, Brevundimonas, Chryseobacterium, and Pseudomonas were the central genera within sub-network I, and the Fusobacterium was the central genus within sub-network II. Significant co-variance existed between the salivary microbiota and the systemic inflammatory markers. IL-1β was positively correlated with Bacteroides and IL-6 was positively correlated with Acidovorax. The NLR, LMR, and AMC were strongly correlated with multiple genera (Figure 5, Table S2).
The co-occurrence network of the salivary microbiota and the systemic inflammatory markers. Each green round node represents an OTU, and each red triangle represents an inflammatory marker. The solid and dashed edge represents a positive and negative correlation, respectively. The whole network could be divided into two subnetworks, where the positive correlation exists within each subnetwork, and the trans-subnetwork correlation was negative.
Altered metabolic pathways and correlations among KEGG pathways and clinical indices
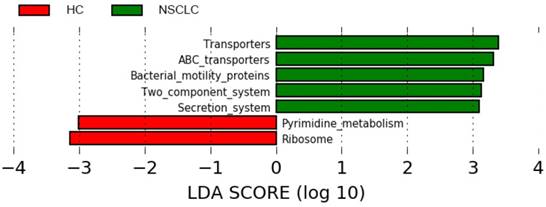
The 16S rDNA sequencing data was further analyzed using the online tool PICRUSt. The genes in each sample of metagenomics data were first classified according to the KEGG gene class, classification level-2 and level-3. After classification, each class of genes with mean relative abundance values > 0.1% was selected for downstream analyses between the two groups, using Wilcoxon rank-sum test with FDR correction, p < 0.05. Overall, 146 level-3 KEGG categories were selected and 92 pathways were significantly regulated (Table S3). Interestingly, pathways related to metabolism, especially xenobiotic biodegradation and metabolism, amino acid metabolism were enriched in the NSCLC group compared with the healthy controls. Further, metabolites of cofactors and vitamins, such as folate biosynthesis and one carbon pool by folate were significantly decreased in the NSCLC group. Inferences of the KEGG pathways for each group were additionally compared using LEfSe with the significance threshold set at α of 0.05 and a LDA score of 3.0. Transporters, ABC transporters, bacterial motility proteins, two component system, and secretion system were significantly overrepresented in the NSCLC group, while pyrimidine metabolites and ribosome were significantly overrepresented in the healthy controls (Figure 6).
Different structures of predicted KEGG pathways between non-small cell lung cancer and healthy control groups. Histogram of the linear discriminant analysis (LDA) scores for differentially abundant bacterial taxa between non-small cell lung cancer patients and healthy controls. Only taxa meeting an LDA significant threshold >3.0 are shown. NSCLC represent non-small cell lung cancer group, and HC, healthy controls.
Among these, correlations were performed to link the clinical indices and KEGG pathways in saliva. Strong correlations with p < 0.001 and ¦r¦ > 0.3 were detected. As Table 3 shows, transporters, the enriched biomarker in the NSCLC group, which participated in the environmental information processing, was negatively correlated with PLR. While, secretion system was positively correlated with AMC, and negatively correlated with LMR. Additionally, the ribosome and pyrimidine metabolism, which took part in the translation and nucleotide metabolism, were positively correlated with LMR, where they were negatively correlated with AMC.
Summary of the significant correlations between systemic inflammatory markers and the predicted KEGG pathways. Spearman correlation coefficients (r) with p <0.05
| From | To | r | p-value | From | To | r | p-value |
|---|---|---|---|---|---|---|---|
| AMC | Secretion system | 0.33 | 0.011 | LMR | Chaperones and folding catalysts | 0.389 | 0.002 |
| AMC | DNA replication proteins | -0.345 | 0.007 | LMR | DNA replication proteins | 0.388 | 0.002 |
| AMC | Homologous recombination | -0.302 | 0.02 | LMR | Homologous recombination | 0.362 | 0.005 |
| AMC | Ribosome | -0.304 | 0.019 | LMR | Transcription factors | -0.306 | 0.018 |
| AMC | Arginine and proline metabolism | 0.336 | 0.009 | LMR | Ribosome | 0.315 | 0.015 |
| AMC | Peptidases | -0.357 | 0.006 | LMR | Methane metabolism | 0.314 | 0.016 |
| AMC | Pyrimidine metabolism | -0.341 | 0.008 | LMR | Peptidases | 0.383 | 0.003 |
| IL-1β | DNA replication proteins | 0.317 | 0.014 | LMR | Pyrimidine metabolism | 0.304 | 0.019 |
| IL-1β | Homologous recombination | 0.302 | 0.002 | NLR | Chaperones and folding catalysts | -0.423 | 0.001 |
| IL-6 | Chaperones and folding catalysts | -0.319 | 0.014 | PLR | Transporters | 0.317 | 0.015 |
| LMR | Secretion system | -0.342 | 0.008 | PLR | Oxidative phosphorylation | -0.301 | 0.02 |
| LMR | Two component system | -0.328 | 0.011 |
Discussion
Altered salivary microbiota compositions have been identified in patients with oral cancer and non-oral disease [8, 11, 14, 34, 35]. To date, only one study conducted by Yan et al. has investigated the correlation between oral bacterial pathogens and NSCLC. The main finding in their work was that the significantly elevated levels of Capnocytophaga and Veillonella in the saliva samples from the NSCLC group, may serve as potential biomarkers for NSCLC detection and classification [19]. In this study, the simple comparison of NSCLC patients and matched healthy subjects revealed a significant difference in the microbial community. Additionally, the interactions among oral bacterial pathogens, clinical inflammatory markers, and the predicted metagenome suggested that the shift in the salivary microbiota plays a potential pathogenic role in the process of NSCLC. These results pointed to a potential target for further interventions to improve the prognosis of NSCLC.
This study also revealed no major shift in microbial richness, but it did show a significantly lower diversity in the NSCLC group, which is consistent with findings by Yu et al. comparing microbiota from malignant versus benign human lung tissues [36]. In contrast, another previous study revealed conflicting results when comparing the buccal samples from NSCLC patients versus healthy subjects [37]. Notably, systemic inflammation and the immune response might be major factors that influence salivary microbial alpha diversity [38]. Diet, lifestyle, and environmental factors also should not be excluded. Given the strict restriction on living area and race in the enrolment criteria of this study, our results merely represent the patients commonly seen in clinical practice in the Jinan district. Therefore, well-conducted large studies are needed for further investigation.
Observation of the overall composition of salivary microbiota showed that the salivary microbiome from the cancer and non-cancer groups were significantly different, based on the measurement of UniFrac distance in the PCoA plots, while the samples from the cancer group did not cluster by patients' clinical stage, T-stage, node positivity, or pathology. This indicates that the alterations seen on the beta diversity analysis are due to the cancer versus non-cancer groups, not to demographic differences. Additionally, several cases in the healthy group resembled the cancer samples more than the samples from healthy subjects, which may imply a predisposition to lung carcinogenesis. These data provided initial insight into the alterations in salivary microbiota associated with NSCLC. Due to the large variation in the salivary microbiota among the population, further longitudinal studies, particularly including the representative patients, are required to further investigate these issues.
Previous studies in saliva microbiome using next-generation sequencing (NGS) have revealed the most abundant phyla were Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria, and Actinobacteria [19]. This is similar to our present findings at the phyla level. However, in our study, significant expansion of Firmicutes and Proteobacteria with decreased Bacteroidetes was found in the NSCLC cases. Note that in patients with oral squamous cell carcinoma, Firmicutes was upregulated compared to that in the matched non-malignant samples [34]. Genus Veillonella and Streptococcus, of the Veillonellaceae and Streptococcaceae families respectively, contributed most to the increase in Firmicutes. Elevated Veillonella in saliva samples was reported as a potential biomarker for disease detection in NSCLC and neck squamous cell carcinoma [19, 39]. Members of the significantly higher genus Streptococcus, act as opportunistic pathogens, and in the tumour samples this was also reported to discriminate tumour samples from the controls in oral cancer [39]. Not limited in habitat, Streptococci in the oral cavity have a close relationship with the entire body [40]. Additionally, as Veillonella and Streptococcus can potentially interact metabolically and frequently co-occur in immunomodulation by pair-wise combinations of strains in the human intestine [41], the combinations of Veillonella and Streptococcus in the oral cavity might elicit an immune response profile that is relevant to the progression of NSCLC. Elevated prevalence of opportunistic pathogen was associated with underlying disease [42]. Decreased abundances of Lautropia and Rothia had been reported to be associated with an increased risk of esophageal squamous carcinoma and oral cancer, respectively [10, 35]. However, genus Lautropia and Rothia, which were contrary to our expectation, showed an increased prevalence in the NSCLC group in this study. Based on the above, our observation showed significance in guiding clinical treatment of NSCLC patients. Maladjusted oral microbiota in NSCLC indicated that opportunistic pathogens from saliva samples in NSCLC patients might be involved in cancer progression, which again requires subsequent in-depth studies.
To further verify the potential role of salivary microbiota in the progression of NSCLC, we investigated the correlation between certain specific bacteria and prognostic factors of NSCLC. Poor prognosis of NSCLC is associated with biomarkers of inflammation, such as elevated NLR, PLR, IL-6, IL-1β concentrations and decreased levels of LMR [43-48]. In this study, we found that Fusobacterium, which was documented to be involved in the pathogenesis of colorectal adenoma [49], was strongly associated with NLR and LMR. Veillonella, a potential biomarker for NSCLC detection and classification [19], was also significantly associated with NLR. The role of salivary microbiota revealed in our network analysis may partially explain the correlation among salivary microbiota, systemic inflammation, and the prognosis of NSCLC patients.
Additionally, most metabolites in the human body are believed not to be of human origin, and thus a change in microbial metabolic activity might directly affect human health [50]. It is not surprising to find an increased metabolic activity involved in xenobiotic biodegradation in the NSCLC group, which is usually caused by the release of industrial compounds, as several reports have revealed a direct association between exposure to poor air quality and increasing rates of morbidity of respiratory cancer [51]. Pathways involved in amino acid metabolism were also significantly overrepresented in the NSCLC group. Previous evidences have documented the important role of commensal microbiota in the extraction and synthesis of the available amino acids [52] and the association between intestinal dysbiosis and protein energy malnutrition [53]. The alterations in amino acid metabolism in our results might be, in part, affected by metabolic dysfunction of bacteria. Additionally, pathways related to folate metabolism were significantly underrepresented in the NSCLC group. It has been reported that increased risk of breast cancer was associated with lower folate levels among women [54]. In this study, the underrepresented folate might, in part, play a role in the occurrence and development of NSCLC. Despite our small sample size, LEfSe analysis did reveal differences between the two groups, as shown in Figure 6. In patients with NSCLC, the pathways involved in transporters and secretion system were overrepresented. This may reflect the activation of salivary microbiota homeostasis mechanisms responding to the effect of NSCLC, which may lead to salivary microbial dysbiosis. Pathways related to pyrimidine metabolism and ribosome were also reported to be correlated with the progression of colorectal cancer [55] and breast cancer [56], respectively. Our result provides a new avenue for potential dietary intervention in NSCLC.
Not only that, correlations between systemic inflammatory biomarkers and the predicted metabolic pathways were also revealed. As the documented inflammatory factors are associated with the prognosis of NSCLC [43-48], these correlations might suggest specific and pronounced interactions between NSCLC and the accumulation of oral metabolites. Additional work identifying the specific mechanisms of the interaction between the progression of NSCLC and the salivary microbiome metabolites is required.
However, it is worth noting that the study described above is a preliminary pilot project with a limited sample size, and more extensive studies are needed to confirm these observations. We also acknowledge that our investigation only detected the bacterial community structure at a single time-point as opposed to dynamic monitoring, which might contribute to a better understanding of the altered salivary microbiota associated with NSCLC. Despite the strict enrolment criteria, the overall oral health parameters of the participants were unidentified. Additionally, specific confounding factors, such as diet lifestyle, air pollution, and family history were not covered in this study. Nevertheless, further studies, preferably longitudinal, in cooperation with mechanistic analyses and animal model systems, are required to thoroughly investigate the causal relationships between NSCLC and salivary microbiota.
To our knowledge, this is the first study to assess the compositional changes and the predicted KEGG pathways in the salivary microbiota between NSCLC patients and healthy subjects, and for the first time, we revealed the cross-link among salivary microbiota dysbiosis, systemic inflammatory markers, and the predicted KEGG pathways that are reportedly associated with NSCLC. Taken together, our study identified distinct salivary microbiome composition in patients with NSCLC. Based on our preliminary findings, further investigation of the potential role of salivary microbiota in the progression of NSCLC is in demand. Our work not only extends this observation to patients with NSCLC, but also might facilitate clinical therapeutic strategies for monitoring and altering salivary dysbiosis in NSCLC patients.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We acknowledge the patients and volunteers for their collaboration and collection of the fecal and serum samples. This study was supported by the Natural Science Foundation of Shandong Province (Grant No. ZR2015HM054), the Key Research and Development Program of Shandong Province (Grant No. 2016ZDJS07A15, 2017GSF22107, 2017GSF218112, and 2017G006028), and the Science and Technology Department Public Welfare Project of Shandong Province (Grant No. 2014kjhm0107).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. Journal of clinical oncology. 2006;24:2137-50
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
3. Moyer VA, Force USPST. Screening for lung cancer: U.S. Preventive Services Task Force recommendation statement. Annals of internal medicine. 2014;160:330-8
4. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010;60:277-300
5. Ilie M, Mazure NM, Hofman V, Ammadi RE, Ortholan C, Bonnetaud C. et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. British journal of cancer. 2010;102:1627-35
6. Wei F, Lin CC, Joon A, Feng Z, Troche G, Lira ME. et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. American journal of respiratory and critical care medicine. 2014;190:1117-26
7. Xiao H, Zhang L, Zhou H, Lee JM, Garon EB, Wong DT. Proteomic analysis of human saliva from lung cancer patients using two-dimensional difference gel electrophoresis and mass spectrometry. Molecular & cellular proteomics: MCP. 2012;11:M111 012112
8. Kumar PS. Oral microbiota and systemic disease. Anaerobe. 2013;24:90-3
9. Le Bars P, Matamoros S, Montassier E, Le Vacon F, Potel G, Soueidan A. et al. The oral cavity microbiota: between health, oral disease, and cancers of the aerodigestive tract. Canadian journal of microbiology. 2017;63:475-92
10. Schmidt BL, Kuczynski J, Bhattacharya A, Huey B, Corby PM, Queiroz EL. et al. Changes in abundance of oral microbiota associated with oral cancer. PloS one. 2014;9:e98741
11. Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM. et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67:120-7
12. Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. Journal of clinical microbiology. 2005;43:5721-32
13. Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy "core microbiome" of oral microbial communities. BMC microbiology. 2009;9:259
14. Gholizadeh P, Eslami H, Yousefi M, Asgharzadeh M, Aghazadeh M, Kafil HS. Role of oral microbiome on oral cancers, a review. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;84:552-8
15. Lee SH, Sung JY, Yong D, Chun J, Kim SY, Song JH. et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung cancer. 2016;102:89-95
16. Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC. et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome biology. 2016:17
17. Cameron SJS, Lewis KE, Huws SA, Hegarty MJ, Lewis PD, Pachebat JA. et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PloS one. 2017;12:e0177062
18. Mao Q, Jiang F, Yin R, Wang J, Xia W, Dong G. et al. Interplay between the lung microbiome and lung cancer. Cancer letters. 2017;415:40-8
19. Yan X, Yang M, Liu J, Gao R, Hu J, Li J. et al. Discovery and validation of potential bacterial biomarkers for lung cancer. American journal of cancer research. 2015;5:3111-22
20. Keller MK, Brandsborg E, Holmstrom K, Twetman S. Effect of tablets containing probiotic candidate strains on gingival inflammation and composition of the salivary microbiome: a randomised controlled trial. Beneficial microbes. 2018;9:487-94
21. Sheth CC, Makda K, Dilmahomed Z, Gonzalez R, Luzi A, Jovani-Sancho Mdel M. et al. Alcohol and tobacco consumption affect the oral carriage of Candida albicans and mutans streptococci. Letters in applied microbiology. 2016;63:254-9
22. Takeshita T, Kageyama S, Furuta M, Tsuboi H, Takeuchi K, Shibata Y. et al. Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Scientific reports. 2016;6:22164
23. Stewart CJ, Auchtung TA, Ajami NJ, Velasquez K, Smith DP, De La Garza R 2nd. et al. Effects of tobacco smoke and electronic cigarette vapor exposure on the oral and gut microbiota in humans: a pilot study. PeerJ. 2018;6:e4693
24. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957-63
25. Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature methods. 2013;10:57-9
26. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335-6
27. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72:5069-72
28. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261-7
29. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS. et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12:R60
30. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31:814-21
31. McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME journal. 2012;6:610-8
32. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M. et al. KEGG for linking genomes to life and the environment. Nucleic acids research. 2008;36:D480-4
33. Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS computational biology. 2012;8:e1002358
34. Pushalkar S, Mane SP, Ji X, Li Y, Evans C, Crasta OR. et al. Microbial diversity in saliva of oral squamous cell carcinoma. FEMS immunology and medical microbiology. 2011;61:269-77
35. Chen X, Winckler B, Lu M, Cheng H, Yuan Z, Yang Y. et al. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. PloS one. 2015;10:e0143603
36. Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC. et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome biology. 2016;17:163
37. Hosgood HD 3rd, Sapkota AR, Rothman N, Rohan T, Hu W, Xu J. et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environmental and molecular mutagenesis. 2014;55:643-51
38. Nowak P, Troseid M, Avershina E, Barqasho B, Neogi U, Holm K. et al. Gut microbiota diversity predicts immune status in HIV-1 infection. Aids. 2015;29:2409-18
39. Guerrero-Preston R, Godoy-Vitorino F, Jedlicka A, Rodriguez-Hilario A, Gonzalez H, Bondy J. et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget. 2016;7:51320-34
40. Faran Ali SM, Tanwir F. Oral microbial habitat a dynamic entity. Journal of oral biology and craniofacial research. 2012;2:181-7
41. van den Bogert B, Meijerink M, Zoetendal EG, Wells JM, Kleerebezem M. Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PloS one. 2014;9:e114277
42. Schuurhuis JM, Stokman MA, Witjes MJ, Langendijk JA, van Winkelhoff AJ, Vissink A. et al. Head and neck intensity modulated radiation therapy leads to an increase of opportunistic oral pathogens. Oral oncology. 2016;58:32-40
43. Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert opinion on biological therapy. 2011;11:1663-8
44. Wang YQ, Zhi QJ, Wang XY, Yue DS, Li K, Jiang RC. Prognostic value of combined platelet, fibrinogen, neutrophil to lymphocyte ratio and platelet to lymphocyte ratio in patients with lung adenosquamous cancer. Oncology letters. 2017;14:4331-8
45. McLoed AG, Sherrill TP, Cheng DS, Han W, Saxon JA, Gleaves LA. et al. Neutrophil-Derived IL-1beta Impairs the Efficacy of NF-kappaB Inhibitors against Lung Cancer. Cell reports. 2016;16:120-32
46. Song YJ, Wang LX, Hong YQ, Lu ZH, Tong Q, Fang XZ. et al. Lymphocyte to monocyte ratio is associated with response to first-line platinum-based chemotherapy and prognosis of early-stage non-small cell lung cancer patients. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:5285-93
47. Chen S, Guo J, Feng C, Ke Z, Chen L, Pan Y. The preoperative platelet-lymphocyte ratio versus neutrophil-lymphocyte ratio: which is better as a prognostic factor in oral squamous cell carcinoma? Therapeutic advances in medical oncology. 2016;8:160-7
48. Wang L, Liang D, Xu X, Jin J, Li S, Tian G. et al. The prognostic value of neutrophil to lymphocyte and platelet to lymphocyte ratios for patients with lung cancer. Oncology letters. 2017;14:6449-56
49. Allen-Vercoe E, Jobin C. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunology letters. 2014;162:54-61
50. Dorrestein PC, Mazmanian SK, Knight R. Finding the missing links among metabolites, microbes, and the host. Immunity. 2014;40:824-32
51. Ghorani-Azam A, Riahi-Zanjani B, Balali-Mood M. Effects of air pollution on human health and practical measures for prevention in Iran. Journal of research in medical sciences: the official journal of Isfahan University of Medical Sciences. 2016;21:65
52. O'Keefe SJ. Nutrition and colonic health: the critical role of the microbiota. Current opinion in gastroenterology. 2008;24:51-8
53. Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J. et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339:548-54
54. Sellers TA, Kushi LH, Cerhan JR, Vierkant RA, Gapstur SM, Vachon CM. et al. Dietary folate intake, alcohol, and risk of breast cancer in a prospective study of postmenopausal women. Epidemiology (Cambridge, Mass). 2001;12:420-8
55. Wang T, Xu H, Liu X, Chen S, Zhou Y, Zhang X. Identification of Key Genes in Colorectal Cancer Regulated by miR-34a. Medical science monitor: international medical journal of experimental and clinical research. 2017;23:5735-43
56. Meng S, Chen B, Yang J, Wang J, Zhu D, Meng Q. et al. Study of Microbiomes in Aseptically Collected Samples of Human Breast Tissue Using Needle Biopsy and the Potential Role of in situ Tissue Microbiomes for Promoting Malignancy. Frontiers in oncology. 2018;8:318
Author contact
![]() Corresponding author: Prof. Xiaogang Zhao, Department of Thoracic Surgery, The Second Hospital of Shandong University, Shandong University, 247 Beiyuan Avenue, Jinan, Shandong 250033, P.R. China. Tel & FAX: 86-531-85875009; E-mail: zhaoxiaogangedu.cn
Corresponding author: Prof. Xiaogang Zhao, Department of Thoracic Surgery, The Second Hospital of Shandong University, Shandong University, 247 Beiyuan Avenue, Jinan, Shandong 250033, P.R. China. Tel & FAX: 86-531-85875009; E-mail: zhaoxiaogangedu.cn