Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(1):105-111. doi:10.7150/jca.27633 This issue Cite
Review
Tuning the Protein Phosphorylation by Receptor Type Protein Tyrosine Phosphatase Epsilon (PTPRE) in Normal and Cancer Cells
Jinping Liang1,2, Jun Shi1, Na Wang1, Hui Zhao3, Jianmin Sun1,4 ![]()
1. School of Basic Medical Sciences, Ningxia Medical University, Yinchuan, P.R.China
2. Ningxia Key laboratory of Clinical and Pathogenic Microbiology, The General Hospital of Ningxia Medical University, Yinchuan, China
3. School of Biomedical Science, Faculty of Medicine, the Chinese University of Hong Kong,
4. Division of Translational Cancer Research, Lund Stem Cell Center, Department of Laboratory Medicine, Lund University, Lund, Sweden
Received 2018-6-3; Accepted 2018-10-24; Published 2019-1-1
Abstract
Tyrosine phosphorylation is an important post-translation modification of proteins that is controlled by tyrosine kinases and phosphatases. Disruption of the balance between the activity of tyrosine kinases and phosphatases may result in diseases. Receptor type protein tyrosine phosphatase epsilon (PTPRE) is closely related with receptor type protein tyrosine phosphatase alpha (PTPRA). PTPRE has been studied in osteoclast cells, nerve cells, hematopoietic cells, cancer cells and others, and it has different functions among various tissues. In this review, we summarized the current knowledge about the regulation of PTPRE on cellular signal transduction and its function under normal and pathological conditions.
Keywords: tyrosine phosphatase, PTPRE, signaling, cancer
Introduction
Tyrosine phosphorylation is an important post-translational modification that can regulate signal transduction in cells [1]. Tyrosine kinases catalyze the transfer of a phosphate group from ATP to certain tyrosine residues in proteins leading to protein tyrosine phosphorylation. Tyrosine phosphorylation is tightly controlled in normal cells, and aberrant activation of tyrosine kinases caused by overexpression, gene rearrangement, and oncogenic mutation has been implicated in various types of cancers [2-4].
In contrary to tyrosine kinases, tyrosine phosphatases catalyze the removal of the phosphate group from tyrosine residues in their substrate proteins, resulting in protein dephosphorylation. The tyrosine kinases and tyrosine phosphatases coordinate to maintain the balance between protein phosphorylation and dephosphorylation in living cells. Tyrosine phosphatases are divided into classical tyrosine phosphatases and dual specificity phosphatases depending on their specificity. Classical tyrosine phosphatases can only catalyze the removal of the phosphate group from tyrosine residues while dual specificity phosphatases can catalyze the dephosphorylation of serine and threonine residues in addition to tyrosine residue of proteins [5]. Among classical tyrosine phosphatases, receptor type tyrosine phosphatases are a group of tyrosine phosphatases that are expressed on the cell surface. They have an extracellular domain, a transmembrane domain and an intracellular domain. Non-receptor type tyrosine phosphatases cannot be expressed on the cell surface due to the lack of extracellular domain and transmembrane domain [6, 7].
Receptor type protein tyrosine phosphatase epsilon (PTPRE) is a receptor type phosphatase that is closely related to receptor type protein tyrosine phosphatase alpha (PTPRA), they share high homology [8-10]. PTPRE was identified in 1990 by hybridization with a Drosophila phosphatase cDNA under non-stringent hybridization situation [8]. Later on, the murine PTPRE gene was mapped to chromosome 7 [11, 12] and human PTPRE gene was mapped to chromosome 10q26 [12]. By comparison of the amino acid sequence, human PTPRE shows 94% homology with the murine and rat counterparts [13].
PTPRE has been studied in osteoclast cells, nerve cells, hematopoietic cells, cancer cells and others, and its function shows a tissue specific manner. In this review, we reviewed literatures searched by using Pubmed and summarized the current knowledge about the regulation of PTPRE on cellular signal transduction and its function in various tissues under normal and pathological situations.
Expression of PTPRE
PTPRE gene has at least two promotors, and transcription of PTPRE starting from two promotors results in two transcripts [14]. The longer transcript is translated into a 100-110 KD protein that can anchor in the cell membrane with the molecule weight varying when expressed in different tissues [15], this isoform is called membrane type PTPRE (memPTPRE). The shorter transcript that lacks the coding sequence for signal sequence, extracellular domain and transmembrane domain is 1926 bp long. It is translated into a 72 KD protein [15, 16] which is mainly localized in cytosol with weak localization in cell membrane [16, 17], it is called cytosol type PTPRE (cytPTPRE) (figure 1).
Schematic structure of PTPRE isoforms.
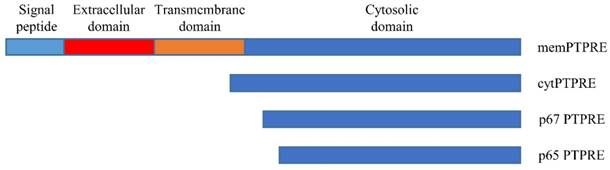
In addition to memPTPRE and cytPTPRE, two other PTPRE isoforms were also identified: p67 and p65. Unlike memPTPRE and cytPTPRE that are translated from their corresponding mRNAs respectively, translation of p67 starts from an internal ATG code at position 258 or 84 in memPTPRE or cytPTPRE mRNA respectively. In contrast to memPTPRE, cytPTPRE and p67 that are translated from mRNA, p65 is generated by proteolytic cleavage of memPTPRE, cytPTPRE and p67 [17]. Both p67 and p65 are localized in the cytosol, and they are active in the inhibition of Src-mediated phosphorylation of Kv2.1 since both of them contain the full length of phosphatase domain [17]. Among all four isoforms of PTPRE, cytPTPRE is the only member that can be localized in cell nuclei. Its entry to cell nuclei is mediated by 10 amino acid residues at its N-terminal with residues R4, K5, and R9 particularly important [18]. Besides the aforementioned four isoforms of PTPRE, a 90KD PTPRE is identified in cells expressing memPTPRE. The 90KD PTPRE is considered as a form of memPTPRE lacking glycosylation [16].
Expression pattern of PTPRE isoforms is tissue specific. memPTPRE is highly expressed in brain, testes, lymph node and lung while cytPTPRE is found in spleen, lung and thymus [15, 16]. In hematopoietic cells, cytPTPRE is the dominant isoform [13]. The difference in the tissue distribution of memPTPRE and cytPTPRE could be explained by the differential usages of its two promoters regulated by different mechanisms. [14]. In addition, PTPRE is also expressed in neural tube during embryo development [19].
Regulation of PTPRE activity
Similar to most receptor type tyrosine phosphatases, PTPRE has two tyrosine phosphatase domains in its intracellular domain: the membrane proximal domain (D1) and the membrane distal domain (D2). D1 domain is the major catalytic moiety of PTPRE while D2 domain has very weak catalytic activity [20-22]. PTPRE can form homodimer through its D2 domain, resulting in inhibition of its catalytic activity. The dimerization of PTPRE can be regulated by extracellular stimuli such as EGFR activation or increased oxidative stress [23].
Microtubules associate with PTPRE and the association inhibits the activity of PTPRE. Disruption of microtubule structures in cells inhibits their binding to PTPRE, alters the subcellular localization of PTPRE, and increases the activity of PTPRE. In addition, activation of EGFR induces phosphorylation of Y638 in PTPRE, which enhances the association between microtubules and PTPRE, and inhibits the activity of PTPRE [24].
Interaction of PTPRE with other proteins
PTPRE is a phosphatase and it can remove the phosphate group from phosphorylated tyrosine residues. A number of cell membrane proteins are the substrates of PTPRE, therefore the signal transduction or activity of these proteins can be regulated by PTPRE. Insulin receptor can physically interacts with both memPTPRE and cytPTPRE, and the association inhibits the activation of insulin receptor and its downstream signaling molecules such as Akt and GSK-3 [25-27]. Similar to the insulin receptor, the signal transduction of the receptor tyrosine kinase PDGFRB signaling can also be inhibited by PTPRE [28]. In addition to the above two receptors, voltage-gated potassium channel Kv2.1 can bind to PTPRE, and PTPRE inhibits Src- or Fyn-stimulated Kv2.1 currents and tyrosine phosphorylation [29].
PTPRE can associate with signaling proteins in the cytosol as well. PTPRE can bind to Jak2 and dephosphorylates Jak2 [30], and the adaptor protein Grb2 can bind to phosphorylated Y695 at the c-terminal of both memPTPRE and cytPTPRE and the binding is necessary for the activation of Src [31, 32]. The Src family kinases are a group of intracellular tyrosine kinases that shares high homology, their functions are redundant but not totally overlapped [33, 34]. Regulation of the activity of Src makes it possible that PTPRE are involved in the regulation of numerous signaling pathways.
In addition to the regulation of the protein activation, the activity of PTPRE can be regulated by other proteins as well. Integrin activation can phosphorylate Y638 of cytPTPRE, and the phosphorylation is necessary for the binding of cytPTPRE with Src and further activation of Src [35]. Neu can phosphorylate Y695 of PTPRE which is necessary for the activation of Src in mammary tumor cells. Furthermore, phosphorylation of PTPRE inhibits its dimerization without affecting its activity significantly [36].
PTPRE in normal cells
Insulin receptor
Insulin receptor is a receptor tyrosine kinase that plays a crucial role in cell metabolism. It is the first reported tyrosine kinase whose phosphorylation can be inhibited by PTPRE [25]. Upon binding of insulin to insulin receptor, cytPTPRE associates with insulin receptor and inhibits the phosphorylation of insulin receptor and insulin receptor substrate-1, leading to the inhibition of Akt and GSK-3 activation that are mediated by insulin receptor and insulin receptor substrate-1. As a consequence, insulin induced glucose uptake is inhibited by cytPTPRE [26].
In addition to cytPTPRE, memPTPRE can regulate insulin receptor signaling as well. Compared with cytPTPRE, memPTPRE is more efficient in the inhibition of insulin induced receptor activation and downstream signaling due to its localization in cell membrane. Forced expression of cytPTPRE on the cell surface makes it equally efficient as memPTPRE in the regulation of insulin receptor signaling [27], suggesting equal catalytic activities between the two isoforms of PTPRE. The memPTPRE shows stronger inhibitory effects on insulin signaling probably due to its easy access to insulin receptor.
The association between PTPRE and insulin receptor is not dependent on the binding of insulin to its receptor although the stimulation of insulin can induce conformational changes within insulin receptor and PTPRE complex [37]. Upon binding of insulin, tyrosine residues in insulin receptor is phosphorylated, and which further activate downstream signaling pathways. memPTPRE can dephosphorylate tyrosines 972, 1158, 1162 and 1163 in insulin receptor and inhibits the activation of downstream signaling molecules such as Akt, Erk and GSK3. Glycogen synthesis and suppression of phosphoenol pyruvate carboxykinase (PEPCK) expression induced by insulin in primary hepatocytes can also be inhibited by memPTPRE [38].
Osteoclasts
Osteoclasts mediate bone destruction and it maintain the bone remodeling together with osteoblasts. The proper function of osteoclasts needs PTPRE, lack of PTPRE adversely affects podosomal arrangement, reduces their association with bone, and inhibits the production of C-terminal collagen telopeptides by osteoclast. The defect of osteoclast function results in increase of trabecular bone mass in PTPRE knockout young female mice [39].
In osteoclasts, loss of PTPRE expression inhibits the phosphorylation and activity of Src, Pyk2, and Rac while Rho activity is upregulated, indicating that integrin signaling is defective. Integrin activation can induce phosphorylation of Y638 in cytPTPRE that is dependent on Src activity, and the phosphorylation is necessary for the binding of cytPTPRE with Src and further activation of Src. Increase of Src activity or inhibition of Rho in PTPRE knockout osteoclasts restores their podosomal stability phenotype [35]. Grb2 is an adaptor that can bind to phosphorylated tyrosine through its SH2 domain. It binds to Y695 at the c-terminal of both memPTPRE and cytPTPRE [31]. The binding of Grb2 to phosphorylated Y695 of PTPRE is necessary for the activation of Src and podosomal organization by PTPRE in osteoclasts [32].
PTPRA is closely related with PTPRE, however it cannot be detected in podosome of osteoclasts. Loss of PTPRA expression has effects on neither bone mass and bone production and resorption, nor production, structure, function, and podosome organization of osteoclasts. The 12 N-terminal residues of cytPTPRE is crucial for podosome regulation, attaching this sequence to cytPTPRA confer a role for PTPRA in osteoclasts. Among the 12 amino acid residues, serine 2 regulates the activity of cytPTPRE and its effects on podosomes [40].
Nerve cells
Voltage-gated potassium (Kv) channels are heterogeneous family of proteins that play key roles in brain and cardiac excitability. Changes in cell membrane potential can activate Kv channels, and the channel activation can be modulated by tyrosine phosphorylation. In cultured primary Schwann cells and sciatic nerve tissues, Kv channel activity was upregulated and the Kv1.5 and Kv2.1 α-subunits were hyperphosphorylated. Kv2.1 can bind to PTPRE, and PTPRE inhibits Src- or Fyn-stimulated Kv2.1 currents and tyrosine phosphorylation [29]. Y124 in Kv2.1 was identified as the phosphorylation site by Src and PTPRE. Loss of phosphorylation of Y124 inhibits Kv2.1 channel activity. In newborn mice, loss of PTPRE expression leads to hypomyelination of sciatic nerve axons [41], and which is further supported by that a catalytically inactive memPTPRE delays optic nerve myelination [42]. In the brain cortices of PTPRE knockout mice, tyrosine phosphorylation of Kv1.1, Kv1.2, Kv7.3, and large-conductance Ca(2+)-activated K(+) channels were enhanced. Loss of PTPRE expression results in decreased I(K) density and enhanced after-depolarization [43].
Vascular endothelium
PTPRE is highly expressed in vascular endothelium, and its expression inhibits the proliferation of umbilical vein endothelial cells [44]. However, the survival and migration of porcine aortic endothelial cells which express memPTPRE but not cytPTPRE can be enhanced by memPTPRE activated Src, while overexpression of cytPTPRE reduces viability of porcine aortic endothelial cells [45]. In addition to its role in vascular endothelium, PTPRE also inhibits PDGF induced proliferation and migration of vascular smooth muscle cells by inhibition of PDGFRB signaling [28].
Leptin signaling
In hypothalamus, PTPRE binds to Jak2 and dephosphorylates Jak2. Leptin can induce phosphorylation of Y695 in PTPRE, which enhances the dephosphorylation of Jak2 by PTPRE. Loss of PTPRE expression makes the mice hypersensitive to leptin and protects mice from weight gain induced by high-fat food. These mice also exhibit increased metabolic rate and decreased adiposity [30].
Mast cells
Mast cells express high-affinity IgE receptor (FcepsilonRI) that can induce mast cell activation and granule release when stimulated by IgE. FcepsilonRI-induced Ca (2+) mobilization, JNK and p38 activation in PTPRE (-/-) mice were enhanced, and degranulation and cytokine production were evoked. Signaling studies showed enhanced phosphorylation of Syk, LAT and SLP-76 [46]. Lipopolysaccharide (LPS) or tumor necrosis factor alpha (TNFalpha) can prime normal macrophages but not PTPRE deficient macrophages for an enhanced respiratory burst. In response to the challenge of LPS, reduced TNFalpha production and enhanced IL-10 production was observed in PTPRE deficient macrophages [47].
PTPRE in cancer
Cancer is a leading cause of death worldwide, and many types of cancer are caused by aberrant activation of tyrosine kinases [48-51]. It is not surprise to see the implication of PTPRE in cancer such as breast cancer. Expression of PTPRE is relatively low in normal non-transformed mammary gland, while memPTPRE expression is upregulated during pregnancy and mammary gland regression [15]. memPTPRE is highly expressed in c-neu and v-Ha-ras induced mammary tumors in mice, but not in those initiated by c-myc or int-2. Ras induced upregulation of PTPRE expression is mammary gland-specific, which is not seen in other Ras initiated tumors [15]. In breast cancer cell lines, PMA, FGF, and serum stimulation enhance expression of PTPRE through EGFR and Erk1/2 activation. PTPRE contributes to Erk1/2 and Akt activation, improve the cell viability and colony formation of breast cancer cell lines. EGFR, Erk and PTPRE forms a positive feedback loop [52]. Overexpression of PTPRE in murine mammary epithelium leads to mammary hyperplasia and higher occurance of tumor [53], suggesting that PTPRE itself is not sufficient to induce cell transformation.
In mammary tumor cells, PTPRE regulates the phosphorylation of the Src family kinases including Src, Yes and Fyn leading to the activation of these kinases which contribute to oncogenesis. Src, Yes and Fyn are closely related, however, expression of Src but not Yes and Fyn can rescue the morphological phenotype of PTPRE deficient tumor cells, indicating the difference in the roles of these related kinases in Neu-induced mammary tumor cells [36, 54]. PTPRE can dephosphorylate Shc in the presence of Src, inhibition of Shc phosphorylation in turn reduces Erk activation. Neu binds to the PTB domain of Shc and protect Shc from dephosphorlation by PTPRE probably by competing the binding to Shc with PTPRE. It might explain why PTPRE contributes to the tumorigenesis only in Neu-drived mammary tumors that the inhibition of Erk activation by PTPRE is blocked by Neu while keeping the function in Src activation [55]. Neu can induce phosphorylation of Y695 in PTPRE which is necessary for the activation of Src in mammary tumor cells. The phosphorylation of PTPRE does not affect its activity toward to Kv2.1. Phosphorylation of PTPRE inhibits its dimerization without affecting its activity significantly [56]. Furthermore, The role of PTPRE in mammary tumor cells cannot be compensated by the close related PTPRA [36].
The similarities and differences of PTPRE in normal and cancer cells
The signal transduction of PTPRE has been studies in various cell types such as osteoclasts, nerve cells, vascular endothelium and mast cells, and its role in the signaling of leptin and insulin were also studied. In contrary to the normal cells, the role of PTPRE in tumorigenesis is not well studied yet. In normal cells, PTPRE inhibits the activation of insulin receptor [26, 27] and PDGFB signaling [28]. Unlike that in normal cells, PTPRE positively regulates the receptor tyrosine kinase EGFR signaling [52]. The underlying mechanisms as to how PTPRE regulates the signaling of different receptor tyrosine kinases in opposite ways between normal and cancer cells remain obscure, and should be further explored.
The Src family kinases are a group of non-receptor tyrosine kinase that regulates multiple cellular processes such as proliferation, differentiation and survival. Dysregulation of Src family kinases csuaes the pathogenesis of diseases including cancer [57]. The regulation of PTPRE on the activity of Src family kinases has been studied in osteoclasts [35], endothelial cells [45] and breast cancer cells [36, 54]. PTPRE was shown to positively regulate Src family kinases in all these cells. So far, no essential difference was found in term of the signal transduction of PTPRE between normal and cancer cells. However, it is worth noting that cancer cells can utilize existing signaling pathways to promote cells transformation and it is necessary to further study the role of PTPRE in different types of cancer.
The scientific and clinic significances of PTPRE
We have 518 tyrosine kinase genes in our genome and dysregulation of the activity of many tyrosine kinases implicated in various diseases including cancer [58]. The activity of tyrosine kinases can be regulated by phosphatases. Due to the substrate specificity and cellular distribution, a phosphatase such as PTPRE cannot regulate the activity of each tyrosine kinase. To figure out the substrates of PTPRE and its regulation on the activity of tyrosine kinase will contribute to a better understanding of its role in various tissues. Dysfunction of tyrosine kinases causes cell transformation, and targeted therapies against tyrosine kinases have been approved for the treatment of various cancers and they have dramatically improved treatment outcome. Study of phosphatase will improve our understanding of tyrosine kinase activation and its regulation in cancer. In addition, although phosphatase dephosphorylate tyrosine kinase, it does not necessarily deactivate tyrosine kinase. For example, PTPRE can activate Src family kinases in mammary tumor cells thus contributes to the oncogenesis of mammary tumor [36, 54], indicating that PTPRE can act as an oncogene in certain situations. Study of the signaling of PTPRE in cancer will expand our understanding of cell transformation and help to develop novel therapeutic approaches targeting tyrosine kinases.
Conclusions
Dysregulation of tyrosine kinases such as gene overexpression or oncogenic mutations leads to over activation of tyrosine kinases and therefore induces cell transformation. Small molecular tyrosine kinase inhibitors or antibodies have been developed to inhibit the activity of tyrosine kinases in the treatment of cancer, and targeting tyrosine kinase has been proved to be efficient in cancer treatment. Compared with tyrosine kinases, tyrosine phosphatases are less studied although they are crucial in the regulation of the activity of tyrosine kinases. PTPRE has been studied in various tissues (figure 2) and its function shows a tissue specific manner. The detailed mechanism that PTPRE regulates its substrates has been partially discovered but still needs further study. The implication of PTPRE in cancer has been investigated in breast cancer, and whether PTPRE plays a role in the oncogenesis of other types of cancer needs to be further elucidated.
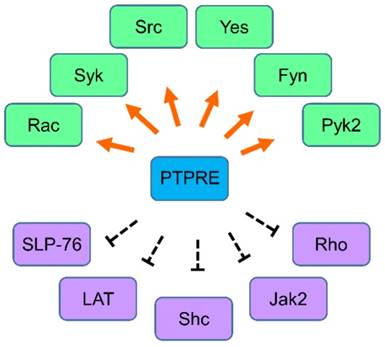
The regulation network of PTPRE. Molecules that are regulated by PTPRE in various cells are summarized together. Green color, the molecule can be activated by PTPRE; Purple color, the molecule can be inhibited by PTPRE.
Acknowledgements
The work is supported by National Natural Science Foundation of China (No. 81660473, No. 81660001), Ningxia Natural Science Foundation (No. 2018AAC02010), Ningxia High Education Scientific Research Program (No. NGY2016120, NGY2016119, NGY2017112) and West China Top Class Discipline Project in Basic Medical Sciences of Ningxia Medical University (No. NXYLXK2017B07).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hunter T. The genesis of tyrosine phosphorylation. Cold Spring Harbor perspectives in biology. 2014;6:a020644
2. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355-65
3. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-34
4. Torkamani A, Verkhivker G, Schork NJ. Cancer driver mutations in protein kinase genes. Cancer letters. 2009;281:117-27
5. Berman-Golan D, Granot-Attas S, Elson A. Protein tyrosine phosphatase epsilon and Neu-induced mammary tumorigenesis. Cancer metastasis reviews. 2008;27:193-203
6. Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH. et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Molecular and cellular biology. 2001;21:7117-36
7. Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A. et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699-711
8. Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. The EMBO journal. 1990;9:3241-52
9. Fischer EH, Charbonneau H, Tonks NK. Protein tyrosine phosphatases: a diverse family of intracellular and transmembrane enzymes. Science. 1991;253:401-6
10. Charbonneau H, Tonks NK. 1002 protein phosphatases? Annual review of cell biology. 1992;8:463-93
11. Watanabe T, Mukouyama Y, Rhodes M, Thomas M, Kume T, Oishi M. Chromosomal location of murine protein tyrosine phosphatase (Ptprj and Ptpre) genes. Genomics. 1995;29:793-5
12. Elson A, Kozak CA, Morton CC, Weremowicz S, Leder P. The protein tyrosine phosphatase epsilon gene maps to mouse chromosome 7 and human chromosome 10q26. Genomics. 1996;31:373-5
13. Wabakken T, Hauge H, Finne EF, Wiedlocha A, Aasheim H. Expression of human protein tyrosine phosphatase epsilon in leucocytes: a potential ERK pathway-regulating phosphatase. Scandinavian journal of immunology. 2002;56:195-203
14. Tanuma N, Nakamura K, Kikuchi K. Distinct promoters control transmembrane and cytosolic protein tyrosine phosphatase epsilon expression during macrophage differentiation. European journal of biochemistry. 1999;259:46-54
15. Elson A, Leder P. Protein-tyrosine phosphatase epsilon. An isoform specifically expressed in mouse mammary tumors initiated by v-Ha-ras OR neu. The Journal of biological chemistry. 1995;270:26116-22
16. Elson A, Leder P. Identification of a cytoplasmic, phorbol ester-inducible isoform of protein tyrosine phosphatase epsilon. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:12235-9
17. Gil-Henn H, Volohonsky G, Toledano-Katchalski H, Gandre S, Elson A. Generation of novel cytoplasmic forms of protein tyrosine phosphatase epsilon by proteolytic processing and translational control. Oncogene. 2000;19:4375-84
18. Kraut J, Volohonsky G, Toledano-Katchalski H, Elson A. Nuclear localization of non-receptor protein tyrosine phosphatase epsilon is regulated by its unique N-terminal domain. Experimental cell research. 2002;281:182-9
19. Mukouyama Y, Kuroyanagi H, Shirasawa T, Tomoda T, Saffen D, Oishi M. et al. Induction of protein tyrosine phosphatase epsilon transcripts during NGF-induced neuronal differentiation of PC12D cells and during the development of the cerebellum. Brain research Molecular brain research. 1997;50:230-6
20. Blanchetot C, Tertoolen LG, Overvoorde J, den Hertog J. Intra- and intermolecular interactions between intracellular domains of receptor protein-tyrosine phosphatases. The Journal of biological chemistry. 2002;277:47263-9
21. Wang Y, Pallen CJ. The receptor-like protein tyrosine phosphatase HPTP alpha has two active catalytic domains with distinct substrate specificities. The EMBO journal. 1991;10:3231-7
22. Lim KL, Lai DS, Kalousek MB, Wang Y, Pallen CJ. Kinetic analysis of two closely related receptor-like protein-tyrosine-phosphatases, PTP alpha and PTP epsilon. European journal of biochemistry. 1997;245:693-700
23. Toledano-Katchalski H, Tiran Z, Sines T, Shani G, Granot-Attas S, den Hertog J. et al. Dimerization in vivo and inhibition of the nonreceptor form of protein tyrosine phosphatase epsilon. Molecular and cellular biology. 2003;23:5460-71
24. Sines T, Granot-Attas S, Weisman-Welcher S, Elson A. Association of tyrosine phosphatase epsilon with microtubules inhibits phosphatase activity and is regulated by the epidermal growth factor receptor. Molecular and cellular biology. 2007;27:7102-12
25. Moller NP, Moller KB, Lammers R, Kharitonenkov A, Hoppe E, Wiberg FC. et al. Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases alpha and epsilon. The Journal of biological chemistry. 1995;270:23126-31
26. Aga-Mizrachi S, Brutman-Barazani T, Jacob AI, Bak A, Elson A, Sampson SR. Cytosolic protein tyrosine phosphatase-epsilon is a negative regulator of insulin signaling in skeletal muscle. Endocrinology. 2008;149:605-14
27. Andersen JN, Elson A, Lammers R, Romer J, Clausen JT, Moller KB. et al. Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling. The Biochemical journal. 2001;354:581-90
28. Shimizu H, Nakagawa Y, Murakami C, Aoki N, Kim-Mitsuyama S, Miyazaki H. Protein tyrosine phosphatase PTPepsilonM negatively regulates PDGF beta-receptor signaling induced by high glucose and PDGF in vascular smooth muscle cells. American journal of physiology Cell physiology. 2010;299:C1144-52
29. Peretz A, Gil-Henn H, Sobko A, Shinder V, Attali B, Elson A. Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. The EMBO journal. 2000;19:4036-45
30. Rousso-Noori L, Knobler H, Levy-Apter E, Kuperman Y, Neufeld-Cohen A, Keshet Y. et al. Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell metabolism. 2011;13:562-72
31. Toledano-Katchalski H, Elson A. The transmembranal and cytoplasmic forms of protein tyrosine phosphatase epsilon physically associate with the adaptor molecule Grb2. Oncogene. 1999;18:5024-31
32. Levy-Apter E, Finkelshtein E, Vemulapalli V, Li SS, Bedford MT, Elson A. Adaptor protein GRB2 promotes Src tyrosine kinase activation and podosomal organization by protein-tyrosine phosphatase in osteoclasts. The Journal of biological chemistry. 2014;289:36048-58
33. Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124:2013-24
34. Amata I, Maffei M, Pons M. Phosphorylation of unique domains of Src family kinases. Frontiers in genetics. 2014;5:181
35. Granot-Attas S, Luxenburg C, Finkelshtein E, Elson A. Protein tyrosine phosphatase epsilon regulates integrin-mediated podosome stability in osteoclasts by activating Src. Molecular biology of the cell. 2009;20:4324-34
36. Gil-Henn H, Elson A. Tyrosine phosphatase-epsilon activates Src and supports the transformed phenotype of Neu-induced mammary tumor cells. The Journal of biological chemistry. 2003;278:15579-86
37. Lacasa D, Boute N, Issad T. Interaction of the insulin receptor with the receptor-like protein tyrosine phosphatases PTPalpha and PTPepsilon in living cells. Molecular pharmacology. 2005;67:1206-13
38. Nakagawa Y, Aoki N, Aoyama K, Shimizu H, Shimano H, Yamada N. et al. Receptor-type protein tyrosine phosphatase epsilon (PTPepsilonM) is a negative regulator of insulin signaling in primary hepatocytes and liver. Zoological science. 2005;22:169-75
39. Chiusaroli R, Knobler H, Luxenburg C, Sanjay A, Granot-Attas S, Tiran Z. et al. Tyrosine phosphatase epsilon is a positive regulator of osteoclast function in vitro and in vivo. Molecular biology of the cell. 2004;15:234-44
40. Finkelshtein E, Lotinun S, Levy-Apter E, Arman E, den Hertog J, Baron R. et al. Protein tyrosine phosphatases epsilon and alpha perform nonredundant roles in osteoclasts. Molecular biology of the cell. 2014;25:1808-18
41. Tiran Z, Peretz A, Attali B, Elson A. Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. The Journal of biological chemistry. 2003;278:17509-14
42. Muja N, Lovas G, Romm E, Machleder D, Ranjan M, Gallo V. et al. Expression of a catalytically inactive transmembrane protein tyrosine phosphatase epsilon (tm-PTP epsilon) delays optic nerve myelination. Glia. 2004;48:278-97
43. Ebner-Bennatan S, Patrich E, Peretz A, Kornilov P, Tiran Z, Elson A. et al. Multifaceted modulation of K+ channels by protein-tyrosine phosphatase epsilon tunes neuronal excitability. The Journal of biological chemistry. 2012;287:27614-28
44. Thompson LJ, Jiang J, Madamanchi N, Runge MS, Patterson C. PTP-epsilon, a tyrosine phosphatase expressed in endothelium, negatively regulates endothelial cell proliferation. American journal of physiology Heart and circulatory physiology. 2001;281:H396-403
45. Nakagawa Y, Yamada N, Shimizu H, Shiota M, Tamura M, Kim-Mitsuyama S. et al. Tyrosine phosphatase epsilonM stimulates migration and survival of porcine aortic endothelial cells by activating c-Src. Biochemical and biophysical research communications. 2004;325:314-9
46. Akimoto M, Mishra K, Lim KT, Tani N, Hisanaga SI, Katagiri T. et al. Protein tyrosine phosphatase epsilon is a negative regulator of FcepsilonRI-mediated mast cell responses. Scandinavian journal of immunology. 2009;69:401-11
47. Sully V, Pownall S, Vincan E, Bassal S, Borowski AH, Hart PH. et al. Functional abnormalities in protein tyrosine phosphatase epsilon-deficient macrophages. Biochemical and biophysical research communications. 2001;286:184-8
48. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S. et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996;2:561-6
49. Shaw AT, Kim TM, Crino L, Gridelli C, Kiura K, Liu G. et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology. 2017;18:874-86
50. Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB. et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2017 JCO2017741025
51. Pegram MD, Lipton A, Hayes DF, Weber BL, Baselga JM, Tripathy D. et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1998;16:2659-71
52. Nunes-Xavier CE, Elson A, Pulido R. Epidermal growth factor receptor (EGFR)-mediated positive feedback of protein-tyrosine phosphatase epsilon (PTPepsilon) on ERK1/2 and AKT protein pathways is required for survival of human breast cancer cells. The Journal of biological chemistry. 2012;287:3433-44
53. Elson A. Protein tyrosine phosphatase epsilon increases the risk of mammary hyperplasia and mammary tumors in transgenic mice. Oncogene. 1999;18:7535-42
54. Granot-Attas S, Elson A. Protein tyrosine phosphatase epsilon activates Yes and Fyn in Neu-induced mammary tumor cells. Experimental cell research. 2004;294:236-43
55. Kraut-Cohen J, Muller WJ, Elson A. Protein-tyrosine phosphatase epsilon regulates Shc signaling in a kinase-specific manner: increasing coherence in tyrosine phosphatase signaling. The Journal of biological chemistry. 2008;283:4612-21
56. Berman-Golan D, Elson A. Neu-mediated phosphorylation of protein tyrosine phosphatase epsilon is critical for activation of Src in mammary tumor cells. Oncogene. 2007;26:7028-37
57. Espada J, Martin-Perez J. An Update on Src Family of Nonreceptor Tyrosine Kinases Biology. International review of cell and molecular biology. 2017;331:83-122
58. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912-34
Author contact
![]() Corresponding author: Jianmin Sun, No. 1160 Shengli Street, Yinchuan 750004, P.R.China jianmin.sunlu.se, jianmin.sunedu.cn, telephone: +86-9516880676, +46-462226451
Corresponding author: Jianmin Sun, No. 1160 Shengli Street, Yinchuan 750004, P.R.China jianmin.sunlu.se, jianmin.sunedu.cn, telephone: +86-9516880676, +46-462226451