Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(1):138-146. doi:10.7150/jca.28500 This issue Cite
Research Paper
Crosstalk between Fisetin-induced Apoptosis and Autophagy in Human Oral Squamous Cell Carcinoma
Bong-Soo Park1,3,4*, Nak-Eun Choi1*, Ji Hye Lee2,3,4, Hae-Mi Kang1,3, Su-Bin Yu1, Hye-Jin Kim5, Hyun-Kyung Kang6, In-Ryoung Kim1,4 ![]()
1. Department of Oral Anatomy, School of Dentistry, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea
2. Department of Oral Pathology, School of Dentistry, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea
3. BK21 PLUS Project, School of Dentistry, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea
4. Institute of Translational Dental Sciences, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea
5. Department of Dental Hygiene, Dongeui University, Gaya 1-dong, Busanjin-gu, Busan, 47230, South Korea
6. Department of Dental Hygiene, Silla University, 140 Baekyang-daero 700 beon-gil, Busan 46958, South Korea
* Both the authors contributed equally to this study.
Received 2018-7-13; Accepted 2018-10-27; Published 2019-1-1
Abstract
Fisetin (3,3-,4-,7-tetrahydroxyflavone), a naturally occurring flavonoid, has antioxidant, anti-inflammatory, and anticancer effects. Oral squamous cell carcinoma (OSCC) has a 5-year survival rate lower than that of most other carcinomas, and can create functional and aesthetic problems for the patient. New therapies for OSCC are necessary, and treatment using plant-derived natural substances has recently become a trend. It has been suggested that autophagy may play an important role in cancer therapy. Several studies demonstrated that autophagy inhibition enhances apoptotic cell death. Therefore, autophagy inhibition might be a promising therapeutic method against OSCC. Our results showed that fisetin induced apoptotic cell death in human tongue squamous cell line Ca9-22 could be enhanced by inhibition of autophagy. Thus, autophagy process in fisetin treated OSCC might presumed to play a role of pro-survival. The combination of fisetin and an effective autophagy inhibitor could be a potentially adjuvant and useful treatment for oral cancer.
Keywords: fisetin, oral squamous cell carcinoma, apoptosis, autophagy
Introduction
Head and neck malignancies constitute approximately 5% of human malignancies [1]. Almost 95% of head and neck cancers are oral squamous cell carcinoma (OSCC), affecting nearly 500,000 individuals each year [2, 3]. The classical treatments for OSCC are surgery, radiotherapy, and/or chemotherapy [4]. The clinical characteristics of OSCC, including carcinogenesis, development, progression, invasion, and metastasis, have not yet been elucidated [5], and this malignancy is difficult to cure despite aggressive therapies [6]. Therefore, new therapies against OSCC are necessary, and treatment using plant-derived natural substances has recently become a trend.
Fisetin (3,3-,4-,7-tetrahydroxyflavone) is a naturally occurring flavonoid commonly found in fruits and vegetables, such as apples, grapes, strawberries, cucumbers, and onions [7, 8]. Fisetin has antioxidative and anti-inflammatory effects [9]. Recently, its anticancer potential has been explored, making it a promising agent for prevention and therapy of various cancers, including prostate, colon, and breast [10-12]. Fisetin interacts with the cell by binding to and interacting with various molecular targets; for example, it disrupts Wnt, mTOR, and NF-κB signaling, resulting in cell-cycle arrest and preventing invasion and migration of cancer cells [13].
The three main features of programmed cell death (PCD) are apoptosis, autophagy, and programmed necrosis, which are easily distinguished by their morphological differences. PCD affects the balance between cell survival and death, and plays a key role in the ultimate outcome of cancer [14]. Type I PCD, apoptosis, is characterized by specific morphological changes (cell shrinkage, nuclear condensation and fragmentation, and dynamic membrane blebbing) [15] and biochemical changes (chromosomal DNA cleavage into internucleosomal fragments, phosphatidylserine externalization, and intracellular substrate cleavage by specific proteolysis) [16]. Type II PCD, autophagy, is a cellular homeostatic, catabolic degradation response. It begins when double-membrane vesicles form and engulf proteins, cytoplasm, protein aggregates, and organelles, which are then delivered to lysosomes, where they are degraded to serve as alternate energy sources [17, 18]. Autophagy allows prolonged survival of tumor cells, with consequential defects in apoptosis [19]. Recent evidence shows that inhibition of autophagy restores chemosensitivity and enhances tumor cell death [20]. Therefore, the inhibition of autophagy by anticancer reagents has been recognized as an important component of cancer therapy [21, 22].
Fisetin has been examined in the context of cancer treatment and has been shown to induce apoptosis in cancer cells. However, no reports have yet examined the effects of fisetin on autophagy in a human OSCC cell lines. The present study was conducted to investigate whether fisetin can induce autophagy in OSCC cells, and to determine its underlying molecular mechanisms.
Materials and Methods
Chemicals
Dulbecco's Modified Eagle's Medium: Nutrient Mixture F-12 (DMEM/F-12), fetal bovine serum (FBS), and trypsin-EDTA were purchased from GE Healthcare Life Sciences (Hyclone, Logan, UT, USA). Fisetin, dimethyl sulfoxide (DMSO), methylthiazolyldiphenyl-tetrazolium bromide (MTT), acridine orange (AO) staining solution, and 3-methyladenine (3-MA) were purchased from Sigma (St. Louis, MO, USA). RIPA Lysis and Extraction Buffer was purchased from Thermo Fisher Scientific (San Jose, CA, USA). Bradford protein assay was purchased from Bio-Rad (Richmond, CA, USA). Polyvinylidene fluoride (PVDF) membranes were purchased from Millipore (Billerica, MA, USA). SuperSignal West Femto was purchased from Pierce (Rockford, IL, USA). All other chemicals and reagents were purchased from Sigma unless otherwise specified.
Cell culture
CAL-27 and Ca9-22 human oral squamous carcinoma cell lines were purchased from ATCC (Rockville, MD, USA). Cells were maintained in DMEM/ F-12 with 1% penicillin streptomycin and 10% FBS at 37°C with 5% CO2 in a CO2 incubator.
Fisetin treatment
Fisetin (100 mM) stock solution was made by dissolving it in DMSO and was then kept frozen at -20°C until use. Various concentrations (10~200 µM) of fisetin were applied when cells had grown to 80~90% confluence, for 24-72 h. Cells grown in medium containing an equivalent amount of DMSO without fisetin served as controls.
MTT assay
CAL-27 and Ca9-22 cells (1.5×104) were seeded in 96-well plates, incubated for 24 h, then treated with fisetin (25-200 μM) for 24-72 h. After fisetin treatment, all cells were treated with 500 μg/ml of MTT solution, then incubated at 37°C with 5% CO2 for 4 h in the dark. The supernatant was aspirated and the formed formazan crystals were dissolved in DMSO. Absorbance was measured with an ELISA reader (Tecan, Männedorf, Switzerland) at the 620-nm excitatory emission wavelength.
Hoechst staining
CAL-27 and Ca9-22 cells were seeded on Falcon® 8-Chambered Cell Culture Slides and incubated for 24 h. After fisetin treatment, cells were fixed in 4% paraformaldehyde (PFA) for 15 min, stained in 1 μg/ml Hoechst 33342 and 1 μg/ml rhodamin phalloidin for 15 min at room temperature, washed three times in PBS, and mounted with 50% glycerol. All samples were observed and photographed under an epifluorescence microscope (Carl Zeiss, Göettingen, Germany).
Detection of mitochondrial membrane potential (MMP)
CAL-27 and Ca9-22 (5×105) cells were placed on 6-well plates, then 50, 100, 150 μM fisetins were treated for 24 h. Then cells were suspended using trypsin-EDTA, washed with PBS, and stained by JC-1 (1 μg/ml) dye for 30 min at CO2 incubator. MMP was measured by CYTOMICS FC500 flow cytometry system (Beckman Coulter). Data were acquired and analyzed using CXP software, version 2.2
Detection of acidic vesicular organelles (AVOs)
To determine the autophagy ratio induced by fisetin in oral cancer cells, fisetin-treated cells were suspended from the culture plate with trypsin-EDTA, stained with AO (1 μg/mL) for 5 min, and analyzed using a Beckman Coulter FC500 Flow Cytometer (Indianapolis, IN, USA). Cells were measured using the 488-nm excitation detector (green fluorescence/ FL1) and the 540-nm emission detector (red fluorescence/FL3), which quantified the development of AVOs. Cellular fluorescence changes were observed using a confocal microscope (LSM 700; Carl Zeiss, Germany). Cells were stained with 1 μg/mL AO for 5 min and washed with PBS.
Western blot analysis
Cells (2×106) were harvested and washed twice in ice-cold PBS, then lysed in 100 μl of RIPA Lysis and Extraction Buffer at 4°C for 60 min. The lysates were centrifuged (13,000 rpm) and acquired total protein. Protein concentration was determined with Bradford protein assay, and 20 μg of proteins were loaded onto 10% SDS/PAGE gel using an electrophoresis gel-running system. The gels were transferred to PVDF membranes using a trans-blot system. Blot membranes were blocked with 5% skim milk for 1 h, reacted with appropriate primary antibodies overnight, and washed 5 times in PBS. Secondary antibodies were reacted for 1 h and the blot was detected using SuperSignal West Femto enhanced chemiluminescence substrate and the Alpha Imager HP (Alpha Innotech, Santa Clara, CA, USA). Primary and secondary antibodies were used as below; antibodies against the cleaved form of caspase-3, Beclin-1, and PARP were purchased from Cell Signaling Technology (Beverly, MA, USA). LC3 and p62/SQSTM1 (Sigma) were also used. ATG5 and mouse anti-β-actin antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), as were mouse anti-rabbit IgG and rabbit anti-mouse IgG antibodies.
Statistical Analysis
Data were expressed ± the standard deviation (SD) from at least three independent experiments. Statistical analyses used GraphPad Prism version 5.0 for Windows (GraphPad Software, San Diego, CA). A one-way ANOVA was used for Dunnett's multiple- comparison test in the statistical analysis.
Results
Fisetin induces apoptosis via the mitochondrial pathway
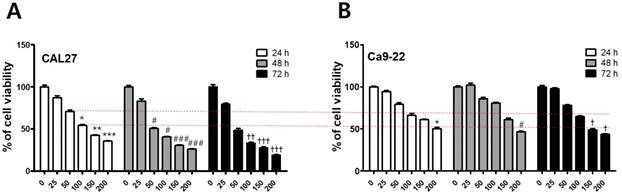
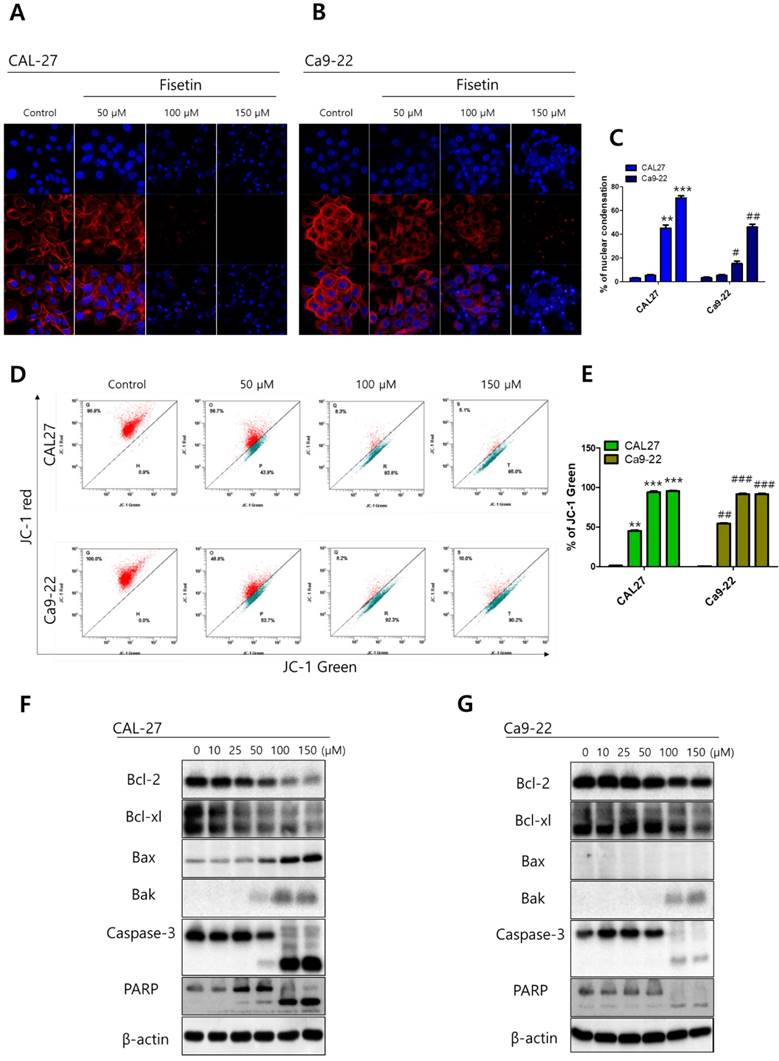
To investigate the cytotoxicity of fisetin in CAL-27 and Ca9-22 OSCC cells, we conducted MTT assays. Each cell line was treated with fisetin concentrations of 0-300 μM for 24-72 h. Fisetin reduced cell viability in a dose- and time-dependent manner in both cell lines. Notably, the Ca9-22 cells were relatively resistant to the same doses compared to the CAL-27 cells (Figure 1). To determine whether fisetin treatment induces apoptosis accompanied by morphological and biochemical changes, as described below, we measured nuclear morphology, mitochondrial membrane potential (MMP), and changes in apoptosis-related proteins. The control cells of the CAL -27 and Ca9-22 lines showed typical round nuclei, whereas both types of cells treated with <100 μM of fisetin displayed condensed and fragmented nuclei (Figure 2A and 2B) and an increased nuclear condensation ratio (Figure 2C). To identify fisetin-induced cell death through the mitochondrial pathway of apoptosis, we examined the MMP and apoptosis-related proteins in both OSCC cell lines. As shown in Figure 2D and 2E, fisetin dramatically reduced the MMP and the proapoptotic members Bax and Bak. It also caused accumulation of the antiapoptotic members Bcl-2 and Bcl-XL. The MMP involves an initial release of cytochrome c, which activates the caspase cascade, resulting in DNA fragmentation [23]. Fisetin activated caspase-3 and PARP, and produced a processed fragment dose-dependently in both cell lines (Figure 2F and 2G). It is noteworthy that the CAL-27 cells showed a more sensitive response to fisetin treatment, compared to the Ca9-22 cells.
Fisetin treatment reduced the viability of OSCC cells. (A and B) Fisetin (25-200 μM) was used on CAL-27 and Ca9-22 OSCC cell lines for 24-72 h, and cell viability was analyzed using the MTT assay. Data were represent the mean ± SD (n=6) of three independent experiments performed in triplicate and analyzed by a one-way ANOVA using Dunnett's multiple-comparison test (24 h, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; 48 h, #p < 0.05, ##p < 0.01, and ###p < 0.001; 72 h, †p < 0.05, ††p < 0.01, and †††p < 0.001 compared with control group).
Fisetin induced mitochondrial apoptosis in Ca9-22 cells. Cells were treated with fisetin (10-150 μM) for 24-72 h. (A and B) Nuclear morphology of CAL-27 and Ca9-22 cells. (C) Ratio of nuclear condensation in both types of fisetin-treated cells. (D) MMP was measured in JC-1 stained cells with flow cytometry and (E) the JC aggregation (red) ratio was constructed graphically. (F and G) Bcl-2, Bcl-XL, Bak, Bax, caspase-3, and PARP were measured with western blot. Data were represent the mean ± SD (n=3) of three independent experiments performed in triplicate and analyzed by a one-way ANOVA using Dunnett's multiple-comparison test (CAL-27 cells, *p<0.05, **p<0.01 and ***p<0.01; Ca9-22 cells, #p<0.05, ##p<0.01 and ###p<0.01 compared with the control group).
Fisetin treatment caused autophagy in Ca9-22 cells
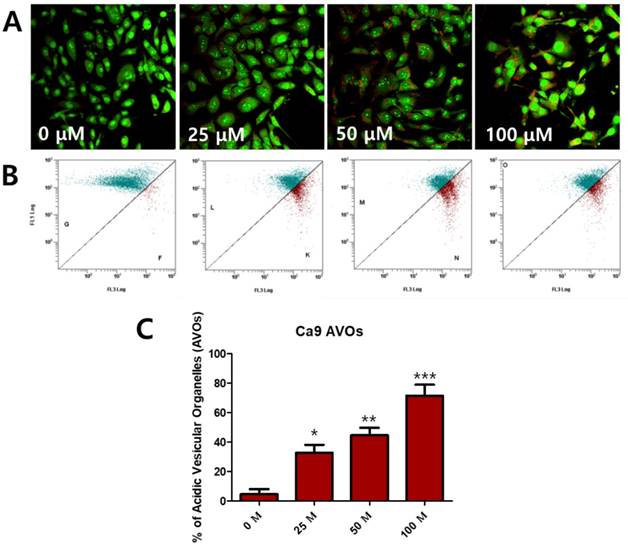
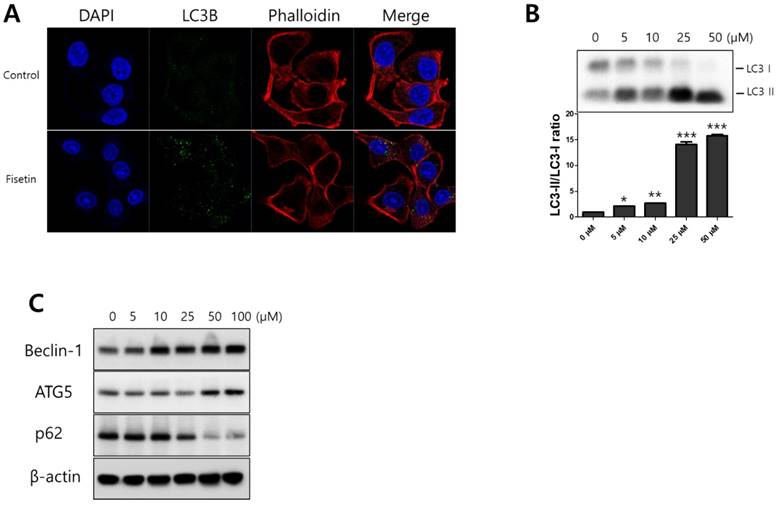
To determine whether fisetin treatment caused autophagy in Ca9-22 cells, we investigated AVO formation, which is characterized by autophagic vacuoles. AVOs were apparent in the Ca9-22 cells (red fluorescence), as detected and measured by vital staining with AO. As shown in Figure 3A, AVO development was observed following treatment with fisetin in a dose-dependent manner. To quantify the AVOs in fisetin-treated Ca9-22 cells, we performed flow cytometry. Fisetin treatment increased the ratio of AVOs with prominent red fluorescence from 3.75% to 78.42% in the Ca9-22 cells (Figure 3B and 3C). These findings indicate that fisetin treatment induces development of autophagic vacuoles in OSCC cells. Fisetin-induced autophagy in Ca9-22 cells was observed via various autophagy markers, such as LC3, Beclin-1, p62/SQSTM1, and ATG5. Fisetin treatment produced LC3B in the cytosol (Figure 4A), converted LC3-I to LC3-II, and caused accumulation of Beclin-1 and ATG5 dose-dependently in the Ca9-22 cells (Figure 4B); however, p62/SQSTM1 was decreased.
Fisetin treatment caused AVO accumulation in Ca9-22 cells. Cells were stained with acridine orange (AO) and detected with fluorescence microscopy and flow cytometry. (A) Fisetin treatment induced AVOs in cytosol and (B and C) increased the ratio of AVOs. Data were represent the mean ± SD (n=3) of three independent experiments performed in triplicate and analyzed by a one-way ANOVA using Dunnett's multiple-comparison test (*p<0.05, **p<0.01 and ***p<0.01 compared with the control group).
Fisetin treatment induced autophagy in Ca9-22 cells. Cells were treated with various concentrations of fisetin (5-100 μM) for 24 h. (A) Fisetin treatment initiated LC3B formation in cytosol and (B and C) increased the expression levels of autophagy-related proteins, such as Beclin-1, p62/SQSTM1, ATG5-ATG12, and LC3, which is evidence of autophagy. Data were represent the mean ± SD (n=3) of three independent experiments performed in triplicate and analyzed by a one-way ANOVA using Dunnett's multiple-comparison test (*p<0.05, **p<0.01 and ***p<0.01 compared with the control group).
Inhibition of autophagy enhances fisetin-induced apoptosis
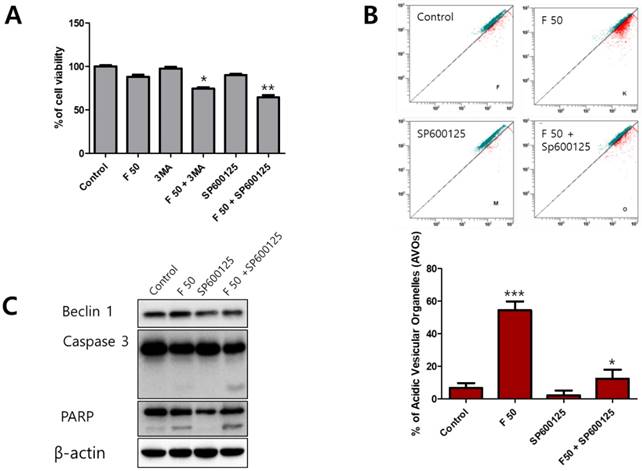
Autophagy is a complicated process that controls the survival or death of cancer cells [24]. To determine the role of autophagy under fisetin treatment, an autophagy inhibitor, 3-methyladenine (3-MA) and a Beclin-1 inhibitor, SP600125, were used as a combination treatment on fisetin-treated Ca9-22 cells. When co-treated with the inhibitors and fisetin, cell viability showed a much greater decrease than in the fisetin-only Ca9-22 cells. In particular, cells co-treated with fisetin and SP600125 showed very low viability (Figure 5A). We also found that AVO accumulation in Ca9-22 cells after SP600125 and fisetin co-treatment was lower than with fisetin-only treatment (Figure 5B). Next, to determine whether blocking of Beclin-1 using SP600125 could promote apoptosis, apoptosis-related proteins were identified with western blot. As shown in Figure 5C, SP600125 led to decreased expression of Beclin-1, cleaved caspase 3, and cleaved PARP compared with fisetin treatment alone. The inhibition of the autophagic process with 3-MA and SP600125 resulted in augmentation of fisetin's cytotoxic activity.
Blocking of autophagy using 3-MA and SP600125 enhanced apoptosis in Ca9-22 cells. Cells were pretreated with 1 mM 3-MA or 10 μM SP600125 for 1 h, then exposed to 50 μM fisetin for 24 h. Cells were analyzed using (A) MTT, (B) AO staining, and (C) western blot. Data were represent the mean ± SD (n=3) of three independent experiments performed in triplicate and analyzed by a one-way ANOVA using Dunnett's multiple-comparison test (*p<0.05, **p<0.01 and ***p<0.01 compared with the control group).
Discussion
In recent years, it has been reported that survival rates for OSCC are lower than for most other carcinomas, and are not substantially improving. Surgery, radiation therapy, and chemotherapy have been proposed as the main treatment modalities for OSCC, but patients experience functional and aesthetic problems and the 5-year survival rate remains low [25]. Hence, it is of great importance to find novel systemic therapies for OSCC. Natural substances from food, plants, and traditional medicine have been widely investigated as powerful therapeutic agents for metabolic disorders, cardiovascular diseases, musculoskeletal diseases, and cancer [26-29]. Many individuals use natural-substance therapies for cancer, which might improve survival [30]. Autophagy is widely known as an important process in cell physiology, involved in both cell survival and death [31]. Many studies have shed light on the importance of autophagy in cancer, but it is still unclear whether autophagy suppresses tumorigenesis or provides cancer cells with a rescue mechanism under unfavorable conditions [32]. Autophagy functions as a tumor suppressor in the early stages of tumor development [33]. It protects some cancer cells against anticancer treatments by blocking apoptosis [34], but induces autophagic cell death in other cancer cells after cancer treatment [35, 36]. Therefore, a double-edged sword of autophagy phenomenon could be considered very important that the anticancer drugs or therapeutic supplements from natural substance involves the pro-survival or pro-death effects. This study demonstrated that fisetin, a natural flavonoid, induced apoptosis and autophagy in OSCC cells, but autophagy induction, which had pro-survival effects, inhibited fisetin's anticancer properties.
Fisetin is the dietary flavonoid and exhibits antioxidant, anti-inflammatory, anti-cancer and neuroprotective activities [37]. Recent studies demonstrated that fisetin induces apoptotic cell death through the endoplasmic reticulum stress in human melanoma [38] and mitochondria-dependent apoptotic pathway human oral cancer cells [39]. Apoptosis is a morphologically, chemically, and pathophysiologically distinct from necrosis, and the development of cancer treatment can be promoted through understanding the mechanism of apoptosis [40]. Specific morphological changes of apoptosis are nuclear condentation, cell volume reduction and DNA fragmentation [41]. In the present study, nuclear condensation and fragmentation of OSCC cells (CAL-27 and Ca9-22) by fisetin treatments were observed by Hoechst staining. The mechanism of apoptosis involves a wide variety of molecular events which have two main apoptotic pathways as extrinsic (death receptor) and intrinsic (mitochondrial) pathway [42]. One of the most important regulators of intrinsic pathway is Bcl-2 family [43]. Bcl-2 family includes pro-apoptotic members as Bax, Bak and Bad, and anti-apoptotic members as Bcl-2, Bcl-XL and Bfl-1 [44]. When the cells are undergoing apoptosis, mitochondrial membrane permeability is increased by inhibition of anti-apoptotic proteins and activation of pro-apoptotic, and then the cytochrome c release into the cytosol from mitochondria [45]. We found that fisetin treatment activates Bax and Bak, inhibits Bcl-2 and Bcl-XL, and up-regulates mitochondrial membrane permeability in OSCC cells. In cytosol, released cytochrome c forms a complex with Apaf-1 and caspase-9 to produce apoptosome, which subsequently triggering a cascade of caspase activation [46]. Activated caspase induces to cleavage of cellular proteins as poly-(ADP-ribose) polymerase (PARP) which protects the genome by functioning in the DNA damage [47]. We clearly found that fisetin treatment formed activated and cleaved caspase-3 and PARP in OSCC cells. These findings suggest that fisetin treatment induces apoptosis via intrinsic pathway and caspase activation, especially, CAL-27 cells showed a more sensitive response to fisetin treatment, compared to the Ca9-22 cells. We hypothesized that the low sensitivity of Ca9-22 cells to apoptosis corresponds to the pro-survival signal of autophagy by fisetin treatment. Autophagy is characterized by AVO formation, which can be detected and measured by vital staining of acridine orange. AO moves freely across biological membranes and accumulates in acidic compartments, where it is seen as red fluorescence [48]. We confirmed the autophagy process in fisetin-treated Ca9-22 cells and found that fisetin caused the formation and accumulation of AVOs in these cells.
We also confirmed the protein expression of p62/SQSTM1, LC3, ATG5-ATG12 complex, and Beclin-1, which are associated with autophagy in fisetin- treated Ca9-22 cells. Rat microtubule‐associated protein 1 light chain 3 (LC3) plays a critical role in the formation of autophagosomes [49]. It has two forms, LC3‐I and LC3‐II, when newly synthesized. This is followed by the conversion of a fraction of LC3‐I into LC3‐II, which is correlated with the extent of autophagosome formation [50]. In this study, fisetin directly affected the conversion of a fraction of LC3‐I into LC3‐II. p62/SQSTM1 is commonly found in inclusion bodies, polyubiquitinated and degraded by autophagy [51]. Beclin-1 plays a critical role in the two fundamentally important cell biological pathways of autophagy and apoptosis [52]. It forms a core complex with pre-autophagosomal structures (PAS) and class III type phosphoinositide 3-kinase (PI3KC3)/Vps34, which regulates the autophagy and membrane- trafficking involved in several physiological and pathological processes [53]. ATG12-ATG5 conjugation systems are necessary for the formation of autophagosomes [54]. Our results showed that there was degradation of p62/SQSTM1 and of the expression of Beclin-1 and ATG5 proteins.
Autophagy can promote cell survival via cancer therapeutic reagents [55], and several studies have demonstrated that autophagy inhibition enhances apoptotic cell death in various cancer cells [55, 56], and suggested that combination treatment with autophagy inhibition agents are useful methods to cancer treatment [57]. Therefore, autophagy inhibition might be a promising therapeutic target for cancer treatment. Very recently, Kim et al demonstrated that 3-methyladeine (3-MA) and 740Y.P impeding on autophagy accelerated an apoptotic response caused by natural compound Deoxypodophyllotoxin. They were showed that the 3-MA and 740Y.P markedly increased apoptosis-positive cells compared with Deoxypodophyllotoxin alone, supported by significantly increased caspase-3 and PARP cleavage [58]. In present study, we were to identify whether autophagy inhibition can enhance apoptotic cell death with fisetin treatment, we used the well-known autophagy inhibitors 3-MA and SP600125, espescially, SP600125 was found to enhance the evidence of apoptotic cell death by cleaving caspase-3 and PARP, and to reduce the evidence of autophagy by Beclin-1 expression compared with fisetin alone. These results strongly suggest that fisetin-induced autophagy protects Ca9-22 cells as a survival signal, and the appropriate inhibition of autophagy is to increase the sensitivity of fisetin-induced apopotsis in Ca9-22 cells. This study clearly demonstrates that fisetin can induce autophagy and is involved in the survival of Ca9-22 cells, and that the combination of fisetin and an effective autophagy inhibitor could be a potentially useful therapy for oral cancer. We believe that the strategy of combining fisetin with autophagy inhibitors will open up new avenues for clinically applicable combination therapies in treating OSCC.
Abbreviations
AO: acridine orange; OSCC: AVOs: acidic vesicular organelles; DMSO: dimethyl sulfoxide; FBS: fetal bovine serum; LC3: microtubule‐associated protein 1 light chain 3; MMP: mitochondrial membrane potential; MTT: methylthiazolyldiphenyl-tetrazolium bromide; OSCC: oral squamous cell carcinoma; PAS: pre-autophagosomal structures; PVDF: Polyvinylidene fluoride, PCD: programmed cell death; 3-MA: 3-methyladenine.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (No. NRF-2017R1C1B5018034).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics, 1996. CA: A cancer journal for clinicians. 1996;46:5-27
2. Laramore G, Scott C, Al-Sarraf M, Haselow R, Ervin T, Wheeler R. et al. Adjuvant chemotherapy for resectable squamous cell carcinomas of the head and neck: report of intergroup study 0034. International Journal of Radiation Oncology* Biology* Physics. 1992;23:705-13
3. Casiglia J, Woo S. A comprehensive review of oral cancer. General dentistry. 2001;49:72-82
4. Bell RB, Kademani D, Homer L, Dierks EJ, Potter BE. Tongue cancer: Is there a difference in survival compared with other subsites in the oral cavity? J Oral Maxil Surg. 2007;65:229-36
5. Lo WY, Tsai MH, Tsai Y, Hua CH, Tsai FJ, Huang SY. et al. Identification of over-expressed proteins in oral squamous cell carcinoma (OSCC) patients by clinical proteomic analysis. Clin Chim Acta. 2007;376:101-7
6. Shen J, Huang CH, Jiang L, Gao F, Wang Z, Zhang YY. et al. Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochem Pharmacol. 2007;73:1901-9
7. Arai Y, Watanabe S, Kimira M, Shimoi K, Mochizuki R, Kinae N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. The Journal of nutrition. 2000;130:2243-50
8. Higa S, Hirano T, Kotani M, Matsumoto M, Fujita A, Suemura M. et al. Fisetin, a flavonol, inhibits T H 2-type cytokine production by activated human basophils. Journal of Allergy and Clinical Immunology. 2003;111:1299-306
9. Suh Y, Afaq F, Johnson JJ, Mukhtar H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis. 2009;30:300-7
10. Chen Y-C, Shen S-C, Lee W-R, Lin H-Y, Ko C-H, Shih C-M. et al. Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Archives of toxicology. 2002;76:351-9
11. Khan N, Afaq F, Syed DN, Mukhtar H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis. 2008;29:1049-56
12. Yang P-M, Tseng H-H, Peng C-W, Chen W-S, Chiu S-J. Dietary flavonoid fisetin targets caspase-3-deficient human breast cancer MCF-7 cells by induction of caspase-7-associated apoptosis and inhibition of autophagy. International journal of oncology. 2012;40:469-78
13. Lall RK, Adhami VM, Mukhtar H. Dietary flavonoid fisetin for cancer prevention and treatment. Mol Nutr Food Res. 2016;60:1396-405
14. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B. et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487-98
15. Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circulation research. 2008;103:343-51
16. Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349-52
17. Liu J-j, Lin M, Yu J-y, Liu B, Bao J-k. Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer letters. 2011;300:105-14
18. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
19. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature Reviews Cancer. 2007;7:961-7
20. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533-41
21. Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Annals of surgical oncology. 2009;16:761-71
22. Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Medical oncology. 2011;28:105-11
23. Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol. 2004;16:647-52
24. Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer treatment development. Cancer Chemotherapy and Pharmacology. 2015;75:439-47
25. Freier K, Engel M, Lindel K, Flechtenmacher C, Mühling J, Hassfeld S. et al. Neoadjuvant concurrent radiochemotherapy followed by surgery in advanced oral squamous cell carcinoma (OSCC): A retrospective analysis of 207 patients. Oral Oncology. 2008;44:116-23
26. Jaiswal P, Kumar P, Singh V, Singh D. Areca catechu L.: A valuable herbal medicine against different health problems. Res J Med Plant. 2011;5:145-52
27. Olaku O, White JD. Herbal therapy use by cancer patients: a literature review on case reports. European journal of cancer. 2011;47:508-14
28. Bent S. Herbal medicine in the United States: review of efficacy, safety, and regulation. Journal of general internal medicine. 2008;23:854-9
29. Bnouham M, Ziyyat A, Mekhfi H, Tahri A, Legssyer A. Medicinal plants with potential antidiabetic activity-A review of ten years of herbal medicine research (1990-2000). International Journal of Diabetes and Metabolism. 2006;14:1
30. Chen Z, Gu K, Zheng Y, Zheng W, Lu W, Shu XO. The use of complementary and alternative medicine among Chinese women with breast cancer. The Journal of Alternative and Complementary Medicine. 2008;14:1049-55
31. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T. et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237-48
32. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature Reviews Cancer. 2005;5:726
33. Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nature Reviews Microbiology. 2004;2:301-14
34. Boya P, González-Polo R-A, Casares N, Perfettini J-L, Dessen P, Larochette N. et al. Inhibition of macroautophagy triggers apoptosis. Molecular and cellular biology. 2005;25:1025-40
35. Ellington AA, Berhow M, Singletary KW. Induction of macroautophagy in human colon cancer cells by soybean B-group triterpenoid saponins. Carcinogenesis. 2005;26:159-67
36. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB. et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer research. 2005;65:3336-46
37. Yen JH, Wu PS, Chen SF, Wu MJ. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int J Mol Sci. 2017:18
38. Syed DN, Lall RK, Chamcheu JC, Haidar O, Mukhtar H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Archives of biochemistry and biophysics. 2014;563:108-17
39. SHIH Y-L, HUNG F-M, LEE C-H, YEH M-Y, LEE M-H, LU H-F. et al. Fisetin Induces Apoptosis of HSC3 Human Oral Cancer Cells Through Endoplasmic Reticulum Stress and Dysfunction of Mitochondria-mediated Signaling Pathways. In Vivo. 2017;31:1103-14
40. Bold RJ, Termuhlen PM, McConkey DJ. Apoptosis, cancer and cancer therapy. Surgical Oncology. 1997;6:133-42
41. Cohen G, Sun X, Snowden R, Dinsdale D, Skilleter D. Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochemical Journal. 1992;286:331-4
42. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicologic Pathology. 2007;35:495-516
43. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA: a cancer journal for clinicians. 2005;55:178-94
44. Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1-6
45. Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clinical cancer research. 2009;15:1126-32
46. Kim H-E, Jiang X, Du F, Wang X. PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Molecular cell. 2008;30:239-47
47. Yu S-W, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ. et al. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science. 2002;297:259-63
48. Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of Autophagic Cell Death in Malignant Glioma Cells by Arsenic Trioxide. Cancer Research. 2003;63:2103-8
49. Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T. et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. The Journal of cell biology. 1999;147:435-46
50. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO Journal. 2000;19:5720-8
51. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. Journal of Biological Chemistry. 2007;282:24131-45
52. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proceedings of the National Academy of Sciences. 2003;100:15077-82
53. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death And Differentiation. 2011;18:571
54. Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14050-5
55. Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI. et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. Journal of Clinical Investigation. 2007;117:326-36
56. Xi G, Hu X, Wu B, Jiang H, Young CY, Pang Y. et al. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer letters. 2011;307:141-8
57. Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Current opinion in investigational drugs (London, England: 2000). 2009;10:1269-79
58. Kim SH, Son KM, Kim KY, Yu SN, Park SG, Kim YW. et al. Deoxypodophyllotoxin induces cytoprotective autophagy against apoptosis via inhibition of PI3K/AKT/mTOR pathway in osteosarcoma U2OS cells. Pharmacological reports: PR. 2017;69:878-84
Author contact
![]() Corresponding author: In-Ryoung Kim, Professor, Department of Oral Anatomy, School of Dentistry, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea Tel: +82515108552, Fax: +82515108241, E-mail: biowoolac.kr
Corresponding author: In-Ryoung Kim, Professor, Department of Oral Anatomy, School of Dentistry, Pusan National University, Busandaehak-ro, 49, Mulguem-eup, Yangsan-si, Gyeongsangnam-do, 50612, South Korea Tel: +82515108552, Fax: +82515108241, E-mail: biowoolac.kr