Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):927-936. doi:10.7150/jca.27160 This issue Cite
Review
5'-Methylthioadenosine and Cancer: old molecules, new understanding
Yaofeng Li1, Yubo Wang2, Ping Wu1 ![]()
1. Department of Pathophysiology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
2. College of Pharmacy, Hubei University of Chinese Medicine, Wuhan 430065, China.
Received 2018-5-8; Accepted 2018-12-17; Published 2019-1-29
Abstract
While the metabolic changes in cancer tissues were first observed by Warburg Otto almost a century ago, altered metabolism has recently returned as a focus of cancer research. 5'-Methylthioadenosine (MTA) is a naturally occurring sulfur-containing nucleoside found in numerous species. While MTA was first isolated several decades ago, a lack of sensitive and specific analytical methodologies designed for its direct quantification has hampered the study of its physiological and pathophysiological features. Many studies indicate that MTA suppresses tumors by inhibiting tumor cell proliferation, invasion, and the induction of apoptosis while controlling the inflammatory micro-environments of tumor tissue. In this review, we assessed the effects of MTA and of related materials on the growth and functions of normal and malignant cells.
Keywords: Cancer, MTA, Metabolism, MTAP, Polyamine
1. Introduction
Metabolic changes in cancer tissues were first observed by Warburg Otto almost a century ago [1]. Although the genetic underpinnings of cancer have dominated cancer research for a long time, altered metabolism has recently returned to the spotlight [2]. Beyond what was firstly observed, as cancer cells primarily use anaerobic glycolysis to produce energy rather than oxidative phosphorylation even in the presence of oxygen, now metabolomics research on tumors covers a relatively broader range of processes involving not only the production of energy but also the generation of cellular building materials from nutrients [2, 3]. It is no doubt that oncogenic transformation reprograms cellular metabolism to sustain elevated rates of growth and division.
5'-Methylthioadenosine (MTA) is a naturally occurring sulfur-containing nucleoside found in numerous species, including prokaryotes, yeast, plants and higher eukaryotes [4]. It was first isolated from yeast in 1912 before Warburg's theory was first proposed [5], with its correct molecular structure was confirmed in 1924 [6]. Over the following 30 years, several studies on the nutritional and pharmacologic effects of MTA have illustrated its physiological significance [7]. With a stronger emphasis placed on tumor metabolism, numerous investigations appear to show that MTA and its associated materials have striking regulatory effects on tumorigenesis. It is the purpose of this article to assess the effects of these metabolites on the growth and functions of normal and malignant cells. We performed a literature search through the PubMed, EMBASE and CNKI (Chinese National Knowledge Infrastructure) databases using the following keywords: MTA, S-adenosylmethionine (AdoMet), methionine (MET), MTA phosphorylase (MTAP), or polyamine. The most recent article found was published on Jan, 2018. Reference lists of the relevant publications were also carefully reviewed to obtain additional information.
2. Biosynthesis and metabolism of MTA
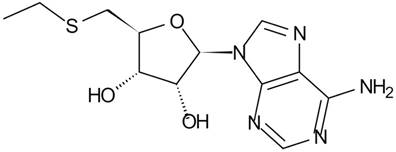
MTA is a hydrophobic sulfur-containing adenine nucleoside in which the hydroxyl group at the 5' position of the ribose is substituted with a methylthio moiety (Figure 1) [4]. Its molecular weight is 297.33. At least five independent pathways of MTA generation have been discovered in mammals, invertebrate animals, bacteria and plants [8]. In many mammalian tissues, MTA is mainly defined as the intermediate metabolite or byproduct of MET cycle and polyamine synthases (Figure 2) [7]. As the provider of the sulfur atom in MTA, MET was first isolated in 1923 [9], and the methyl-group of MTA derives from AdoMet (also abbreviated as SAMe and SAM), which is synthesized from MET and ATP through the catalysis of MET Adenosyltransferases (MAT, EC 2.5.1.6). During the synthesis of polyamines, the aminopropyl group of decarboxylated AdoMet is transferred to putrescine to form spermidine and then to spermidine to form spermine as MTA is generated. Although inverse reactions are not insignificant, MTA has a powerful inhibitory effect on several phases involved in its generation [10].
Structure of 5'-Methylthioadenosine (MTA)
Biosynthesis and metabolism of 5'-Methylthioadenosine (MTA). The left circle shows the MET cycle. The right circle illustrates polyamine synthesis. Green lines denote the MET salvage pathway from MTA to MET. Enzymes are shown in italics.
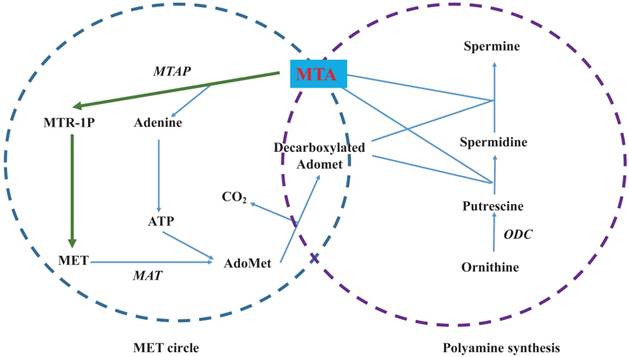
According to in vitro and in vivo reports, the concentration of cellular MTA only falls within the nM range or lower. As in human hepatic adenocarcinoma cell line Sk-Hep1, there are nearly 0.3 nM MTA in every one million cells [11] while in both human hepatocytes and several HCC cell lines, concentrations of MTA range from 2 to 10 pmol/mg protein [12]. Similar levels of MTA have also been found in rat tissue (liver 0.8-3.4; lung 1.1-1.7; kidney 1.0-2.3; testis 0.9-1.7 and heart 1.1-2.5 nmol per gram) [13]. In many mammalian tissues, MTAP (EC 2.4.2.28) is the main enzyme that divides MTA into 5-methylthioribose-1-phosphate (MTR-1P) and adenine (Figure 2), maintaining relatively low levels of cellular MTA [14]. It is well known that tumor cells present lower levels of MTAP or losses of MTAP activity. This phenomenon may explain why tumor tissues often accumulate more MTA than normal tissue. In health people, MTA levels are only valued at 10 to 20 nM in the skin, but in melanoma patients, levels are measured approximately 140 nM [15]. In another two studies comparing normal and HCC liver tissue, MTA was found to increase from roughly 1 pmol/ mg of tissue to 3 or 4 pmol/mg [12, 16].
After MTR-1P is generated, a series of reactions sequentially salvages its meythl-thio group to form MET and to finally complete the MET cycle, which is also called the MET salvage pathway [17]. As the other cleavage product of MTA, adenosine is then converted into AMP. This of course creates a pathway for the metabolic salvaging of the purine portion of ATP used for the synthesis of AdoMet, which is cyclically utilized in MTA synthesis. Other pathways of MTA catabolism are found in bacteria, including the degradation to adenine and 5-mythylthio-α-D-ribose and deamination to methylthioinosine [18]. These metabolic pathways apparently contribute to further DNA and protein synthesis and to cell energy generation.
It remains unclear that how MTA can be excreted into the extracellular fluid, however, the in vitro evidence shows that some tumor cells can secrete large levels of MTA and particularly the cells devoid of MTAP. In the culture medium of leukemic cells, MTA can be detected after 1h of culturing and it presents a time-dependent pattern of elevation after 1 to 6 hours [19]. In melanoma cell lines, levels range from 15 to 30 nM [15], and in HCC cell lines, levels range from 10 to 42 pM [12]. More interestingly, cells treated with conditioned media collected from MTAP-siRNA transiently transfected HCC cells presented similar changes with cells treated with MTA [12]. At the same time, the detection of MTA in the urine from both health people and cancer patients provided in vivo and clinical evidence to its cellular excretion [20]. This phenomenon may be viewed as a compensatory response to decreased levels of MTAP activity for the prevention of cell MTA accumulation.
3. Measurement of MTA
Since the identification of MTA, a lack of sensitive and specific analytical methodologies for its direct quantification has hampered the study of its physiological and pathophysiological features. In 1953, Smith et al. measured concentrations of MTA via nitroprusside testing and ultraviolet spectrophotometry [21]. The authors show that concentrations less than 0.2 umol per gram of tissue cannot be measured accurately. In 1980, Seidenfeld et al. successfully elevated measurement sensitivity levels to nmol per gram via reversed-phase high-performance liquid chromatography (HPLC)-UV detection at 254 nm after sample concentration. Their study shows that MTA is ubiquitously distributed in rat tissues at concentrations of 0.8 to 7 nmol per gram of tissue [13]. Several other reports have been conducted on the HPLC separation of MTA and on its detection by means of UV absorbance [22, 23]. It is well accepted that LC/UV is not sensitive or selective enough for the direct analysis of MTA. However, according to Kiyoko and his colleagues, with acetylated samples obtained from the sequential passage through a boronate column and reversed-phase HPLC, the UV absorbance peak can accurately reflect the elutin of MTA [20]. From this method, both normal subjects and patients with different tumors presented an average of total urinary MTA level of approximately 175 ug per day.
Aside from the above modified HPLC assays, technical developments have made it possible to apply mass spectrometry (MS) for the profiling of metabolites. In 2005, a research group in Germany developed a LC-coupled ion trap MS method that can separate 15 standard nucleosides in urine, including MTA. The assay not only ensures high levels of sensitivity (in the lower of pmol range) and reproducibility but also generates valuable information on the structure of the targeted nucleoside [24]. The same group further modified this method by HPLC-separation, matrix-assisted laser desorption/ionization time-of-flight MS and post source decay to successfully yield a detection limit to 100 fmol of MTA for urine [25]. Later, the LC electrospray ionization tandem MS method was developed by Bosserhoff's group in 2008 [15, 26]. Using stable isotope MTA as an internal standard, the absolute quantitative determination of MTA in cell culture media and cell extracts was achieved. The authors also successfully quantified more than ten other intermediates of MET and polyamine generation from cultured human HCC cell line and clinical HCC tissue samples [16].
4. MTA and tumor
As can be inferred from the above, MTA is located at the crossroads of cellular metabolism. It not only serves as an intermediary of MET circling and polyamines synthesis but also acts as a starting point for both MET and the purine salvage pathway. Its physiologic and pathophysiologic functions include the control of gene expression, cell proliferation, lymphocyte activation, apoptosis, even tumor development and invasiveness. As shown in a chemical hepatocarcinogenesis model, endogenous MTA is greatly reduced and that administration of MTA has an inhibitory effect on tumor growth [27]. Similar inhibitory effects of MTA have been observed in a xenograft melanoma mouse model [28]. By contrast, from benefits of the abovementioned LC electrospray ionization tandem MS detection method, Bosserhoff and his colleagues directly qualified higher levels of MTA in cultured melanoma cells and in clinical malignant melanoma and HCC tissues. Based on these results and the correlation between MTA and tumor metastasis, the authors hypothesized that MTA itself has a tumor-promoting effect [15, 16].
4.1. MTA and cell proliferation
As a key organ governing the body's metabolism, the liver attracted much attention in regard to how it is affected by MTA. In rats with partial hepatectomy, endogenous MTA levels in the remaining liver are reduced coinciding with the replicative responses of hepatocytes [29]. At the same time, the exogenous administration of MTA inhibits hepatocyte DNA synthesis [27]. Similar effects of MTA have been observed in in vitro cultured rat hepatocytes treated with HGF [30]. These observations suggest that MTA participates in the reverse regulation of the liver proliferative response.
In addition to impacting on the proliferation of normal liver cells, MTA is also known to interfere with the proliferation of a variety of tumor cells. In a chemically induced hepatocarcinogenesis rat model, both endogenous and exogenous MTA levels were found to be negatively correlated with preneoplastic liver lesions and DNA synthesis [27, 29]. Similar inhibitory effects of MTA have also been observed in cultured HCT116 human colon cancer cells, colorectal cancer cells, HeLa cervical cancer cells, Daudi and Raji lymphoma cells, erythroleukemia cells, promyelocytic cells and mitogen stimulated human peripheral blood lymphocytes [8, 31-35].
For most of the above in vitro studies, MTA working conditions range from 10-4 to 10-3 M. Obviously, this is much higher than the physiologic levels of MTA. In 2011, a research group from Germany found out that, at the concentration of 5 umol/L, a dosage similar to those determined in HCC tissue, MTA induced the HCC cell proliferation [12]. In another experiment, SK-Hep1 HCC cells were engineered to express MTAP and reduce MTA level. After that, it was observed that the proliferation rate became lower [11]. This experiment provided us with an indirect proof that MTA at nM scale, which mirrored endogenous tumor levels, could promote proliferation.
In most cases, inhibitory effects of MTA are associated with a marked inhibition of the activity of ornithine decarboxylase (ODC, EC 4.1.1.17), a key enzyme of polyamine synthesis. The correlation between MTA and ODC is discussed below. A second mechanism may be related to MAT, the main cellular enzyme catalyzing the formation of MET to AdoMet. This enzyme is also required for survival as AdoMet is the principal biological methyl donor. In mammals, homologous MAT catalytic subunits are encoded by two genes, MAT1A and MAT2A. The latter is associated with increased growth and malignant degeneration [36]. A clear switch from MAT1A to MAT2A is observed in human liver cancer and colon cancer tissues [36-38]. MTA not only lowers MAT2A baseline expression but also prevents a variety mitogens from inducing the up-expression of MAT2A in cultured colon cancer cells [36]. As a result, it effectively prevents mitogens from stimulating the proliferation of colon [36] and liver cancer cells [39]. Another study of Hct116 colon cancer cells provides a third interpretation of the inhibitory effects of MTA on tumor cells. The authors found that MTA treatment can downregulate RNA Pol III-dependent transcription [40], which is crucial to tumorigenesis. Although DNA methylation modification is increasingly recognized as a crucial factor for carcinogenesis, MTA has not been found to affect DNA methylation.
4.2. MTA and apoptosis
It is noteworthy that most of these studies indicate that MTA had the opposite effect on the apoptosis of normal versus cancerous cells. In vitro, it enhances the apoptosis of human RKO and SW620 colon cancer cells [35, 41] and of HuH7 liver cancer cells [42]. However, it does not have such effects on normal colon epithelial cells or even has anti-apoptotic effects on normal hepatocytes [41, 42].
Increased ratios of pro-apoptotic to anti-apoptotic genes in tumor cells may explain this effect of MTA. For example, such ratios may affect the cellular phosphorylation states and the alternative splicing of genes, resulting in the selective induction of pro-apoptotic protein Bcl-xS but not anti-apoptotic protein Bcl-xL in HepG2 cells [43]. In colon cancer cells, this can selectively decrease the mRNA levels of cFLIPs, an anti-apoptotic gene, by 50-60% [41], or downregulate anti-apoptotic regulators (bcl and bax-xl) with an upregulation of pro-apoptotic regulators (bax and bak) [31]. At the same time, several other essential steps of apoptosis, including the activation of procaspase-8 and the release of cytochrome c from mitochondria, are also activated by MTA [41]. Recently, Maria and his colleagues compared the threshold dosage for MTA's pro-apoptotic and anti-proliferative effects for a human CRC cell line. Their results show that it has an anti-proliferative effect at lower dosages (in the uM range) [35].
4.3. MTA and tumor invasiveness and metastasis
In addition to the above articles on direct effects of MTA on tumor cells, several studies have focused on tumor invasiveness and metastasis. In 2009, Stevens and his colleagues explored how angiogenesis features change in melanoma cells after MTA treatment. Vascular endothelial growth factor (VEGF), basis fibroblast growth factor (bFGF) and two invasion and metastasis related enzymes, matrix metalloproteinase (MMP) 9 and MMP14, were studied. All four target genes were upregulated. The authors found out that the stromal cells exhibited stronger contractile capacities with elevated levels of collagen types I, type III and MMP within cells. More importantly, despite finding no obvious change in apoptosis or proliferation patterns, hyper-invasive potential and vessel-like structures formed around tumor cells [15]. Similar results are also found in cultured HCC cells. MTA induced the expression of MMP1 and FGF2 [12].
Opposite to above result, in a metastasis liver expansion mice model reported recently, MTA completely abrogated any sign of colorectal cancer in the liver and in other organs [35]. As a class of small noncoding RNAs regulating gene expression at the posttranscriptional level and whether and how microRNAs shape regulatory effects of MTA on tumor cells have also attracted much attentions. In this study, the in vitro data confirm that MTA increases miR-34a/b levels and then down-regulates the expression of MAT2A, as the later contains a binding sequence in its 3'UTR for miR-34a/b [35]. MiR-34a has previously been confirmed as one of the tumor suppressor miRNA that is down-regulated in multiple human cancers, including colorectal cancer. Mice lacking miR-34a developed more obvious EMT-mediated colorectal cancer invasive and metastasis [44].
Although these two studies draw opposing conclusions, both present us with a new role of MTA with profound impact on the tumor microenvironment that extends beyond direct effects on tumor cells. The authors consider this difference to be related to different doses of MTA used in these experiments. For our opinion, in most of the in vitro experiments mentioned above, final MTA concentrations range from 10-7 M to 10-3 M. We believe that the various cell types used may have led to most to these differences and that more experiments must be conducted to identify more specific reasons.
4.4. MTA and tumor-related inflammation & oxidation
An increasing number of reports show that MTA is a powerful inhibitor of inflammation both in vitro and in vivo. In a lipopolysaccharide (LPS) challenged mice model, MTA administration completely lowered animal lethality rates accompanied with the suppression of circulating pro-inflammatory cytokines, including TNF-alpha, inducible NO synthesis, and the stimulation of anti-inflammatory cytokines IL-10. Anti-inflammatory effects of MTA have also been observed in LPS-treated RAW264.7 cells and isolated hepatocytes [45]. Similar responses have also seen in in vitro cultured human peripheral blood lymphocytes. MTA inhibits both the mitogen-induced blastogenesis and the pokeweed mitogen-driven immunoglobulin synthesis [46]. It also inhibits LPS-induced TNF production in macrophages and IL-1 activated ICAM-1 expression in endothelial cells [47]. In addition to the above well-known inflammation cytokines, a newly identified inflammatory form of programmed cell death, pyroptosis, is also elevated by MTA. It is very interesting that relative to the exogenous addition of MTA, its depletion has a less powerful influence on pyroptosis [48]. These findings established a link between nucleoside metabolism and immunodeficiency. MTA may serve as a signal of the nutritional status of the host of sulfur-containing amino acids. Under infection and other pro-inflammatory states, MET use is enhanced to support inflammation-related cells proliferation and the synthesis of acute phase protein [48, 49]. This increase heightens levels of MTA and flux through the MET salvage pathway to support a more robust pyroptotic and anti-inflammatory response.
Direct correlations between MTA and tumor-related inflammation have been identified in a mice colon cancer model, in which chronic inflammation is believed as a crucial underlying risk factor. In vivo, the treatment of MTA induced apoptosis and inhibited proliferation and the pro-inflammatory signaling pathway involving β-catenin, NK-κB and IL-6. More importantly, tumor loads were also reduced [50]. Similar effects of MTA have also been observed for liver cancer [51]. Given the well-established hypothesis that smoldering inflammation, the seventh hallmark of cancer, contributes to the proliferation and survival of malignant cells [52] and especially to similarities in inflammation and tumor metabolism [53], anti-inflammatory effects should help to explain how MTA may exert its anti-tumor functions at least partially. As is well accepted, chronic tissue injury and inflammation are considered to be drivers of liver disease progression from fatty liver disease and fibrosis to HCC. In a CCl4-induced rat liver injury and fibrosis model, MTA exhibits an inhibitory effect on oxidation, a phenomenon that can damage DNA and spur liver cirrhosis or even carcinogenesis [54]. This shows that MTA may interfere with the liver's inflammation-fibrosis-cancer axis. However, at present, there is no direct proof that these effects of MTA are involved in tumor prevention.
5. Role of MTA related materials in tumor
5.1. MTA and AdoMet
In addition to being the precursor for polyamine and MTA, AdoMet is essential for life as the principal biological methyl donor for biosynthetic methylation reactions. For many years it has received much more attention than its metabolite, MTA. The liver serves as the main source of AdoMet biosynthesis and consumption, turning over nearly 8 g per day in a healthy adult. In mammals, nearly half of the daily intake of MET is converted into AdoMet. For example, cells expressing higher levels of AdoMet grow much slower while exogenous AdoMet treatment inhibits liver cancer cell growth in vitro[55]. In vivo mounting evidences supports the remarkable chemopreventive effects of AdoMet on liver carcinogenesis induced by a variety of hepatocarcinogenesis [42, 56-58]. At the same time, chronic AdoMet depletion results in spontaneous mice hepatic cellular carcinoma [59]. In addition to cell growth, AdoMet also induces apoptosis in the liver cancer cells and colon cancer cells [41, 42].
The restoration of depleted GSH in the hepatocytes has been proposed to play an important role in AdoMet's hepatoprotective role [60]. Interestingly, although MTA is not a GSH precursor nor a methylating agent, some beneficial effects of AdoMet on liver functions like the inhibition of preneoplastic lesions and HCC cell proliferation, inhibition apoptosis in normal hepatocytes and the induction of apoptosis in liver cancer cells, are mimicked by MTA [27, 42]. Expression pattern changes of the MAT subtype, an essential cellular enzyme catalyzing the formation of MET to AdoMet, may be another mechanism of AdoMet. In case of human liver cancer, a change in MAT subtypes from MAT1A-dominated to MTA2A-dominated occurs [61]. In vitro cultured colon cancer cells, a variety of mitogens selectively induced MAT2A expression [36]. More importantly, clinical data showed that the percentages of MTA2A are correlated with the stages of colorectal tumor development [36, 37]. Both AdoMet and MTA maintain MAT1A expression and suppress MTA2A expression and thus slow tumor growth [62, 63]. AdoMet also affects tumor apoptosis by maintaining antiapoptotic gene cFIP expression in colon cells [41].
Based on its current use in human for the treatment of liver injuries including those of ethanol and contraceptive toxicity, hepatitis, and liver cirrhosis, it is easy to anticipate its therapeutic application as a chemopreventive agent. Compared with MTA, AdoMet is highly unstable with a short half-time (12 h at 37 in culture medium). At the same time, the rate of its conversion to MTA is 0.013 mM per hour per mM AdoMet under above condition [36]. It suggests that the pharmacological actions of AdoMet may actually depend at least in part on its transformation into MTA.
5.2. MTA and MTAP
Another protein of MTA metabolism that has received more interest as of late is MTAP, the main enzyme that keeps cellular MTA levels consistently low by catalyzing the phosphorylation of MTA to MTR-1P. MTAP was first identified in rat prostate in 1969 and has since been detected and purified from various tissues. In normal human cells and tissues, MTAP is expressed abundantly, but the liver presents the highest expression levels [64]. Even in rats it has presented considerable levels of activity in every organ examined thus far, including the prostate, liver, lungs, spleen, kidneys and heart [65, 66]. In contrast, a loss of MTAP activity is very common in a variety of cancers as shown by two clinical investigations of patients with non-small cell lung cancer [67, 68]. Similar phenomena have also been observed in case of glioma [69], pancreatic cancer [70, 71], T-cell acute leukemia [72], bladder cancer, breast cancer [73], osteosarcoma [74], endometrial cancer [75], biliary tract cancer [76], HCC [77] and melanoma [78].
The frequent loss of MTAP activity observed is easily understood by determining its chromosomal location. From the centromeric end, the gene order of human chromosome 9p21 is mapped as p15—p16—MTAP—INF-α—INF-β [79], forming a common region involving the selective deletion of many kinds of tumors. Dr. Zappia and his colleagues screened 35 different human malignant cell lines of several tumor types via direct radiochemical assay and immunochemical techniques. They found a full correlation between a lack of MTAP and a lack of p161NK4 [80]. MTAP gene deletion was further found across a wide range of tumor types [73-75, 81, 82].
Gene silencing by DNA methylation also contributes to a loss of MTAP activity. DNA CpG methylation has been extensively investigated for over 50 years. It is implicated in the expression controlling of a series genes involved in cell development and homeostasis and in cancer-driving activities [83]. A clinical investigation shows that MTAP promoter hyper-methylation is observed in nearly 23.1% of gastric adenocarcinoma patients [84]. Several experimental studies of human melanoma and the HCC cell line provide in vitro evidence of a significant correlation between the down-regulation of MTAP mRNA expression and the hyper-methylation of its promoter [78, 85].
A clinical screening of tissue microarray samples drawn from 99 non-small cell lung cancer patients showed that patients with lower MTAP levels had poorer survival rates. This was even confirmed to constitute an independent prognostic factor of lung cancer [64]. However, what interests us more is the clinical significance of the strong inverse correlation between MTAP protein levels and clinical progression from benign melanocytic nevi to melanomas formation and to the further development of metastatic melanomas. This phenomenon directly shows that a loss of MTAP expression in malignant melanoma results in a higher levels of invasive potential [78]. At the same time, stable in vitro transfection for re-inducing MTAP expression in breast cancer cell and HCC has no effect on cell proliferation while reducing cell invasiveness [73, 85]. These results show that MTAP is a tumor suppressor gene and especially in terms of metastasis.
In contrast to the results of most related studies, one clinical study of 50 patients with colorectal cancer found that MTAP protein levels are higher in tumor tissues than in adjacent normal tissues and that this is associated with higher hepatic metastasis susceptibility [86]. In reference to mouse xenografts of lung cancer, the authors found a robust suppression of lung cancer growth and of metastases via the systemic inhibition of MTAP, leading to increased MTA levels in blood, tumors and urine [87].
In consideration of the phenomena that tumor cells with lower level of MTAP activity can excrete out more MTA, it is not clear whether MTA is one factors involved in the carcinogenesis appeared in MTAP deficiency. We must also note that its functions are also linked to polyamine synthesis, AdoMet recycling from MTA, methionine re-synthesis and adenine salvaging. It may be speculated that different MTAP levels and thus different capacities to metabolize normal and tumor cell MTA may help explain the opposite effect of MTA on these cells.
5.3. Polyamines and tumors
The generation of MTA is accompanied by the synthesis of polyamines, which, including putrescine, spermidine, and spermine, are small and aliphatic amines. Naturally occurring polyamines derive from the amino acid ornithine, which is decarboxylated in the initial rate-limiting reaction by ODC to yield putrescine (Figure 2) [16].
In as early as 1988 researchers found that polyamines are essential for cell growth and that the enhanced capabilities of polyamine synthesis may reflect enhanced levels of cell proliferation. Increases in polyamine biosynthesis and intracellular polyamine content levels are of the most consistent biochemical alterations observed in cancer cells of all types, denoting their importance for tumorigenesis. A recent study highlights the dependence of melanoma cells not only on higher levels of polyamine biosynthesis but also on upregulated polyamine transport systems [88]. They are also involved in carcinogenesis and tumor biology properties such as those of breast cancer [89] and lung adenocarcinoma invasiveness and metastasis [90]. It is evident that the polyamine-biosynthetic pathway serve as an inviting target for the development of agents that inhibit carcinogenesis and tumor growth [91]. Inhibitory effects of MTA on polyamine generation contribute to its anti-tumor effects as observed for colon cells subjected to MTA treatment [36].
Levels of ODC, the first rate-limiting enzyme involved in polyamine biosynthesis, have also been examined in tumor research. Its expression and activity are regulated normally but are remarkably enhanced after stimulation by growth factors, carcinogens, viruses or oncogenes. Moreover, it is closely correlated with carcinogenesis both experimentally and clinically. For example, the overexpression of ODC in NIH/3T3 cells can spur transformation in vitro [92], as is the case for lymphomas [93] and skin tumors [94]. More strong correlations have been found from two clinical investigations of breast cancer, in which ODC activity levels were found to be significantly higher than those of normal tissues and to be correlated with higher levels of polyamine content and with histological grade, peritumoral lymphatic or blood vessel invasion [95]. ODC activity levels have also been found to be closely negatively correlated with both disease-free and overall survival, showing that ODC serves as a poor prognostic factor [96]. Data for HCC patients also reveal similar effects of ODC [97, 98]. More importantly, ODC expression was found to be associated with worsened survival outcomes and with increased recurrence in patients with endometrial cancer [99].
The above studies suggest that ODC may play both a prognostic role and acts as a chemoprevention target. In fact, the capacity for an agent to inhibit ODC activity is commonly considered to be a predictor of chemopreventive activity. Some endometrial cancers appear to be particularly sensitive to a specific inhibitor of ODC, resulting in reduced cell proliferation, cell viability and colony formation in cell line models [99].
Curcumin, which is traditionally known for its antioxidant, anti-inflammatory, and anti-carcinogenic properties, presents potential uses for both chemoprevention and chemotherapy since being confirmed to decrease ODC activity. The effects of curcumin on polyamine metabolism were first investigated from the CD-1 skin carcinogenesis mouse model [100]. Several studies also show that pretreatment with curcumin can abrogate carcinogen-induced ODC activity and tumor development in rodent tumorigenesis models targeting various organs. Similar effects have been observed from the xenograft model for esophageal squamous cell carcinoma [101] and for human lung cancer A549 cells [102]. Alpha-difluoromethylornithine (DFMO), an inhibitor of ODC, not only markedly reduces in vitro invasiveness of the human breast cancer cell line, but also inhibits the pulmonary metastasis of orthotopically implanted breast cancer xenografts in nude mice [103].
6. Conclusion
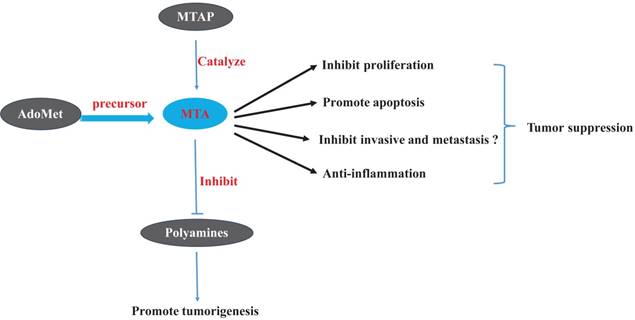
In addition to the fields of transcriptomic, genomics and proteomics, studied have shown that the discipline of metabolomics is becoming an important field of cancer research. Early perturbations of cellular metabolism are a hallmark of cancer. The field of metabolomics has provided us with new insights into the pathology of cancer and has revealed new biomarkers that may prove useful for diagnosis and prognosis. Cancer metabolomics research has not only focused on gleaning fundamental mechanistic insight into carcinogenesis but has also focused on obtaining more diagnostic biomarkers of patients with cancer. As indicated by most of such studies (Figure 3), at therapeutic concentrations, MTA suppresses tumors through the inhibition of tumor cell proliferation and invasion, through the induction of apoptosis, and by controlling the inflammatory micro-environment of tumor tissue. AdoMet, as the precursor of MTA, has the most anti-tumor effects which could be mimicked by MTA. Polyamines, which act as the essential nutrients of tumorigenesis, together with ODC, are targets of several chemoprevention medicines. MTA can also perform its role as a tumor-suppressor partially through inhibiting the activity of ODC and preventing the synthesis of polyamines. We believe that the MTA has potential therapeutic application for tumor treatment, especially for the tumors promoted by chronic inflammation.
Effects of 5'-Methylthioadenosine (MTA) and of related materials on tumors
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81272313).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Warburg O, Posener K, Negelein E. On the metabolism of carcinoma cells. Biochem Z. 1924;152:309-44
2. Vermeersch KA, Styczynski MP. Applications of metabolomics in cancer research. J Carcinog. 2013;12:9
3. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ. et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol. 2017;8:248
4. Avila MA, Garcia-Trevijano ER, Lu SC, Corrales FJ, Mato JM. Methylthioadenosine. Int J Biochem Cell Biol. 2004;36:2125-30
5. John A. Mandel EKD. Preliminary note on a purine-hexose compound. J Biol Chem. 1912;11:85-6
6. Suzuki U, Odake S, Mori T. Uber einen neuen schwefelhaltigen bestandteil der hefe. Biochem Z. 1924;154:278-89
7. Williams-Ashman HG, Seidenfeld J, Galletti P. Trends in the biochemical pharmacology of 5'-deoxy-5'-methylthioadenosine. Biochem Pharmacol. 1982;31:277-88
8. Vandenbark AA, Ferro AJ, Barney CL. Inhibition of lymphocyte transformation by a naturally occurring metabolite: 5'-methylthioadenosine. Cell Immunol. 1980;49:26-33
9. Mueller JH. A new sulfur-containing amino-acid isolated from the hydrolatic products of protein. J Biol Chem. 1923;56:157-69
10. Bowman WH, Tabor CW, Tabor H. Spermidine biosynthesis. Purification and properties of propylamine transferase from Escherichia coli. J Biol Chem. 1973;248:2480-6
11. Bigaud E, Corrales FJ. Methylthioadenosine (MTA) Regulates Liver Cells Proteome and Methylproteome: Implications in Liver Biology and Disease. Mol Cell Proteomics. 2016;15:1498-510
12. Kirovski G, Stevens AP, Czech B, Dettmer K, Weiss TS, Wild P. et al. Down-regulation of methylthioadenosine phosphorylase (MTAP) induces progression of hepatocellular carcinoma via accumulation of 5'-deoxy-5'-methylthioadenosine (MTA). Am J Pathol. 2011;178:1145-52
13. Seidenfeld J, Wilson J, Williams-Ashman HG. Androgenic regulation of 5'-deoxy-5'-methylthioadenosine concentrations and methylthioadenosine phosphorylase activity in relation to polyamine metabolism of rat prostate. Biochem Biophys Res Commun. 1980;95:1861-8
14. Toohey JI. Methylthioadenosine nucleoside phosphorylase deficiency in methylthio-dependent cancer cells. Biochem Biophys Res Commun. 1978;83:27-35
15. Stevens AP, Spangler B, Wallner S, Kreutz M, Dettmer K, Oefner PJ. et al. Direct and tumor microenvironment mediated influences of 5'-deoxy-5'-(methylthio)adenosine on tumor progression of malignant melanoma. J Cell Biochem. 2009;106:210-9
16. Stevens AP, Dettmer K, Kirovski G, Samejima K, Hellerbrand C, Bosserhoff AK. et al. Quantification of intermediates of the methionine and polyamine metabolism by liquid chromatography-tandem mass spectrometry in cultured tumor cells and liver biopsies. J Chromatogr A. 2010;1217:3282-8
17. Subhi AL, Diegelman P, Porter CW, Tang B, Lu ZJ, Markham GD. et al. Methylthioadenosine phosphorylase regulates ornithine decarboxylase by production of downstream metabolites. J Biol Chem. 2003;278:49868-73
18. Guan R, Ho MC, Frohlich RF, Tyler PC, Almo SC, Schramm VL. Methylthioadenosine deaminase in an alternative quorum sensing pathway in Pseudomonas aeruginosa. Biochemistry. 2012;51:9094-103
19. Kamatani N, Carson DA. Abnormal regulation of methylthioadenosine and polyamine metabolism in methylthioadenosine phosphorylase-deficient human leukemic cell lines. Cancer Res. 1980;40:4178-82
20. Kaneko K, Fujimori S, Kamatani N, Akaoka I. 5'-Methylthioadenosine in urine from normal subjects and cancer patients. Biochim Biophys Acta. 1984;802:169-74
21. Smith RL, Anderson EE Jr, Overland RN, Schlenk F. The occurrence formation and isolation of thiomethyladenosine. Arch Biochem Biophys. 1953;42:72-82
22. Della Ragione F, Oliva A, Gragnaniello V, Fioretti M, Fioretti A, Menna LF. et al. Chromatographic and radioimmunological methods for the determination of 5'-deoxy-5'-methylthioadenosine in biological fluids. J Chromatogr. 1988;440:141-9
23. Porcelli M, Cacciapuoti G, Oliva A, Zappia V. High-performance liquid chromatographic determination of a naturally occurring xylosyl thioether, 9-[5'-deoxy-5'(methylthio)-beta-D-xylofuranosyl]adenine. J Chromatogr. 1988;440:151-5
24. Kammerer B, Frickenschmidt A, Muller CE, Laufer S, Gleiter CH, Liebich H. Mass spectrometric identification of modified urinary nucleosides used as potential biomedical markers by LC-ITMS coupling. Anal Bioanal Chem. 2005;382:1017-26
25. Kammerer B, Frickenschmidt A, Gleiter CH, Laufer S, Liebich H. MALDI-TOF MS analysis of urinary nucleosides. J Am Soc Mass Spectrom. 2005;16:940-7
26. Stevens AP, Dettmer K, Wallner S, Bosserhoff AK, Oefner PJ. Quantitative analysis of 5'-deoxy-5'-methylthioadenosine in melanoma cells by liquid chromatography-stable isotope ratio tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:123-8
27. Pascale RM, Simile MM, Satta G, Seddaiu MA, Daino L, Pinna G. et al. Comparative effects of L-methionine, S-adenosyl-L-methionine and 5'-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res. 1991;11:1617-24
28. Andreu-Perez P, Hernandez-Losa J, Moline T, Gil R, Grueso J, Pujol A. et al. Methylthioadenosine (MTA) inhibits melanoma cell proliferation and in vivo tumor growth. BMC Cancer. 2010;10:265
29. Ferioli ME, Scalabrino G. Persistently decreased hepatic levels of 5'-deoxy-5'-methylthioadenosine during regeneration of and chemical carcinogenesis in rat liver. J Natl Cancer Inst. 1986;76:1217-21
30. Latasa MU, Boukaba A, Garcia-Trevijano ER, Torres L, Rodriguez JL, Caballeria J. et al. Hepatocyte growth factor induces MAT2A expression and histone acetylation in rat hepatocytes: role in liver regeneration. FASEB J. 2001;15:1248-50
31. Liu YH, Liu GW, Ye HP, Wang XS, Ren WD, Dang WL. Effect and mechanism of 5'-methythioadenosine on the prolieration of NCM460 cells and colon cancer HCT116 cells. Chin J Exp Surg. 2015;32:3049-52
32. de Ferra F, Baglioni C. Correlation between growth inhibition and presence of 5'-methylthioadenosine in cells treated with interferon. Cancer Res. 1984;44:2297-301
33. Wolford RW, MacDonald MR, Zehfus B, Rogers TJ, Ferro AJ. Effect of 5''-methylthioadenosine and its analogs on murine lymphoid cell proliferation. Cancer Res. 1981;41:3035-9
34. Shafman TD, Sherman ML, Kufe DW. Effect of 5'-methylthioadenosine on induction of murine erythroleukemia cell differentiation. Biochem Biophys Res Commun. 1984;124:172-7
35. Tomasi ML, Cossu C, Spissu Y, Floris A, Ryoo M, Iglesias-Ara A. et al. S-adenosylmethionine and methylthioadenosine inhibit cancer metastasis by targeting microRNA 34a/b-methionine adenosyltransferase 2A/2B axis. Oncotarget. 2017;8:78851-69
36. Chen H, Xia M, Lin M, Yang H, Kuhlenkamp J, Li T. et al. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology. 2007;133:207-18
37. Ito K, Ikeda S, Kojima N, Miura M, Shimizu-Saito K, Yamaguchi I. et al. Correlation between the expression of methionine adenosyltransferase and the stages of human colorectal carcinoma. Surg Today. 2000;30:706-10
38. Cai J, Sun WM, Hwang JJ, Stain SC, Lu SC. Changes in S-adenosylmethionine synthetase in human liver cancer: molecular characterization and significance. Hepatology. 1996;24:1090-7
39. Ramani K, Yang H, Xia M, Ara AI, Mato JM, Lu SC. Leptin's mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology. 2008;47:521-31
40. Zhang QS, Cao LY, Wang MJ, Wen YW, Fei LX, Gao JC. et al. Role of methylthioadenosine in cell proliferation of colon cancer Hct116 cells. Anhui Medical and Pharmaceutical Journal. 2017;21:646-9
41. Li TW, Zhang Q, Oh P, Xia M, Chen H, Bemanian S. et al. S-Adenosylmethionine and methylthioadenosine inhibit cellular FLICE inhibitory protein expression and induce apoptosis in colon cancer cells. Mol Pharmacol. 2009;76:192-200
42. Ansorena E, Garcia-Trevijano ER, Martinez-Chantar ML, Huang ZZ, Chen L, Mato JM. et al. S-adenosylmethionine and methylthioadenosine are antiapoptotic in cultured rat hepatocytes but proapoptotic in human hepatoma cells. Hepatology. 2002;35:274-80
43. Yang H, Sadda MR, Li M, Zeng Y, Chen L, Bae W. et al. S-adenosylmethionine and its metabolite induce apoptosis in HepG2 cells: Role of protein phosphatase 1 and Bcl-x(S). Hepatology. 2004;40:221-31
44. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D. et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853-67
45. Hevia H, Varela-Rey M, Corrales FJ, Berasain C, Martinez-Chantar ML, Latasa MU. et al. 5'-methylthioadenosine modulates the inflammatory response to endotoxin in mice and in rat hepatocytes. Hepatology. 2004;39:1088-98
46. Di Padova F, Di Padova C, Stramentinoli G, Tritapepe R. Inhibition of lymphocyte function by a naturally occurring nucleoside: 5'-methylthioadenosine (MTA). Int J Immunopharmacol. 1985;7:193-8
47. Cerri MA, Beltran-Nunez A, Bernasconi S, Dejana E, Bassi L, Bazzoni G. Inhibition of cytokine production and endothelial expression of adhesion antigens by 5'-methylthioadenosine. Eur J Pharmacol. 1993;232:291-4
48. Ko DC, Gamazon ER, Shukla KP, Pfuetzner RA, Whittington D, Holden TD. et al. Functional genetic screen of human diversity reveals that a methionine salvage enzyme regulates inflammatory cell death. Proc Natl Acad Sci U S A. 2012;109:E2343-52
49. Grimble RF, Grimble GK. Immunonutrition: role of sulfur amino acids, related amino acids, and polyamines. Nutrition. 1998;14:605-10
50. Li TW, Yang H, Peng H, Xia M, Mato JM, Lu SC. Effects of S-adenosylmethionine and methylthioadenosine on inflammation-induced colon cancer in mice. Carcinogenesis. 2012;33:427-35
51. Li TW, Peng H, Yang H, Kurniawidjaja S, Panthaki P, Zheng Y. et al. S-Adenosylmethionine and methylthioadenosine inhibit beta-catenin signaling by multiple mechanisms in liver and colon cancer. Mol Pharmacol. 2015;87:77-86
52. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073-81
53. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017;26:49-70
54. Simile MM, Banni S, Angioni E, Carta G, De Miglio MR, Muroni MR. et al. 5'-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J Hepatol. 2001;34:386-94
55. Cai J, Mao Z, Hwang JJ, Lu SC. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998;58:1444-50
56. Pascale RM, Simile MM, De Miglio MR, Feo F. Chemoprevention of hepatocarcinogenesis: S-adenosyl-L-methionine. Alcohol. 2002;27:193-8
57. Pascale RM, Simile MM, De Miglio MR, Nufris A, Daino L, Seddaiu MA. et al. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis. 1995;16:427-30
58. Pascale RM, Marras V, Simile MM, Daino L, Pinna G, Bennati S. et al. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: a long-term study. Cancer Res. 1992;52:4979-86
59. Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang ZZ, Chen L. et al. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292-4
60. Mato JM, Alvarez L, Ortiz P, Pajares MA. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73:265-80
61. Maldonado LY, Arsene D, Mato JM, Lu SC. Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp Biol Med (Maywood). 2018;243:107-17
62. Garcia-Trevijano ER, Latasa MU, Carretero MV, Berasain C, Mato JM, Avila MA. S-adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: a new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. FASEB J. 2000;14:2511-8
63. Yang H, Huang ZZ, Zeng Z, Chen C, Selby RR, Lu SC. Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am J Physiol Gastrointest Liver Physiol. 2001;280:G184-90
64. Su CY, Chang YC, Chan YC, Lin TC, Huang MS, Yang CJ. et al. MTAP is an independent prognosis marker and the concordant loss of MTAP and p16 expression predicts short survival in non-small cell lung cancer patients. Eur J Surg Oncol. 2014;40:1143-50
65. Ferro AJ, Wrobel NC, Nicolette JA. 5-methylthioribose 1-phosphate: a product of partially purified, rat liver 5'-methylthioadenosine phosphorylase activity. Biochim Biophys Acta. 1979;570:65-73
66. Garbers DL. Demonstration of 5'-methylthioadenosine phosphorylase activity in various rat tissues. Some properties of the enzyme from rat lung. Biochim Biophys Acta. 1978;523:82-93
67. Watanabe F, Takao M, Inoue K, Nishioka J, Nobori T, Shiraishi T. et al. Immunohistochemical diagnosis of methylthioadenosine phosphorylase (MTAP) deficiency in non-small cell lung carcinoma. Lung Cancer. 2009;63:39-44
68. Nobori T, Szinai I, Amox D, Parker B, Olopade OI, Buchhagen DL. et al. Methylthioadenosine phosphorylase deficiency in human non-small cell lung cancers. Cancer Res. 1993;53:1098-101
69. Nobori T, Karras JG, Della Ragione F, Waltz TA, Chen PP, Carson DA. Absence of methylthioadenosine phosphorylase in human gliomas. Cancer Res. 1991;51:3193-7
70. Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996;56:1083-90
71. Subhi AL, Tang B, Balsara BR, Altomare DA, Testa JR, Cooper HS. et al. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin Cancer Res. 2004;10:7290-6
72. Hori Y, Hori H, Yamada Y, Carrera CJ, Tomonaga M, Kamihira S. et al. The methylthioadenosine phosphorylase gene is frequently co-deleted with the p16INK4a gene in acute type adult T-cell leukemia. Int J Cancer. 1998;75:51-6
73. Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002;62:6639-44
74. Garcia-Castellano JM, Villanueva A, Healey JH, Sowers R, Cordon-Cardo C, Huvos A. et al. Methylthioadenosine phosphorylase gene deletions are common in osteosarcoma. Clin Cancer Res. 2002;8:782-7
75. Wong YF, Chung TK, Cheung TH, Nobori T, Chang AM. MTAP gene deletion in endometrial cancer. Gynecol Obstet Invest. 1998;45:272-6
76. Karikari CA, Mullendore M, Eshleman JR, Argani P, Leoni LM, Chattopadhyay S. et al. Homozygous deletions of methylthioadenosine phosphorylase in human biliary tract cancers. Mol Cancer Ther. 2005;4:1860-6
77. Berasain C, Hevia H, Fernandez-Irigoyen J, Larrea E, Caballeria J, Mato JM. et al. Methylthioadenosine phosphorylase gene expression is impaired in human liver cirrhosis and hepatocarcinoma. Biochim Biophys Acta. 2004;1690:276-84
78. Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R. et al. Characterization of methylthioadenosin phosphorylase (MTAP) expression in malignant melanoma. Am J Pathol. 2003;163:683-90
79. Olopade OI, Pomykala HM, Hagos F, Sveen LW, Espinosa R 3rd, Dreyling MH. et al. Construction of a 2.8-megabase yeast artificial chromosome contig and cloning of the human methylthioadenosine phosphorylase gene from the tumor suppressor region on 9p21. Proc Natl Acad Sci U S A. 1995;92:6489-93
80. Della Ragione F, Russo G, Oliva A, Mastropietro S, Mancini A, Borrelli A. et al. 5'-Deoxy-5'-methylthioadenosine phosphorylase and p16INK4 deficiency in multiple tumor cell lines. Oncogene. 1995;10:827-33
81. M'Soka T J, Nishioka J, Taga A, Kato K, Kawasaki H, Yamada Y. et al. Detection of methylthioadenosine phosphorylase (MTAP) and p16 gene deletion in T cell acute lymphoblastic leukemia by real-time quantitative PCR assay. Leukemia. 2000;14:935-40
82. Dreyling MH, Roulston D, Bohlander SK, Vardiman J, Olopade OI. Codeletion of CDKN2 and MTAP genes in a subset of non-Hodgkin's lymphoma may be associated with histologic transformation from low-grade to diffuse large-cell lymphoma. Genes Chromosomes Cancer. 1998;22:72-8
83. Long MD, Smiraglia DJ, Campbell MJ. The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules. 2017:7
84. Leal M, Lima E, Silva P, Assumpcao P, Calcagno D, Payao S. et al. Promoter hypermethylation of CDH1, FHIT, MTAP and PLAGL1 in gastric adenocarcinoma in individuals from Northern Brazil. World J Gastroenterol. 2007;13:2568-74
85. Hellerbrand C, Muhlbauer M, Wallner S, Schuierer M, Behrmann I, Bataille F. et al. Promoter-hypermethylation is causing functional relevant downregulation of methylthioadenosine phosphorylase (MTAP) expression in hepatocellular carcinoma. Carcinogenesis. 2006;27:64-72
86. Zhong Y, Lu K, Zhu S, Li W, Sun S. Characterization of methylthioadenosin phosphorylase (MTAP) expression in colorectal cancer. Artif Cells Nanomed Biotechnol. 2018;46:2082-7
87. Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X. et al. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5'-methylthioadenosine phosphorylase. J Biol Chem. 2011;286:4902-11
88. Peters MC, Minton A, Phanstiel Iv O, Gilmour SK. A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Med Sci (Basel). 2018:6
89. Manni A. Role of polyamines in breaset cancer growth, development and progression. Current Cancer Therapy Reviews. 2005;1:207-15
90. Fahrmann JF, Grapov DD, Wanichthanarak K, DeFelice BC, Salemi MR, Rom WN. et al. Integrated Metabolomics and Proteomics Highlight Altered Nicotinamide- and Polyamine Pathways in Lung Adenocarcinoma. Carcinogenesis. 2017;38:271-80
91. Murray-Stewart T, Casero RA. Regulation of Polyamine Metabolism by Curcumin for Cancer Prevention and Therapy. Med Sci (Basel). 2017:5
92. Auvinen M, Paasinen A, Andersson LC, Holtta E. Ornithine decarboxylase activity is critical for cell transformation. Nature. 1992;360:355-8
93. Nilsson JA, Keller UB, Baudino TA, Yang C, Norton S, Old JA. et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell. 2005;7:433-44
94. O'Brien TG, Megosh LC, Gilliard G, Soler AP. Ornithine decarboxylase overexpression is a sufficient condition for tumor promotion in mouse skin. Cancer Res. 1997;57:2630-7
95. Canizares F, Salinas J, de las Heras M, Diaz J, Tovar I, Martinez P. et al. Prognostic value of ornithine decarboxylase and polyamines in human breast cancer: correlation with clinicopathologic parameters. Clin Cancer Res. 1999;5:2035-41
96. Manni A, Mauger D, Gimotty P, Badger B. Prognostic influence on survival of increased ornithine decarboxylase activity in human breast cancer. Clin Cancer Res. 1996;2:1901-6
97. Gan FY, Gesell MS, Alousi M, Luk GD. Analysis of ODC and c-myc gene expression in hepatocellular carcinoma by in situ hybridization and immunohistochemistry. J Histochem Cytochem. 1993;41:1185-96
98. Kubo S, Tamori A, Nishiguchi S, Omura T, Kinoshita H, Hirohashi K. et al. Relationship of polyamine metabolism to degree of malignancy of human hepatocellular carcinoma. Oncol Rep. 1998;5:1385-8
99. Kim HI, Schultz CR, Buras AL, Friedman E, Fedorko A, Seamon L. et al. Ornithine decarboxylase as a therapeutic target for endometrial cancer. PLoS One. 2017;12:e0189044
100. Huang MT, Smart RC, Wong CQ, Conney AH. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1988;48:5941-6
101. Tung LN, Song S, Chan KT, Choi MY, Lam HY, Chan CM. et al. Preclinical Study of Novel Curcumin Analogue SSC-5 Using Orthotopic Tumor Xenograft Model for Esophageal Squamous Cell Carcinoma. Cancer Res Treat. 2018;50:1362-77
102. Liu F, Gao S, Yang Y, Zhao X, Fan Y, Ma W. et al. Antitumor activity of curcumin by modulation of apoptosis and autophagy in human lung cancer A549 cells through inhibiting PI3K/Akt/mTOR pathway. Oncol Rep. 2018;39:1523-31
103. Manni A, Washington S, Griffith JW, Verderame MF, Mauger D, Demers LM. et al. Influence of polyamines on in vitro and in vivo features of aggressive and metastatic behavior by human breast cancer cells. Clin Exp Metastasis. 2002;19:95-105
Author biography
Dr. Ping Wu is an Associated Professor of Pathophysiology at Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. She obtained her Ph.D. degree in 2008 and has trained as a Postdoctoral Fellow in the School of Medicine, Utah University, USA. Her current research focuses on the role of uncontrolled inflammation in tumor development.
![]() Corresponding author: Ping Wu, Email: wpingwptjmu.edu.cn, Phone: 86-18971666873
Corresponding author: Ping Wu, Email: wpingwptjmu.edu.cn, Phone: 86-18971666873