Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):874-884. doi:10.7150/jca.27635 This issue Cite
Research Paper
MYH9 Promotes Growth and Metastasis via Activation of MAPK/AKT Signaling in Colorectal Cancer
Bin Wang1,2, Xiaolong Qi1, Jian Liu3,4, Rui Zhou4, Chuang Lin3,4, Junjie Shangguan5, Zhuoli Zhang5,6, Liang Zhao3,4 ![]() , Guoxin Li1
, Guoxin Li1 ![]()
1. Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangdong provincial Engineering Technology Research Center of Minimally Invasive Surgery, Guangzhou, China.
2. Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
3. Department of Pathology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, P.R. China
4. Department of Pathology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, 510515, P.R. China
5. Department of Radiology, Feinberg School of Medicine, Northwestern University, Chicago, IL, 60611, USA
6. Robert H. Lurie Comprehensive Cancer Center, Chicago, IL, 60611, USA
Received 2018-6-4; Accepted 2019-1-4; Published 2019-1-29
Abstract
The contractile protein MYH9 (non-muscle myosin IIA) is an actin-binding protein that plays a fundamental role in cell adhesion, migration, and division. However, its distinct role in colorectal cancer (CRC) still remains unidentified. In this study, we detected significant MYH9 overexpression in CRC samples compared with paired normal tissues using western blotting and tissue microarray immunohistochemistry (IHC). Moreover, analysis of patient clinical information demonstrated that MYH9 overexpression was strongly correlated with lymph node metastasis and poor overall survival. Endogenous overexpression of MYH9 enhanced the ability of cell proliferation and migration in vitro, and accelerated CRC growth in mouse models. Silencing of MYH9 revealed repressive effects on CRC cells in vitro and in vivo. Furthermore, primary biomechanics that involved MAPK/AKT signaling mediated epithelial-mesenchymal transition (EMT) was uncovered underlying MYH9 dependent cell behaviors. Collectively, our data showed that MYH9 significantly promoted tumorigenesis by regulating MAPK/AKT signaling, and was remarkably correlated with poor prognosis in CRC. MYH9 may thus be a novel biomarker and drug target in the diagnosis and treatment of CRC.
Keywords: MYH9, CRC, Cancer Metastasis, EMT, Tumor Biomarker.
Introduction
Colorectal cancer (CRC) has the third highest cancer incidence and is the fourth most common cause of cancer death globally [1]. In China, the so called westernized lifestyle is believed to be a contributing cause of the increased prevalence of obesity and physical inactivity and thus has an effect on the objective increase in CRC morbidity during the past decades [2]. Although tremendous advances have been achieved in the comprehensive treatment of CRC including surgical resection, chemotherapy, radiotherapy, and immunotherapy, the clinical outcomes and overall survival are still unsatisfactory. Especially in developing countries, a large portion of the patients are diagnosed at late stage with metastasis that accounts for almost 90% mortality of cancer. Cancer metastasis is a multistep biological process and is incurable at present [3]. Although a large number of studies in both clinical and preclinical research have been carried out, the molecular mechanisms underlying tumor metastasis remain largely unknown.
MYH9 is an isoform of the non-muscle II (NM II) family of proteins that includes NM IIA, NM IIB, and NM IIC [4]. It is comprised of two heavy chains (230 kDa), two regulatory light chains (RLCs) (20 kDa), and two essential light chains (ELCs) (17 kDa) that mediates actin-based contractile motion. MYH9 has been reported to have a central role in cell adhesion, polarity, and motility as a skeleton protein [5]. However, the exact function of MYH9 in cancer is controversial. A few studies claimed it as a tumor suppressor in squamous cell carcinomas (SCCs) [6], while other studies demonstrated oncogenic properties of MYH9 in MDA-MB-231 breast carcinoma cells [7], esophageal squamous cancer [8], non-small cell lung cancer (NSCLC) [9], bladder cancer [10], and gastric cancer [11]. Thus, it is essential to determine the distinct role of MYH9 in a more expansive range of human cancers.
In this study, the associations between MYH9 expression and CRC progression were examined and the unique characteristics of MYH9 in CRC cell lines were determined in vitro and in vivo. We proposed a preliminary molecular mechanism of MYH9-regulated CRC progression, which may inspire a new method for CRC prognosis and allow early interventions to prevent disease progression in CRC patients.
Material and Methods
Cell lines and reagents
CRC cell lines HCT116, HT29, LS174T, RKO, SW480 and SW620 were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China). Cells were cultured with RPMI 1640 (Gibco, Waltham, USA) medium containing 10% fetal bovine serum (FBS) (Gibco, Waltham, USA) at 37oC with 5% CO2. MEK inhibitor U0126 that specifically blocks the phosphorylation of ERK, and AKT inhibitor LY29004 (both from Cell Signaling Technology, Danvers, USA) were both dissolved in DMSO and then added to the cultured cells every 24 hours at the recommended concentrations.
Clinical samples and tissue microarrays
Clinical CRC samples and paired normal tissues used in the exploration of MYH9 expression by western blot were supplied by tissue bank of Department of General Surgery, Nanfang Hospital (Guangzhou, China). Tissue microarray, including 100 human CRC samples and 80 normal ones, was purchased from Outdo Biotech (Shanghai, China) for immunohistochemistry (IHC) analysis.
Western blotting
Protein expression was detected by immunoblot analysis with mouse antibodies against β-tubulin (1:1000; Santa Cruz, Dallas, USA), and MYH9 (1:1000; Abcam, Cambridge, USA); rabbit antibodies to p-P44/42, P44/42, p-AKT, AKT, and p-PTEN (1:500; Cell Signaling Technology, Danvers, USA); and rabbit antibody to ZO-1, N-cadherin, β-catenin, Fibronectin, and MMP9 (1:1000; Cell Signaling Technology, Danvers, USA). U0126 and LY29004 were added to the cultured cells for 48 hours before protein extraction.
Immunohistochemistry (IHC)
IHC was performed as described previously [12]. Briefly, the slides were incubated at 4oC overnight with primary antibodies against MYH9 (1:300), ZO-1 (1:250), β-catenin (1:300), and Ki67 (1:300) after high pressure antigen retrieval. After mounting by coverslip with organic resin, these slides were reviewed by three blinded pathologists. The intensity of MYH9 staining was scored as below to quantitatively group expression levels: 0 (no staining), 1 (weak staining, faint yellow), 2 (moderate staining, light brown), and 3 (strong staining, brown). Scores >2 were regarded as high expression. Multiple simultaneous evaluations were conducted to resolve the discrepancies (<5%).
Immunofluorescence
CRC cells were cultured in confocal glass bottom dishes (BD biotech, San jose, USA) overnight. Cells were fixed with 4% paraformaldehyde for 30 min, treated with 0.25% Triton X-100 for 5 min, and then washed with PBS 3 times. After dishes were blocked in 5% Bovine Serum Albumin (BSA) for 30 min and washed, cells were first incubated with primary antibodies rabbit anti-ZO-1 (1:200) or anti-β-catenin (1:300) at 4oC overnight and then incubated with secondary Alexa Fluor 488 and Alexa Fluor 594 anti-rabbit antibodies, respectively, at room temperature for 30 min. Finally, cells were stained with DAPI and observed under fluorescence microscope (Olympus, Tokyo, Japan).
RNA extraction, RT-PCR and real-time PCR
Total RNAs of transfected CRC cells were extracted with Trizol reagent (Invitrogen Life Technologies, Grand Island, USA) as described on manufacturer's protocol. Reverse transcription was performed with SuperScript II RNase H-RT kit (Invitrogen Life Technologies, Grand Island, USA) using 2µg of RNA from each sample to acquire corresponding cDNAs. All primers were designed by Invitrogen, and the primer sequences used in real-time PCR were listed in Supplementary Table S1. A 20µl total-volume reaction system including primers, cDNAs, SYBR master mix, and ddH2O was used for real-time PCR. GAPDH was selected as a normalized control. A method of 2-∆∆CT described previously was utilized to compare the changes in gene expression [13].
siRNA and lentivirus vectors construction
Four siRNA sequences were designed to knock down MYH9 expression as described in Supplementary Table S2, and the most effective one - MYH9-#4 -was selected to construct a stably expressed lentiviral vector (Genepharma, Shanghai, China). A stable lentiviral vector with overexpression of MYH9 was also constructed with human full-length MYH9 cDNA (GeneCopoeia, Rockville, USA). Transfection was described as previously [14].
Cell proliferation assays
Cell proliferation was measured by Cell Counting Kit-8 (CCK-8) assay. Transfected CRC cells were digested by trypsin and counted, and then a total of 1000 cells per well were planted to a 96-well plate. After the cells were cultured overnight, CCK-8 reagent (Dojindo, Shanghai, China) was added to each well every day at the same time point, and the absorbance value (OD) was detected by a microplate reader after 2 hours. In addition to the empty well with mixed medium as control, each group has five duplicate wells.
Cell migration assays
Cell migration was assessed by transwell assay. After digestion and counting, varying numbers of CRC cells (1x105 to 2x105) in RPMI without FBS were added to the upper chamber of the inserts (8 µm pore size, BD Biotech, San jose, USA). RPMI 1640 containing 10% FBS was added to the lower chamber, and the attached cells to the underside of the membrane were considered as migrated cells. After fixation and staining, migrated cells were imaged and counted under a microscope.
Cell invasion assays
Cell invasion assay was a modified transwell migration assay. Matrigel mixed with empty medium (1:3) was added to the membrane of upper chamber, and the chambers were then placed in the incubator for 2 hours before adding cells. The following steps were similar except the longer time for cells to invade across the matrigel. All the experiments were repeated at least 3 times.
Colony formation assay
Cells were harvested and seeded to 6-well plates at a concentration of 200 cells per well (n=6/group). Cells were cultured for two weeks before fixation in 4% paraformaldehyde for 30 min and staining in Giemsa solution for 10 min. The colonies were counted by Adobe Photoshop.
Animals and tumor growth assays
All mice experiments were approved by Animal Research Ethics Committee of Southern Medical University and in accordance to the guidelines on the care and use of animals for scientific purposes. Stable cells (2x106) in 50 µl PBS mixed with Matrigel (4:1) were injected subcutaneously into the left or right hind limb flank of 6-week-old female nude mice (n = 6/group). Tumor size was measured by longest diameter (L) and shortest diameter (S) every week with a caliper. Mice were sacrificed after 4 weeks or when the tumors grew too fast to ulcerate.
Statistical analysis
Data were presented as mean ± SD and were analyzed using SPSS (version 19.0, SPSS, Armonk, USA). Student's t test was used to compare the differences between groups to be statistically significant. Chi-square test was used to investigate whether MYH9 expression level is correlated with specific clinicopathologic characteristics. Kaplan-Meier estimation method was performed to analyze the overall survival of CRC patients. Cox proportional hazards model was displayed to evaluate whether the clinical parameters had statistically significant correlation with overall survival. P<0.05 was considered as statistically significant.
Results
MYH9 is upregulated in CRC tissues compared with normal mucosa
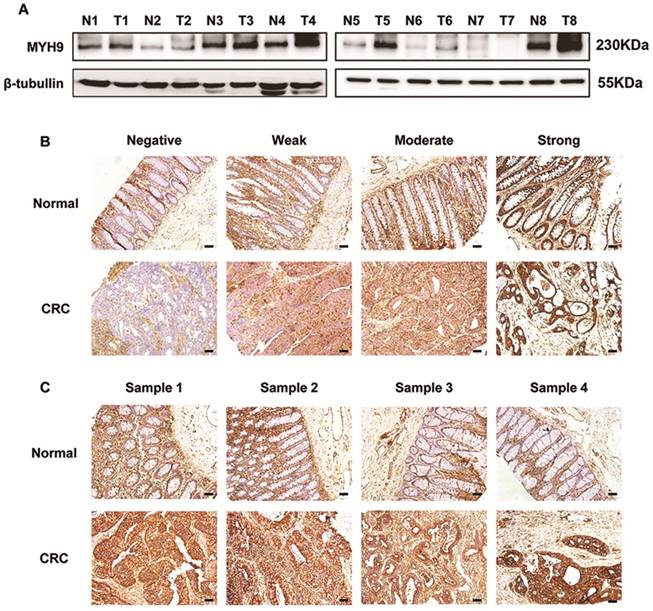
Immunoblotting assays showed a higher expression of MYH9 in 6 of 8 clinical CRC samples than in paired normal tissues (Figure 1A). The staining intensity of MYH9 in immunohistochemistry was semi-quantified and classified into four groups (Figure 1B). IHC of tissue microarray also revealed that CRC tissues had significantly more samples with MYH9 overexpression (72%, 72/100) compared with the 14/80 (17.5%) with MYH9 overexpression in normal colorectal tissue group (P<0.001). The representative figures are shown in Figure 1C.
MYH9 is upregulated in CRC tissues compared with normal mucosa. A, MYH9 expression in CRC samples and paired non-neoplastic tissues was detected by western blot. β-tubulin was selected as a control for normalization. B, MYH9 staining intensity defined into four groups as negative, weak, moderate and strong in clinical CRC samples and adjacent normal tissues by IHC. C, MYH9 expression in the tumors and normal samples was examined by IHC. Representative figures are shown. Scale bars represent 50 µm. N, normal tissues. T, tumor tissues. The raw blots/gels are presented in Supplementary Figure S1.
MYH9 is positively correlated with progression and poor survival in CRC patients
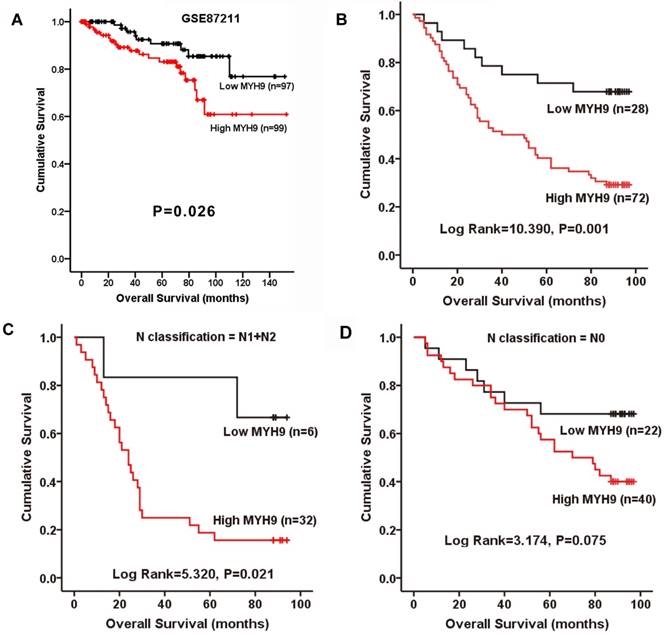
The relationship between MYH9 expression and the clinical characteristics of CRC was then analyzed. Chi-square test indicated that MYH9 expression level was only correlated with N classification (Supplementary Table S3, P=0.033). To further examine whether the findings above could be supported by observation in human primary samples, the clinical significance of MYH9 was analyzed in a microarray data of CRC cohort accessed at GEO Series GSE87211. The median relative expression level of MYH9 in all 203 CRC samples was chosen as the cut-off point for separating tumors with low expression (< median, n=97) and high expression (≥median, n=99) of MYH9. Kaplan-Meier survival analysis and log-rank tests revealed that patients with high MYH9 expression had shorter survival times (Figure 2A). In our validate cohort, Kaplan-Meier estimation showed that high expression of MYH9 in CRC patients correlated with a shorter overall survival in not only in N1+N2 patients, but also all other enrolled patients (Figure 2B-D). Cox proportional hazards model demonstrated MYH9 as an independent prognostic factor for overall survival of CRC patients in both univariate (P=0.002) and multivariate (P=0.007) analysis (Supplementary Table S4).
MYH9 is positively correlated with progression and poor survival in CRC. A, Kaplan-Meier survival curves and univariate analyses (log-rank) for CRC patients with low expression (< median, n=97) and high expression (≥median, n=99) of MYH9 in the testing cohorts derived from a published high-throughput microarray dataset (NCBI/GEO/GSE87211; n=203). B, Kaplan-Meier plot of overall survival of 100 CRC patients (log rank=10.390, P=0.001). C, Overall survival of 38 CRC patients with N1 and N2 stage (log rank=5.320, P=0.021). D, Overall survival of 62 CRC patients with N0 stage (log rank=3.174, P=0.075).
Endogenous overexpression of MYH9 accelerates CRC cell growth and metastasis in vitro and in vivo
To investigate the effect of MYH9 on CRC cell biological characteristics, MYH9 protein expression level in CRC cell lines was examined by western blot. SW480 cells with high levels of endogenous expression and SW620 cells with low expression levels were selected for the following experiments (Figure 3A). Lentivirus vector containing human MYH9 cDNA was constructed and transfected into SW620 cells. The stable overexpression of MYH9 was confirmed by real-time PCR and western blot (Figure 3B). CCK-8 and colony formation assays were performed to test the role of MYH9 in cell proliferation. Results showed that MYH9 overexpression accelerated CRC cell growth in vitro (Figure 3C, P<0.01). Transwell and Matrigel membrane transwell assays were then displayed to examine MYH9 function in cell migration and invasion capability. Results proved the enhanced cellular aggressiveness with higher MYH9 expression (Figure 3D, P<0.01).
Endogenous overexpression of MYH9 accelerates CRC cell growth and metastasis in vitro and in vivo. A, MYH9 expression level in 6 CRC cell lines were explored by western blot (left panel) and quantified (right panel). B, MYH9 mRNA expression (left panel) and protein expression (right panel) in SW620 cells infected with lentivirus including human full-length MYH9 cDNA (LV-MYH9) and empty vector (LV-Con). C, CCK-8 (left panel) and plate colony formation assay (right panel) in indicated cells. D, Cell migration (left panel) and invasion assay (right panel) in indicated cells. E, Nude mice tumorigenicity assay using indicated cells injected into hind limb flank. Tumor size and weight were shown in the right. F. H&E and MYH9 along with Ki67 staining of the tumors collected from the nude mice. Scale bars represent 50 µm. Error bars represented mean ± SD. Three independent experiments were carried out for each assay. (*), P<0.05; (**), P<0.01. The raw blots/gels are presented in Supplementary Figure S2.
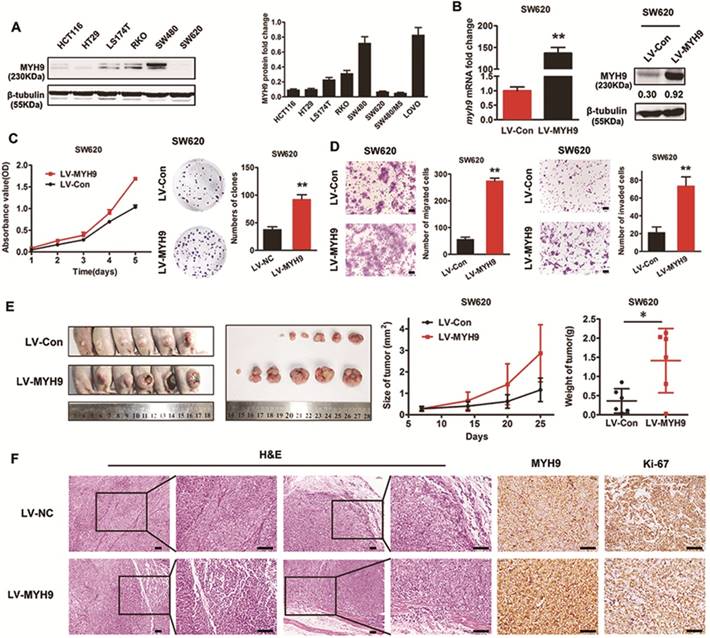
In vivo tumor growth was evaluated by subcutaneous injection into nude mice with CRC cells. Compared with controls, the average tumor volume and weight in MYH9 overexpressing cells was increased (Figure 3E, P<0.05). H&E staining showed that control group tumors had a clear boundary with the surrounding regions. In contrast, the primary tumors derived from MYH9 overexpressing cells showed infiltrative growth, thereby invading the surrounding fat and muscle tissues (Figure 3F). The intensity of Ki-67 staining in the MYH9 overexpressed group was also stronger than controls (Figure 3F). Taken together, these results suggested that MYH9 is essential for CRC cell growth and metastasis in vitro and in vivo.
Silencing of MYH9 inhibits proliferation and migration of CRC cells in vitro and in vivo
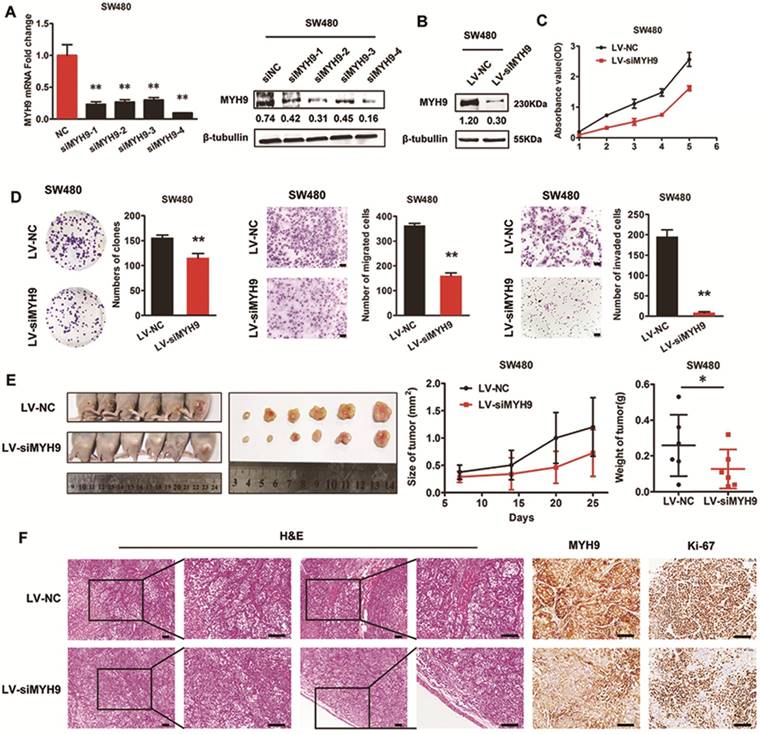
Four siRNA sequences were designed to knock down MYH9 expression in SW480 cells. The effectiveness was assessed by real-time PCR and immunoblotting assays (Figure 4A), and siMYH9-4 was selected to construct a stable CRC cell line with MYH9 knockdown through lentivirus vector transfection (Figure 4B). In vitro loss-of-function assays revealed that MYH9 depletion in CRC cells repressed aggressive phenotypes by decreasing cell proliferation, migration, and invasion (Figure 4C, D, P<0.01). In vivo tumor formation assay also drew a similar conclusion that the injected CRC cells grew more slowly with MYH9 depletion (Figure 4E, P<0.05). Compared with the control group, MYH9 silenced CRC tissues also displayed decreased muscle invasion and diminished Ki-67 index (Figure 4F).
Silencing of MYH9 inhibits proliferation and migration of CRC cells in vitro and in vivo. A, Knockdown effectiveness of four siRNAs were tested by real-time PCR (left panel) and western blot (right panel). B, Knockdown effectiveness of lentivirus (LV-siMYH9), which was derived from siMYH9-4 was validated by western blot. C, CCK-8 assay in indicated cells. D, Colony formation, migration and invasion assays in indicated cells. E, In vivo tumor formation assay using nude mice with tumor size and weight in the right. F, Representative figures of the tumors developed in nude mice with H&E, MYH9 and Ki67 staining. Scale bars represent 50 µm. Error bars served as mean ± SD for three independent experiments. Each experiment was repeated for three times. (*), P<0.05; (**), P<0.01. The raw blots/gels are presented in Supplementary Figure S3.
EMT is required for MYH9-mediated CRC cell aggressive phenotypes
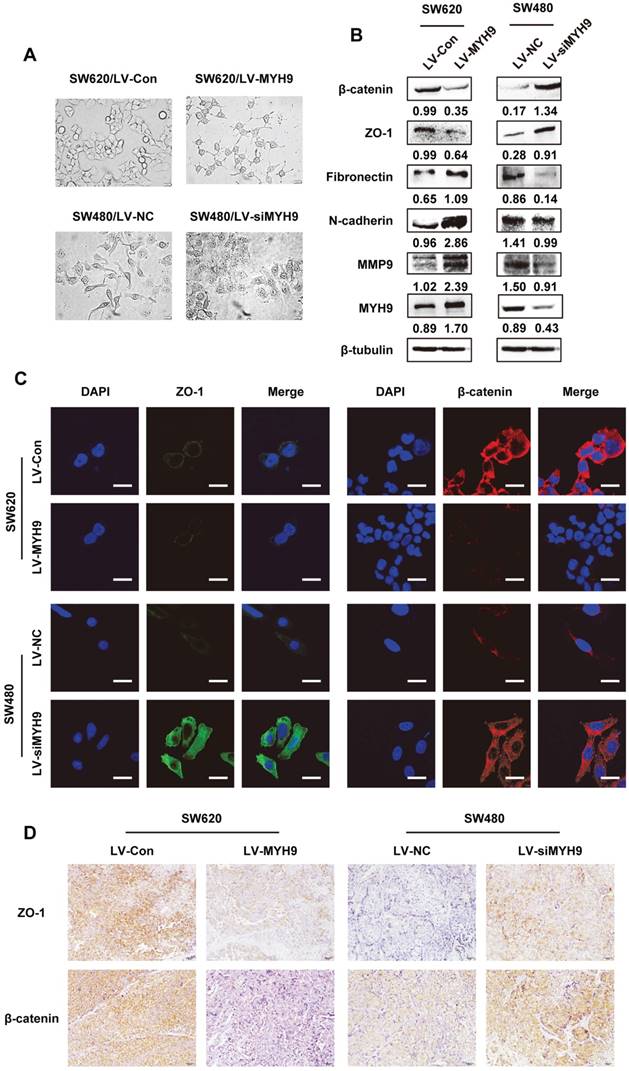
Morphological changes of stable CRC cells with altered MYH9 expression were observed under microscope. SW620 cells with stable MYH9 overexpression were more likely to display vimineous pseudopodia stretching out from a fusiform cell shape compared with the control; in contrast, SW480 cells with MYH9 knockdown appeared to have tight cell junctions with a rounded shape (Figure 5A), which suggested a typical EMT phenotype. Hence, we screened EMT markers as well as MMP9, a well-characterized excetral cellular matrix (ECM)-degrading enzyme, and found that MMP9 was strongly upregulated when MYH9 was overexpressed in SW620 cells in addition to overexpression of mesenchymal markers such as fibronectin and N-cadherin. In comparison, the epithelial markers were significantly upregulated in MYH9-knockdown SW480 cells compared to the control (Figure 5B). Immunofluorescence and IHC assays both validated the negative correlation between MYH9 and the expression of representative epithelial markers, ZO-1 and β-catenin (Figure 5C, D).
EMT is required for MYH9-mediated CRC cell aggressive phenotypes. A, Cell morphology under inverted microscope in indicated cells (scale bars represent 20 µm). B, Western blot of EMT markers and MMP9 in indicated cells. Immunosignals of different protein expression level were quantified by greyscale scanning software (Quantity one) and relative protein abundance was normalized by β-tubulin. C, Immunofluorescence of EMT markers ZO-1 and β-catenin in indicated cells (scale bars represent 10 µm). D, EMT markers ZO-1 and β-catenin expression in nude mice derived tumors with MYH9 overexpression and knockdown by IHC. Scale bars represent 20 µm. The raw blots/gels are presented in Supplementary Figure S4.
MYH9 induces EMT and promotes CRC progression through MAPK and AKT signaling
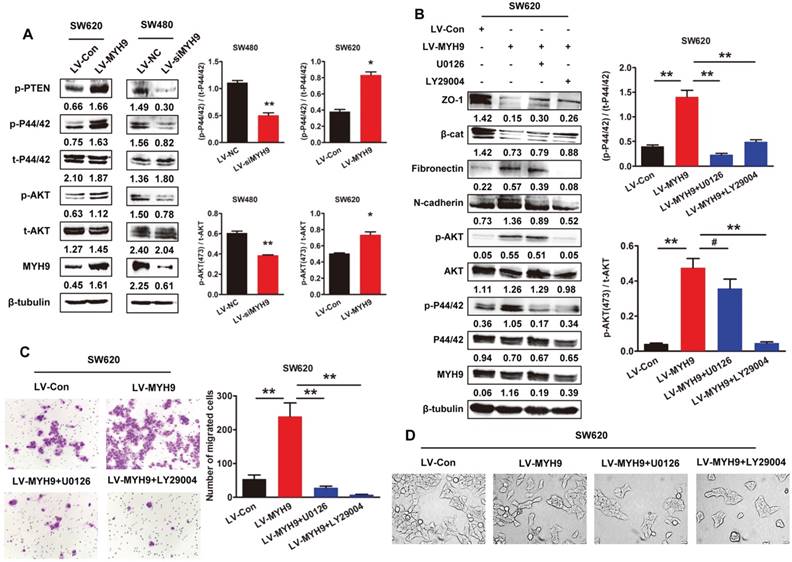
Our previous studies have found that MAPK and AKT signaling pathways were significantly involved in EMT inducement and tumor progression [15, 16]. In this study, western blot analysis showed MYH9 overexpression remarkably phosphorylated AKT and ERK in SW620 cells (Figure 6A, P<0.05). As expected, the phosphorylation was also significantly suppressed in SW480 cells with MYH9 knockdown (Figure 6A, P<0.01). Treatment of AKT inhibitor LY29004 and MEK inhibitor U0126 reversed the MYH9-induced alteration of EMT markers expression (Figure 6B) and cell morphology (Figure 6D), thus also subsequently reversing the enhanced capacity for cell migration and invasion (Figure 6C). These results indicated MAPK and AKT signaling pathway activation is required in the MYH9-induced EMT of CRC cells.
MYH9 induces EMT and promotes CRC progression through MAPK and AKT signaling. A, Western blot analysis of protein expression related to AKT/ERK signaling pathway. B, Western blot analysis of AKT/ERK signaling pathway and EMT markers in indicated cells treated with AKT inhibitor LY29004 and MEK inhibitor U0126. Immunosignals of different protein expression level were quantified by greyscale scanning software (Quantity one) and relative protein abundance was normalized by β-tubulin. C, Transwell assay in indicated cells. D, Cell morphology of indicated cells was observed under inverted microscope. Scale bars represent 50 µm. Error bars represented mean ± SD from three independent experiments. (*), P<0.05; (**), P<0.01. The raw blots/gels are presented in Supplementary Figure S5&6.
Discussion
In the current study, overexpression of MYH9 in CRC samples compared to adjacent normal tissues, and the positive correlation between MYH9 overexpression and poor prognosis in CRC patients were determined for the first time. Subsequently, the impact of MYH9 on cellular phenotype and function of CRC cell lines was investigated in vitro and in vivo. Furthermore, we explored the biomechanism underlying MYH9-mediated cellular behaviors of CRC. AKT/ERK signaling pathway was activated while MMP9 stimulated and EMT developed, which was reversed by treatment with AKT and MEK inhibitors. All the results suggested that MYH9 was a promotive factor in CRC carcinogenesis, and could be a reliable biomarker and target in CRC diagnosis and treatment.
Many previous studies have reported the role of MYH9 in cell motility and division, as it is crucial in the development and differentiation of skeletal muscle, as well as the maintenance of the smooth muscle tension, podocyte cytoskeletal structure, and mechanical function [5, 17]. Ablation of MYH9 resulted in the alteration of podocyte actin cytoskeletal structure and focal adhesion distribution, decreased cell attachment and contractility as well as increased cell motility [4].
Likewise, MYH9 siRNA induced expression change altered cell shape, actin cytoskeleton and adhesion properties [18]. It was reported that Mg2+ -ATP hydrolysis and MYH9 filament formation is regulated by RLC and heavy chain reversible phosphorylation, which involves various kinases including myosin light chain kinase (MLCK), Rho-associated, coiled coil-containing kinase (ROCK), protein phosphatase 1 (PP1) and protein kinase C (PKC), et al [19-22]. Phosphorylation of MYH9 may affect the binding of S100A4 [23], which is a metastasis factor for invasive cancer [24]. Together, they promote directional cell motility via direct interaction. Recent studies have also found that MYH9 interacts with Deleted in liver cancer 1 (Dlc1) gene and Rac 1 activation [25], which in turn accelerates cell movement [26]. Similarly, MYH9 is involved in the MRTF (Myocardin-related transcription factors)-SRF (Serum response factor) pathway contributing to cell adhesion, spreading and invasion [27]. Moreover, MYH9 assembly can be inhibited through direct interaction of myosin binding protein H (MYBPH) with non-muscle myosin heavy chain IIA (NMHC IIA) to reduce cell motility [28]. The disruption of EGFR interactions with MYH9 and β-actin lead to suppression on proliferation and induction of apoptosis in NSCLC cells [29]. Similar MYH9 functions in CRC were identified in our study. Endogenous overexpression of MYH9 promoted CRC cell proliferation and motility (Figure 3), while MYH9 knockdown showed opposite impact (Figure 4). In vivo assays also revealed parallel results. All these interactions with tumor metastasis promotion factors may indicate the overexpression of MYH9 in metastatic cancer.
Since cell proliferation and movement are primarily connected to cancer metastasis and progression [30, 31], the association between MYH9 and malignant tumors has been studied and expounded. Evidence showed that MYH9 acts as a tumor suppressor in squamous cell cancer through an in vivo RNAi screen [6], which is also verified in tissue samples of head and neck squamous cell carcinoma with low-risk p53 mutations [32]. However, other researchers claimed a totally contradictory role of MYH9 in a variety of other cancers. A study reported that myosin IIA (MYH9) was recruited at the lamellar margin when MDA-MB-231 breast carcinoma cells spreading on fibronectin and colocalized at cell leading edge with the metastasis-promoting metastasin-1. Their results showed inhibition of MLCK and Rho kinase as well as siRNA depletion of myosin II blocked cell migration and lamellar spreading [7]. MYH9 was observed to be overexpressed in esophageal squamous cancer and was significantly correlated with malignant clinicopathological features. In vitro cell migration study using MYH9 siRNA revealed attenuated cell migration ability [8]. Similar results were also seen in both clinical samples and preclinical studies of NSCLC [9], bladder cancer [10], and gastric cancer [11]. However, the molecular mechanism needs to be further elucidated. Herein, we identified the distinct cancer promoting effects of MYH9 in CRC cells and also illuminated the signaling pathway and biological processes implicated in the related cancer aggressiveness.
Future researches regarding this topic should consider sampling from larger patient populations with more diverse cancers, examining the use of therapeutics that directly target MYH9 for use in combination therapy, and further investigating the comprehensive molecular mechanism underlying MYH9 regulation network in cancer progression. The interaction of MYH9 with other metastasis promoting proteins [23] and the target regulation by microRNA [33] inspired us to orchestrate our previous work to investigate whether MYH9 directly interacts with LASP1 [24, 34] and whether MYH9 is subjected to regulation by specific microRNAs [14, 35].
In conclusion, we observed significant MYH9 overexpression in CRC samples, and MYH9 overexpression was found to have a positive correlation with poorer prognosis. Gain- and loss-of-function assays certified MYH9 as an enhancer of cancer aggressiveness in vitro and in vivo by promoting EMT via MAPK/AKT signaling activation. Taken together, MYH9 could serve as an independent prognostic factor for CRC patients and is a promising target for future therapy.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81201635; 81672446, 81572813, 81773082, 81872423), Guangdong Natural Science Foundation (S2012040006418, S2013010014254, 2014A030313490), Science and Technology Program of Planning Project of Guangdong Province (2014A020212345), Science and Technology Program of Guangzhou (1563000235). Guangdong Provincial Science and Technology Key Project (2014A020215014), the Research Fund of Public Welfare in the Health Industry, the National Health and Family Planning Commission of China (201402015) and the Key Clinical Specialty Discipline Construction Program.
Ethical approval
This study was approved by the Ethics Committee of Nanfang Hospital, Southern Medical University, Guangzhou, China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global Burden of Disease Cancer C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M. et al. The Global Burden of Cancer 2013. JAMA oncology. 2015;1:505-27
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
3. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275-92
4. Bondzie PA, Chen HA, Cao MZ, Tomolonis JA, He FF, Pollak MR. et al. Non-Muscle Myosin-IIA Is Critical for Podocyte F-Actin Organization, Contractility, and Attenuation of Cell Motility. Cytoskeleton. 2016;73:377-95
5. Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778-90
6. Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D. et al. Direct in Vivo RNAi Screen Unveils Myosin IIa as a Tumor Suppressor of Squamous Cell Carcinomas. Science. 2014;343:309-13
7. Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer research. 2006;66:4725-33
8. Xia ZK, Yuan YC, Yin N, Yin BL, Tan ZP, Hu YR. Nonmuscle myosin IIA is associated with poor prognosis of esophageal squamous cancer. Dis Esophagus. 2012;25:427-36
9. Katono K, Sato Y, Jiang SX, Kobayashi M, Nagashio R, Ryuge S. et al. Prognostic significance of MYH9 expression in resected non-small cell lung cancer. PLoS One. 2015;10:e0121460
10. Xiong D, Ye YL, Chen MK, Qin ZK, Li MZ, Zhang H. et al. Non-muscle myosin II is an independent predictor of overall survival for cystectomy candidates with early-stage bladder cancer. Oncol Rep. 2012;28:1625-32
11. Liu D, Zhang L, Shen Z, Tan F, Hu Y, Yu J. et al. Clinicopathological significance of NMIIA overexpression in human gastric cancer. Int J Mol Sci. 2012;13:15291-304
12. Zhao L, Wang H, Li JM, Liu YW, Ding YQ. Overexpression of Rho GDP-dissociation inhibitor alpha is associated with tumor progression and poor prognosis of colorectal cancer. J Proteome Res. 2008;7:3994-4003
13. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402-8
14. Wang B, Li W, Liu H, Yang L, Liao Q, Cui S. et al. miR-29b suppresses tumor growth and metastasis in colorectal cancer via downregulating Tiam1 expression and inhibiting epithelial-mesenchymal transition. Cell death & disease. 2014;5:e1335
15. Xu L, Zhang Y, Wang H, Zhang G, Ding Y, Zhao L. Tumor suppressor miR-1 restrains epithelial-mesenchymal transition and metastasis of colorectal carcinoma via the MAPK and PI3K/AKT pathway. J Transl Med. 2014;12:244
16. Wang H, An H, Wang B, Liao Q, Li W, Jin X. et al. miR-133a represses tumour growth and metastasis in colorectal cancer by targeting LIM and SH3 protein 1 and inhibiting the MAPK pathway. Eur J Cancer. 2013;49:3924-35
17. Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121:11-8
18. Li Y, Friedmann DR, Mhatre AN, Lalwani AK. MYH9-siRNA and MYH9 mutant alleles: Expression in cultured cell lines and their effects upon cell structure and function. Cell motility and the cytoskeleton. 2008;65:393-405
19. Hirata N, Takahashi M, Yazawa M. Diphosphorylation of regulatory light chain of myosin IIA is responsible for proper cell spreading. Biochem Bioph Res Co. 2009;381:682-7
20. Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends in cell biology. 2005;15:371-7
21. Totsukawa G, Wu Y, Sasaki Y, Hartshorne DJ, Yamakita Y, Yamashiro S. et al. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J Cell Biol. 2004;164:427-39
22. Nishikawa M, Sellers JR, Adelstein RS, Hidaka H. Protein kinase C modulates in vitro phosphorylation of the smooth muscle heavy meromyosin by myosin light chain kinase. J Biol Chem. 1984;259:8808-14
23. Li ZH, Bresnick AR. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer research. 2006;66:5173-80
24. Wang H, Shi J, Luo Y, Liao Q, Niu Y, Zhang F. et al. LIM and SH3 protein 1 induces TGFbeta-mediated epithelial-mesenchymal transition in human colorectal cancer by regulating S100A4 expression. Clin Cancer Res. 2014;20:5835-47
25. Sabbir MG, Dillon R, Mowat MR. Dlc1 interaction with non-muscle myosin heavy chain II-A (Myh9) and Rac1 activation. Biol Open. 2016;5:452-60
26. Xia N, Thodeti CK, Hunt TP, Xu QB, Ho M, Whitesides GM. et al. Directional control of cell motility through focal adhesion positioning and spatial control of Rac activation. Faseb Journal. 2008;22:1649-59
27. Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257-68
28. Hosono Y, Usukura J, Yamaguchi T, Yanagisawa K, Suzuki M, Takahashi T. MYBPH inhibits NM IIA assembly via direct interaction with NMHC IIA and reduces cell motility. Biochem Biophys Res Commun. 2012;428:173-8
29. Chiu HC, Chang TY, Huang CT, Chao YS, Hsu JT. EGFR and myosin II inhibitors cooperate to suppress EGFR-T790M-mutant NSCLC cells. Mol Oncol. 2012;6:299-310
30. Feitelson MA, Arzumanyan A, Kulathinal RJ, Blain SW, Holcombe RF, Mahajna J. et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin Cancer Biol. 2015;35:S25-S54
31. Wells A, Grahovac J, Wheeler S, Ma B, Lauffenburger D. Targeting tumor cell motility as a strategy against invasion and metastasis. Trends Pharmacol Sci. 2013;34:283-9
32. Coaxum. SD, Tiedeken. J, Garrett-Mayer. E, Myers. J, Rosenzweig. SA, Neskey. DM. The tumor suppressor capability of p53 is dependent on non- muscle myosin IIA function in head and neck cancer. Oncotarget. 2017;8:22991-3007
33. Liang S, He L, Zhao X, Miao Y, Gu Y, Guo C. et al. MicroRNA let-7f inhibits tumor invasion and metastasis by targeting MYH9 in human gastric cancer. PLoS One. 2011;6:e18409
34. Zhao LA, Wang H, Liu C, Liu YW, Wang XY, Wang SA. et al. Promotion of colorectal cancer growth and metastasis by the LIM and SH3 domain protein 1. Gut. 2010;59:1226-35
35. Wang H, An HY, Wang B, Liao Q, Li WD, Jin XJ. et al. miR-133a represses tumour growth and metastasis in colorectal cancer by targeting LIM and SH3 protein 1 and inhibiting the MAPK pathway. European journal of cancer. 2013;49:3924-35
Author contact
![]() Corresponding authors: Liang Zhao, MD, PhD. Department of Pathology, Nanfang Hospital, Southern Medical University, Guangzhou, China. Tel./fax: +86 2062789365. E-mail address: liangsmucom. Guoxin Li, MD, PhD, Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangdong provincial Engineering Technology Research Center of Minimally Invasive Surgery, Guangzhou, China. Tel: +86 2061641681; fax: +86 2061641683, E-mail: gzliguoxincom.
Corresponding authors: Liang Zhao, MD, PhD. Department of Pathology, Nanfang Hospital, Southern Medical University, Guangzhou, China. Tel./fax: +86 2062789365. E-mail address: liangsmucom. Guoxin Li, MD, PhD, Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangdong provincial Engineering Technology Research Center of Minimally Invasive Surgery, Guangzhou, China. Tel: +86 2061641681; fax: +86 2061641683, E-mail: gzliguoxincom.