Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(24):4596-4610. doi:10.7150/jca.27993 This issue Cite
Research Paper
Decoding Somatic Driver Gene Mutations and Affected Signaling Pathways in Human Medulloblastoma Subgroups
Charles J. Robbins*, Mayassa J. Bou-Dargham*, Kevin Sanchez, Matthew C. Rosen, Qing-Xiang Amy Sang ![]()
Department of Chemistry & Biochemistry, Institute of Molecular Biophysics, Florida State University
*These authors contributed equally to this work.
Received 2018-6-19; Accepted 2018-9-8; Published 2018-11-17
Abstract
Medulloblastoma is the most common malignant pediatric brain tumor. Prior studies have concentrated their efforts studying the four molecular subgroups: SHH, Wnt, group 3, and group 4. SHH and Wnt are driven by their canonical pathways. Groups 3 and 4 are highly metastatic and associated with aberrations in epigenetic regulators. Recent developments in the field have revealed that these subgroups are not as homogenous as previously believed. The objective of this study is to investigate the involvement of somatic driver gene mutations in these medulloblastoma subgroups. We obtained medulloblastoma data from the Catalogue of Somatic Mutations in Cancer (COSMIC), which contains distinct samples that were not previously studied in a large cohort. We identified somatic driver gene mutations and the signaling pathways affected by these driver genes for medulloblastoma subgroups using bioinformatics tools. We have revealed novel infrequent drivers in these subgroups that contribute to our understanding of tumor heterogeneity in medulloblastoma. Normally SHH signaling is activated in the SHH subgroup, however, we determined gain-of-function mutations in ubiquitin ligase (CUL1) that inhibit Gli-mediated transcription. This suggests a potential hindrance in SHH signaling for some patients. For group 3, gain-of-function in the inhibitor of proinflammatory cytokines (HIVEP3) suggests an immunosuppressive phenotype and thus a more hostile tumor microenvironment. Surprisingly, group 4 tumors possess mutations that may prompt the activation of Wnt signaling through gain-of-function mutations in MUC16 and PCDH9. These infrequent mutations detected in this study could be due to subclonal or spatially restricted alterations. The investigation of aberrant driver gene mutations can lead to the identification of new drug targets and a greater understanding of human medulloblastoma heterogeneity.
Keywords: pediatric brain cancer, infrequent mutations, tumor heterogeneity, bioinformatics, subclonal architecture, lysine methyl transferases
Introduction
Human embryonal tumors of the central nervous system (CNS) are tumors that arise from embryonal cells that remain in the brain after birth. CNS tumors are the most common solid neoplasms of childhood [1,2]. Although survival rates of pediatric brain tumor patients have significantly increased over the years, brain tumors are still a significant cause of cancer-related deaths in children [1-3].
Medulloblastoma is the most common malignant pediatric brain tumor, with the highest incidents reported at ages 3-4 and 8-10 years. Annually, 500 cases of medulloblastoma are diagnosed in the United States, and the 5-year survival rates are about 60% in patients with high risk disease [3,4]. In 2016, the WHO defined four molecular medulloblastoma subgroups based on the transcriptional profile and immunohistochemistry for certain biomarkers: SHH, Wnt, group 3, and group 4 [5,6]. The SHH-activated subgroup has aberrant activation of the SHH pathway and originates from granule neuron precursor cells of the developing cerebellum [7]. Activating mutations in the Wnt pathway effector beta-catenin 1 (CTNNB1) are prevalent in Wnt-activated medulloblastoma and arises outside the cerebellum from cells of the lower rhombic lip located at the dorsal brainstem during early hindbrain development [7]. Group 3 is characterized by elevated levels of MYC amplification or overexpression and appears to derive from cerebellar stem cells [8]. The cells-of-origin of group 4 medulloblastoma have long been unknown, but a recent study suggests that they are progenitors of the upper rhombic lip [9]. Current standard treatments such as surgery, radiation, and chemotherapy fail in more than 20% of patients, and survivors have to deal with the long-term side effects of these invasive treatments [10]. Recent studies have identified subclonal somatic mutations that result in intra-tumoral heterogeneity [11]. To complicate matters, treatment by targeting apparent clonal driving events can give rise to subclones that lack these targets from the original molecular subgroup [12,13]. This heterogeneity within the same molecular subgroup has led to the identification of 12 subtypes within the current molecular subgroups [14], demonstrating the motivation to better characterize these infrequent driving events that contribute to subgroup heterogeneity as well as subclonal architecture.
The majority of medulloblastoma cases are caused by sporadic mutations with only 13.5% of patients associated with germline mutations [15]. Germline mutations in MSH2, RAD50, FGFR2, and FGFR3 have been reported in pediatric medulloblastomas [16,17]. Also, specific hereditary syndromes associated with germline mutations in TP53, PTCH, APC, and CBP predispose individuals to develop medulloblastoma [3]. Syndromes linked with germline mutations include Li-Fraumeni syndrome for TP53 mutations, Gorlin syndrome for PTCH mutations, Turcot syndrome for APC mutations, and Rubenstein-Taybi syndrome for CBP mutations [18-21]. Thus, although the factors that contribute to pediatric brain tumors remain unclear, the early incidence of medulloblastoma and low prevalence of germline mutations associated with these tumors hint at in utero and early life exposures that result in the acquisition of somatic mutations [4]. These mutations and alterations can be induced by maternal endogenous (hormone levels or immune status) or maternal, paternal, and childhood exogenous (nutritional and chemical exposure) factors [22-24].
As an evolutionary process, cancer development occurs when cancer cells accumulate somatic mutations and epigenetic modifications causing phenotypic diversity by natural selection [25]. However, not all acquired somatic mutations contribute to tumorigenesis. Based on their consequence for cancer development, somatic mutations are categorized into driver and passenger mutations. As the names imply, driver mutations are those that confer growth advantages on cells carrying them and have been preserved by selection during cancer evolution, whereas passenger mutations confer no growth advantage [25].
This study focuses on investigating somatic driver gene mutations in the four medulloblastoma subgroups. We used COSMIC medulloblastoma data, which contains samples that have not been analyzed before in a large comprehensive study (in Supplementary Material: S1 file). We implemented several algorithms that interpret mutation patterns, functional impact, and occurrence. We used gene set enrichment analysis (GSEA) to determine aberrant signaling pathways amongst these putative somatic driver gene mutations. Our analyses reveal several novel infrequent driving events that may contribute to subclonal or spatial heterogeneity [11,26]; however, data with higher sequence coverage is a necessity. Overall, this study unravels the somatic driver gene mutations in medulloblastoma subgroups and the affected pathways.
Results
Sonic hedgehog (SHH) subgroup
Twenty-two driver genes were identified in the SHH subgroup (S1 Table). The most significantly affected pathways based on the GSEA results were those involved in the regulation of gene expression, cellular biosynthesis, and developmental processes (Supplementary Material: S2 File). Most transcription regulators and development-involved driver genes were characterized by loss-of-function (truncating) mutations, indicating that transcription regulation and developmental processes were likely inhibited (Fig. 1).
Enriched pathways in molecular medulloblastoma subgroups. This figure demonstrates the enriched pathways in the molecular medulloblastoma subgroups (SHH-activated, Wnt-activated, group 3, and group 4). Green indicates gain-of-function mutations and red suggests loss-of-function. The darker the color, the higher the percentage in the sample. An asterisk "*" is used to indicate the enriched pathways (q < 0.05) per subgroup as specified by the GSEA methods.
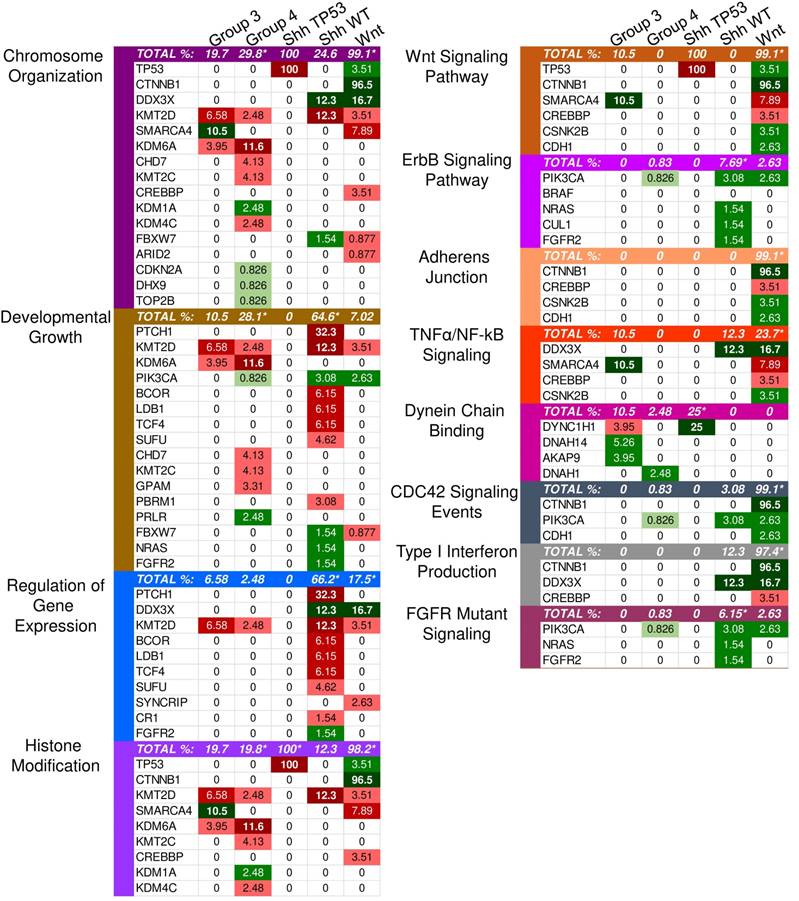
Although the SHH pathway was not significantly enriched (q ≤ 0.05), we identified four SHH genes that possess driver gene mutations. Pathway repressors PTCH1 (32.3%) and SUFU (4.62%) were associated with loss-of-function mutations. Infrequent gain-of-function mutations were identified in the ubiquitin ligases CUL1 (1.54%) and FBXW7 (1.54%). These are members of the Skip-Cullin-F-box (SCF) complex that completely degrades Gli1 but partially degrades Gli2/3 into transcriptional repressors [27-29] (Fig. 2). The activating mutations of CUL1 in the SCF complex downstream from other SHH pathway members suggests that SHH signalizing is suppressed or hindered in these patients. These results shed light on infrequent cases of SHH pathway inhibition in certain SHH-activated medulloblastomas.
Affected pathways in SHH-activated medulloblastoma. Loss-of-function mutations in PTCH1 and SUFU result in activation of the SHH pathway. The SCF complex partially degrades Gli2/Gli3 into transcriptional repressor forms and completely degrades Gli1 when sequestered by SUFU. Shh ligand binds to PTCH1 and releases its inhibition of SMO. SMO activation decreases SUFU sequestration, allowing Gli family transcription factors to localize to the nucleus to promote downstream SHH transcriptional events. Gain-of-function mutations in CUL1 of the SCF complex may promote the ubiquitination of Gli transcription factors and result in transcriptional inhibition of downstream SHH activity. In the somatic TP53 mutant, TP53 loss-of-function and DYNC1H1 gain-of-function promote proliferation of cancer cells.
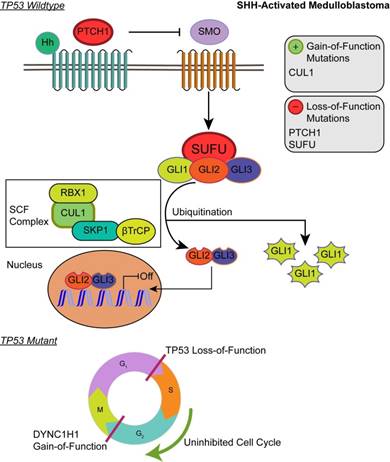
Patients with TP53 somatic mutations in the SHH subgroup had two driver gene mutations, none of which were involved in the SHH pathway: loss-of-function mutations in TP53 (tumor suppressor gene) and gain-of-function mutations in DYNC1H1 (oncogene) (S1 and S2 Tables). Thus, SHH activation is not driven by somatic mutations in the somatic TP53 mutant subgroup. Significantly enriched GO annotations were histone deacetylase regulator activity (q = 0.0383) and dynein light intermediate chain binding (q = 0.0383) (Supplementary Material: S2 File). The dynein chain, DYNC1H1, is involved in retrograde transport along microtubules and cell division by mediating the G2/M transition of the mitotic cycle [30,31]. P53 is known to undergo cytoplasmic dynein-dependent nuclear translocation in response to DNA damage. The 8-kDa light chain (LC8) of dynein binds to p53-binding protein 1 (53BP1). 53BP1 can potentially act as an adaptor to assemble p53 in the dynein complex. Disruption of the interaction between LC8 and 53BP1 in vivo prevents DNA damage-induced nuclear accumulation of p53 [32]. Additionally, truncating p53 mutations impair nuclear localization and association with dynein [33]. In some cases, the somatic driver gene mutations in TP53 mutant SHH likely confer growth advantage by dysregulating the control of cellular mitosis through the missense mutations of the dynein coding gene and the loss-of-function of TP53 (Fig. 2).
Wingless (Wnt) subgroup
Identified driver genes were significantly enriched in gene expression and histone modification, Wnt signaling and adherens junctions, regulation of mitosis and cell cycle transitioning, developmental processes, apoptosis, and interferon type I (IFN-I) signaling (Supplementary Material: S2 File).
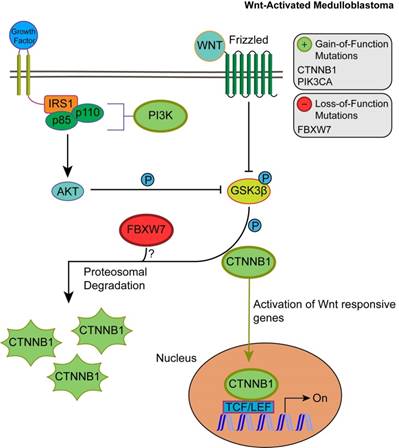
Unlike SHH, TP53 gene mutations were gain-of-function, rendering TP53 an oncodriver. TP53 gain-of-function mutations have been associated with chronic inflammation, regulation of gene expression, and upregulated expression of chromatin regulators [34,35]. Upon investigating β-catenin driver gene mutations, which were identified in 96.5% of Wnt-activated medulloblastoma patients, we found 33 missense mutations at the 32nd amino acid residue. This residue lies within the ubiquitination recognition motif of β-catenin and its mutation negatively impacts the phosphorylation of adjacent S33. Moreover, we found several mutations at S33 and S37 phosphorylation sites (Supplementary Material: S3 File) [36]. These mutations indicate that the phosphorylation and consequent ubiquitination of β-catenin were inhibited, resulting in β-catenin accumulation in the nucleus [36]. In addition, a gain-of-function mutation in PIK3CA (2.63%) was identified, indicating an indirect activation of the canonical Wnt pathway by enhancing the activity of Akt. Since Akt can phosphorylate and inactivate glycogen synthase kinase 3 beta (GSK-3β), the canonical Wnt pathway is enhanced due to β-catenin not being destabilized and degraded [37,38]. Thus, gain-of-function mutations in β-catenin (CTNNB1), loss-of-function mutations in the ubiquitin ligase FBXW7 (0.87%) that degrades β-catenin, and the downstream activation of adherens junctions may result in an uncontrolled activation of the canonical Wnt pathway and consequent increase in cellular proliferation and growth (Fig. 3) [39,40]. On the other hand, the non-canonical planar cell polarity Wnt pathway activates JNK cascades [41]. The identified loss-of-function mutations in the negative regulator of JNK pathway, FBXW7, also signify Wnt/JNK pathway activation [42]. Furthermore, the activation of the PI3K-Akt-mTORC1 pathway has been shown to activate the Wnt pathway [43,44]. Thus, activating driver gene mutations in PIK3CA identified in Wnt-activated medulloblastoma and the inactivating mutations in the negative regulator, FBXW7, presumably indicate an increased activation of the Wnt pathway and a consequent increase in growth and survival of the tumor (Fig. 4, S1 Table) [42]. Thus, Wnt activation seems to be driven by somatic mutations in the majority of cases.
The consequences of driver gene mutations in canonical Wnt signaling. Canonical Wnt activation results in CTTNB1 accumulation by inhibiting the destruction complex through phosphorylation of GSK3β. Activation of Akt from the PIK3CA/Akt pathway also enhances the phosphorylation of GSK3β, which in turn inhibits the phosphorylation and proteosomal degradation of CTNNB1. This permits CTNNB1 to enter the nucleus and activate transcription of Wnt responsive genes. FBXW7 loss-of-function mutations may enhance Wnt activation through CTNNB1 accumulation.
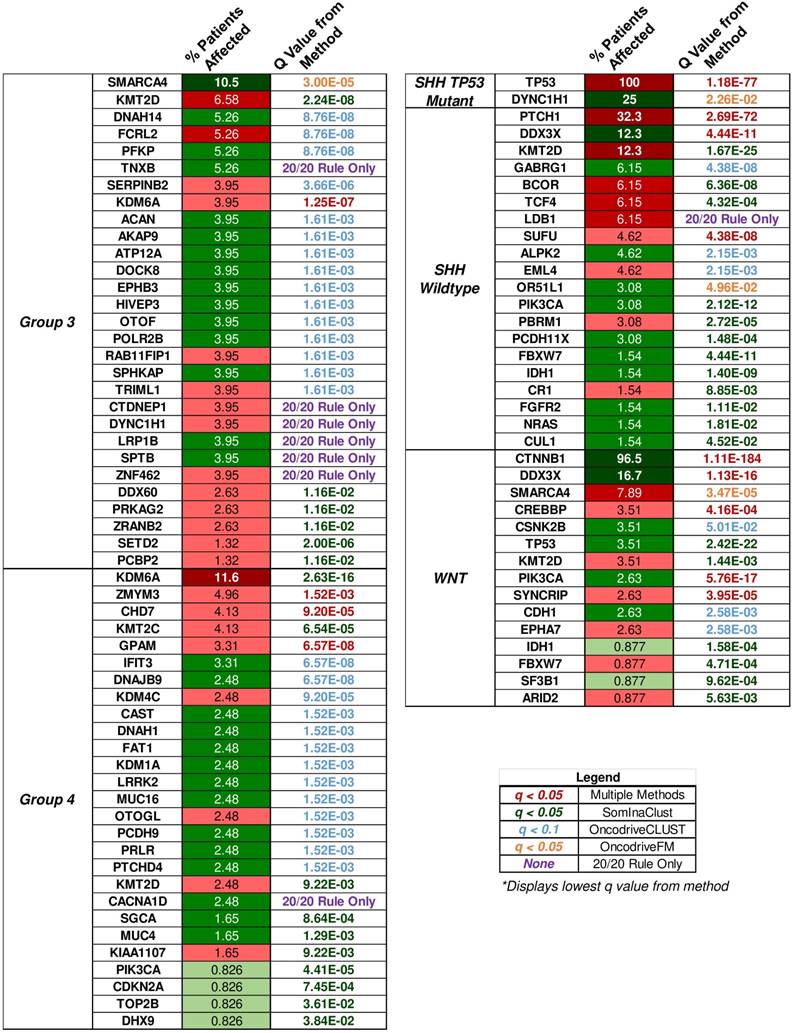
“20/20” rule classification of driver genes in medulloblastoma subgroups. The percent of patient tumors affected by candidate driver gene mutations alongside the methodology q value that determined the candidate driver. The “20/20” rule classification of driver genes into tumor suppressors (TSG), oncogenes (OG), or not classified (NC) is represented as red, green, or yellow colored boxes for the percent of patients affected column. A column of q values is displayed for the methods that identified candidate driver gene mutations. Brownish-yellow to red boxes represent significant driver gene mutations (q < 0.1 for OncodriveCLUST, q < 0.05 for others) identified by the method. Blue boxes represent q values that were not available or not significant. Rows of purple boxes represent candidate driver genes identified by the “20/20” rule only found in three or more patient tumors.
IFN-I signaling was enriched in GSEA. However, gain-of-function mutations in the negative regulator, CTNNB1, and loss-of-function mutations in the transcriptional coactivator CREBBP (3.51%) (Fig. 1) render the signaling inactive [45,46]. Interferon-α (IFN-α), a type I interferon, can inhibit the activation of β-catenin and the canonical Wnt pathway. This induces apoptosis and inhibits proliferation in cancerous cells [47]. Therefore, the inhibition of type I interferons activates the Wnt pathway.
Other driver mutations noted in the Wnt-subgroup were within the enriched TNF-α signaling pathway, which included loss-of-function in EPHA7 (2.6%) and gain-of-function in CSNK2B (3.5%). EPHA7 was identified with loss-of-function frameshift insertions upstream of its catalytic domain. EPHA7 encodes the EphA7 protein, which is a transmembrane receptor for ephrin A5. The EphA7-ephrinA5 complex was shown to recruit tumor necrosis factor receptor 1 (TNFR1) to form a multiprotein complex and induce extrinsic apoptotic signaling cascades [48]. The identified loss-of-function variant in EPHA7 may protect against apoptosis-mediated cell death. CSNK2B, which is part of the TNF-α/NF-κB signaling pathway in Wnt (Fig. 1), was identified with missense mutations in our sample. CSNK2B expresses the regulatory beta subunit of casein kinase 2 (CK2) [49-51] - a negative regulator of caspase activity and involved in phosphorylating several substrates that regulate the cell cycle [52]. Cell survival is enhanced by both CSNK2B gain-of-function mutations and loss of function mutation in EPHA7 through the inhibition of apoptosis.
Group 3
The only enriched GO annotations in group 3 were the catalytic complex (q = 0.00337) and MLL3/4 methyltransferase complex (q = 0.0134). Genes annotated with the catalytic complex included enzymes for hydrolysis, metabolism, histone modification, ATPase, and polymerases. This complex also included genes involved in the histone methyltransferase complex (MLL3/4) (Supplementary Material: S2 File). Mutations in MLL3/4 methyltransferase complex genes, KMT2D (6.58%) and histone demethylase, KDM6A (3.95%), were mostly inactivating indicating the alteration of histone methylation at H3K4 and H3K27 sites (S2 Table). By investigating individual driver genes we identified novel gain-of-function mutations in HIVEP3 (3.95%), which inhibits NFκB-mediated and JNK/SAPK-mediated responses such as apoptosis and pro-inflammatory cytokine gene expression (S2 and S3 Tables) [53,54].
Group 4
The most significantly enriched GO annotations were histone modification, posttranslational modification, and growth (Supplementary Material: S2 File). The majority of driver genes involved in histone and posttranslational modification were characterized by loss-of-function mutations that likely result in decreased expression of some gene and altered posttranslational modifications. Genes associated with growth, however, were mostly characterized by gain-of-function mutations that likely increase growth.
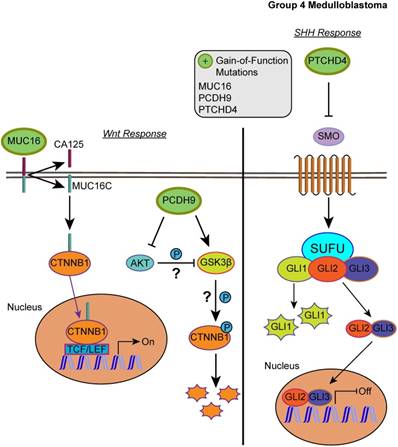
Our list of group 4 mutations included novel infrequent driver genes involved in the Wnt and SHH pathways. Wnt pathway-related driver genes were the Wnt activator MUC16 (2.48%) and the calcium-dependent cell adhesion molecule PCDH9 (2.48%) [55,56]. The SHH-related driver gene is the repressor of the canonical hedgehog signaling - PTCHD4 (2.48%) [57]. All three driver genes were characterized by gain-of-function mutations, which may activate the Wnt pathway and inactivate the SHH pathway (Fig. 5, S2 and S3 Tables). It is worth mentioning, however, that the coding DNA sequence for MUC16 is large; thus, the functional significance of these mutations may be uncertain as they could have accumulated by chance [58].
Affected pathways in group 4 medulloblastoma. The SHH pathway is inactivated and Wnt pathway is activated for group 4. Gain-of-function mutations in MUC16 can result in activation of the Wnt pathway by enhancing the transcriptional activity of CTNNB1. PCDH9 gain-of-function mutations can activate GSK3β and suppress Akt, however, the effect was unable to suppress the Wnt/β-catenin signaling. In the SHH pathway, gain-of-function in PTCHD4 leads to greater inhibition of SMO that can result in repressed transcription of SHH response genes.
KMT2D: A Shared Driver Gene Mutation in Medulloblastoma
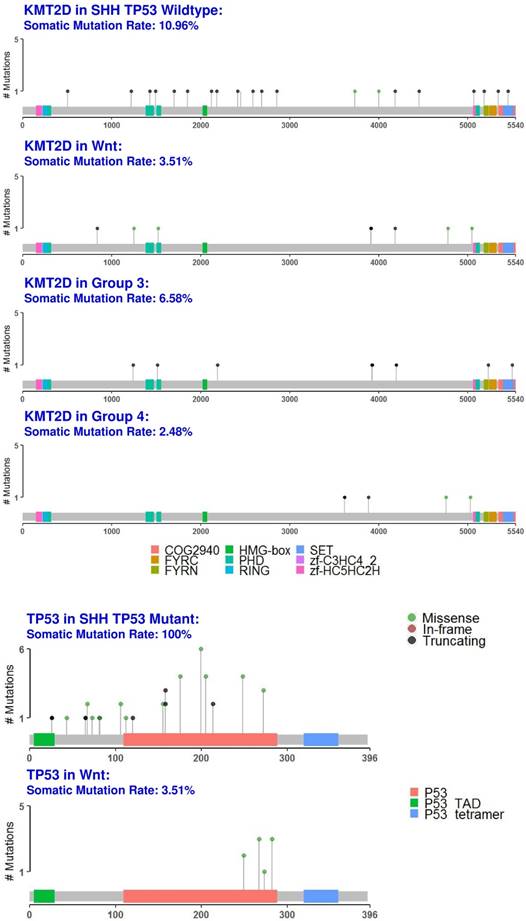
We identified the tumor suppressor gene, KMT2D, as a common medulloblastoma driver gene in all medulloblastoma subgroups (Fig. 1 and fig. 6, S2 Table). KMT2D was recently identified as one of the frequently mutated genes in lymphoma, medulloblastoma, gastric cancer, and Kabuki syndrome [59]. KMT2D targets histone H3 lysine 4 (H3K4), whose methylations act as a gene activation marker [59]. Specifically, group 3 and 4 medulloblastomas had a significant enrichment of the pathways that control epigenetic modification, which corroborate with previous findings [60].
Protein domain graphs of KMT2D and TP53. Protein domain graphs (PDGs) of KMT2D in medulloblastoma subgroups, and TP53 in Wnt and somatic TP53 mutant SHH medulloblastoma. Mutations on the PDG are color-coded based on the variant classification of the mutation: bright red for frameshift deletions/insertions, dark red for nonsense mutations, pink for splice-site mutations, blue for nonstop mutations, green for missense mutations, and light green for in-frame deletions/insertions. Although certain mutations like TP53 in Wnt have only missense mutations, they were less than 20%, which rendered TP53 unclassified.
Patient Demographic and Distribution of Driver Gene Mutations in Medulloblastoma
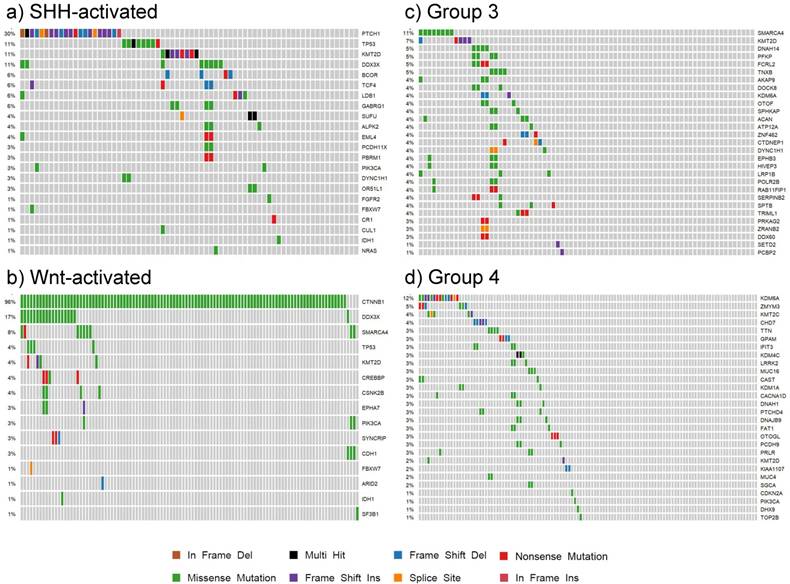
Our findings for each subgroup are summarized in the patient demographic table (S4 Table) and the somatic mutation oncoplots (Fig. 7). In the Wnt subgroup, all tumors possessed somatic driver mutations as identified by our analysis (e.g. CTNNB1). Sixty-six percent of SHH-activated medulloblastoma had distinguishable somatic driver mutations. Less than half of the tumors in groups 3 and 4 had somatic driver gene mutations. The oncoplots of Fig. 7 show that the percentage of shared driver genes amongst the cohort is underwhelming: only 30% of SHH-activated tumors, less than 11% of group 3, and less than 12% of group 4 tumors have shared driver mutations (in the form of PTCH1, SMARCA4, and KDM6A, respectively). This reiterates the value of studying these low frequency driving events that contribute to the inter-tumor heterogeneity observed in medulloblastoma as these are tractable driving events for future therapy development.
Oncoplots of somatic driver genes in each subgroup. Panels a), b), c) and d) are oncoplots of the putative somatic driver mutations for SHH-activated, Wnt-activated, group 3, and group 4 medulloblastoma, respectively. The gene names are the somatic driver mutations found to be significant in each subgroup from the analysis. The percentages on the left represent the percentage of patients with the somatic driver gene mutated. Each column represents a patient in the cohort, and each mutation type for a patient is represented by the colors in the legend.
Discussion
Despite being embryonic and early childhood tumors, medulloblastomas are rarely driven by germline mutations. Embryonic somatic mutations and epigenetic alterations commonly occur during development and contribute to clonal diversity [61]. Accumulation of these mutations - especially in genes that regulate cell proliferation, the cell cycle, and epigenetic machinery - can contribute to disease and result in the clonal expansion of neoplasia [24,61,62]. With different bioinformatics approaches, we identified putative somatic driver gene mutations in a medulloblastoma sequencing cohort of 384 samples (Fig. 7 and S4 Table). Our cohort, derived mainly from the COSMIC database, has distinct medulloblastoma samples that have not been evaluated in a comprehensive study (as depicted by the Venn diagrams in S1). These samples curated in COSMIC have been classified into subgroups by datamining the original features from smaller medulloblastoma studies [58,63,72,73,64-71]. In addition to somatic driver mutations identified previously, our analysis reveals low frequency somatic driver mutations and describes medulloblastoma inter-tumor heterogeneity. The residual disease could be due to subclonal diversity that manifests in these low frequency driving events. Using this comprehensive medulloblastoma cohort, we decode these somatic driver gene mutations.
Our results of the SHH subgroup pinpointed cases of somatic mutations that antagonize the SHH pathway. CUL1, a member of the SCF complex that may inhibit Gli family transcription in the SHH pathway, was identified with a missense mutation in 1.54% of patients. Here we found that the missense mutation in CUL1 was independent of the variants of PTCH1, SUFU, and SMO that activate the SHH pathway. If this is a bona fide activating mutation in CUL1, this would suggest cases of SHH signaling hindrance may be present within the SHH molecular subgroup. Another study reported a higher frequency of FBXW7 variants (6.87%) but these variants included nonsense and splice site mutations [59]. While in our study gain-of-function mutations of FBXW7 was identified in SHH medulloblastoma, other data suggest FBXW7 loss-of-function mutations. The ubiquitin-proteasome system can drive tumor development by elevated degradation of tumor suppressors or impaired destruction of oncoproteins [74]. Most SCF-FBW7 substrates are proto-oncogenes that can create chromosomal instability when not degraded [75,76]. As an example, FBXW7 degrades a master transcription factor regulator of stem cell development, SOX9 [77]. Gain-of-function mutations of FBXW7 may prevent SOX9 from promoting migration, metastasis, and resistance to treatment. The function of these missense mutations in FBXW7 is controversial and is worth further investigation.
Proliferation inducing somatic mutations that directly or indirectly activate the Wnt pathway were present in our Wnt-activated medulloblastoma samples. Previous reports have shown that the interaction between PI3K/Akt and Wnt pathway is crucial for tumor survival [78]. Here, we demonstrate that Wnt pathway activation was enabled specifically by activating PIK3CA mutations in addition to β-catenin mutations. We found driver gene mutations in PIK3CA not only occur in Wnt, but also in SHH and group 4 subgroups. Furthermore, the upregulated expression of IFN-I could be an antiviral response induced by cytomegalovirus (CMV) and this is a rare event in the Wnt subgroup [79]. However, our results showed that IFN-I signaling is significantly inhibited by somatic driver gene mutations, indicating a potential immune evasion [80,81].
Components of the TNF-α signaling pathway, EPHA7 and CSNK2B, are putative drug targets for the Wnt subgroup. EPHA7, an Eph/ephrin gene family member, was identified with loss-of-function frameshift mutations that potentially alter the intracytoplasmic domain containing the catalytic and sterile alpha motif (SAM) domains. Eph/ephrin gene families regulate cerebellar development during embryogenesis and were implicated in regulating apoptosis after birth [48,82]. Thus, the frameshift insertion upstream of the EPHA7 catalytic domain may protect against caspase-mediated death by causing a failure in the recruitment of TNFR1 by the EphA7-ephrinA5 complex [45]. CK2, expressed by CSNK2B, is a negative regulator of caspase activity and phosphorylates many substrates in the cell cycle [52]. Inhibition of CK2 phosphorylation events result in the sensitization of TRAIL-induced apoptosis [83,84], and its activation increases cell survival through the NF-κB pathway [85-89]. This corroborates an anti-apoptotic role for CK2 and indicates that gain-of-function of CK2 through missense CSNK2B mutations likely promotes cell survival. Northcott et al. also observed EPHA7 mutations in 8% and CSNK2B mutations in 14% of Wnt; however, EPHA7 was not determined as a loss-of-function driver gene mutation in this study [60]. Both EphA7 and CK2 can be potential drug targets in the Wnt-activated subgroup.
We have identified a novel molecular mechanism that could explain group 3 metastatic potential and uncovered limitations of group 4 classification. Novel and infrequent driving gain-of-function mutations were identified in the inhibitor of pro-inflammatory cytokines, HIVEP3 in group 3 (S2 and S3 Tables). These activating mutations corroborate with a previous report showing an immunosuppressive phenotype, whereby mouse myeloid-derived suppressor cells infiltrated patient-derived group 3 medulloblastoma xenografts [10]. Although this HIVEP3 mutation is not very common (3.95%), this could indicate that in addition to aberrant regulation of epigenetic modification, the aggressiveness in this subgroup may also be driven by immune suppression in certain cases or subclones. In addition to the altered regulation of epigenetic modification, groups 4 had infrequent driving mutations that may result in Wnt pathway activation (MUC16 and PCDH9) but SHH pathway inhibition (PTCHD4). PCDH9 was shown to activate GSK3β and suppress Akt and ERKs activity, but unlike other protocadherins was unable to suppress the Wnt/β-catenin signaling [90]. A protocadherin that was unable to inhibit Wnt/β-catenin signaling but actually activates it was PCDH11Y [91,92]. Thus, whether PCDH9 only lacks the ability to inhibit the Wnt/β-catenin signaling or has an activating effect remains to be investigated. PCDH9 has not been reported as a driver in medulloblastoma before, whereas PTCHD4 and MUC16 mutations have been previously mentioned but not in the context of driver gene mutations [60]. Although transcriptional profiling and immunohistochemistry may be sufficient to accurately subgroup medulloblastoma patients, the intra-subgroup heterogeneity observed at the level of somatic driving events and the infrequent driving events in certain subgroups urge the need for further studies with deep sequencing.
Due to the limitations of sequencing coverage from tumors [11], variants that were confidently identified are major clonal events or mutations shared within the same tumor. Although these clonal driving events exist, there is still a striking heterogeneity of driver mutations within the same subgroup, especially in group 3 and 4 tumors. Roughly 20% of group 3 and 4 tumors cannot be assigned a single shared driving event, suggesting that the currently defined medulloblastoma subgroups are not able to capture the heterogeneous molecular substructure [60]. Additionally, these genetic alterations could be restricted to subclones at certain regions within the tumor [26]. Through a deep sequencing study of Wnt-activated medulloblastoma, the primary Wnt molecular subgroup also had secondary SHH activation within the subclones [11]. Further complicating this picture is the heterogeneity of the epigenetic landscape. A total of 12 DNA methylation subtypes associated with distinct genetic alterations were proposed to refine this molecular substructure [60,93]. There are some patients that do not have driving events associated with these DNA methylation subtypes for group 3 and 4 tumors warranting further investigation [60].
In summary, revealing the infrequent somatic driving events will describe the genetic architecture of pediatric brain tumors leading to a deeper understanding of the molecular mechanisms driving these tumors. This study of low frequency driver genes suggests the need for single cell approaches in medulloblastoma to identify subclones in the primary tumor that are contributing to tumor heterogeneity and residual diseases. These driver gene mutations may serve as novel therapeutic targets. In the future, to fully characterize these mutationally “quiet” tumors [94] we must further investigate cancer evolution, epigenetic regulation through histone modifiers, structural variation, non-coding RNA, and other regulatory mechanisms. By understanding this dysregulated landscape of events in the tumor, we can aide in the development of therapy for the treatment of these devastating diseases that relapse in children.
Materials and Methods
Retrieving COSMIC data and formatting
Medulloblastoma somatic coding mutations were retrieved from the COSMIC complete coding mutation database (v79) [95]. Formatting, data handling, and analysis were performed in the statistical coding environment of R [96]. Ensembl gene IDs as well as Entrez gene IDs were added to the COSMIC coding mutations and duplicated associated gene names (such as MLL2 and KMT2D) were corrected using the BioMart-Ensembl database version 86 [97]. Using COSMIC mutation IDs, variant call format (VCF) information from the COSMIC server was retrieved for the medulloblastoma samples. The GRCh38/hg38 coordinates VCF files were annotated with the genetic variant annotator ANNOVAR for variant classification and functional score prediction [98]. The respective ANNOVAR outputs were then appended to the original COSMIC coding mutations through the COSMIC mutation IDs to create a master file containing both COSMIC data and ANNOVAR annotations. The master file attached in the supporting information (Supplementary Material: S3 File) were used for subsequent analysis through algorithms and packages listed below. Mutation annotation format (MAF) files necessary for the maftools, and SomInAClust packages in R were constructed from the information in the master file. All tools were run in default mode using instructions provided by the developer. Unknown or unlisted CDS mutations with no VCF information nor variant classifications were discarded in the analysis to determine candidate driver genes (DGs) of the medulloblastoma samples.
COSMIC data downloaded from the medulloblastoma samples contained histological classifications of the samples and did not include the current molecular subgroups, such as the groups 3 and 4 subgroups found in medulloblastoma. Due to COSMIC being a literature-curated database, clinical data was mined from the original studies and was used to identify the COSMIC data with the current molecular subgroup schema [58,63,72,73,64-71]. The “Sample.name” in the COSMIC data is the sample identifier used in the referenced studies which had the molecular subgroup classifications of the data. The molecular subgroup classifications corresponding to the matching “Sample.name” were combined with the prior medulloblastoma samples classified as Wnt or SHH subgroup by COSMIC. These molecular classifications appear in the “Molecular.Subgroup” column of S2 Table. The COSMIC mutation data had only 94 tumor samples that were classified as either Wnt or SHH from the total 937 medulloblastoma tumor samples. Using the classifications from the original studies yielded 114 Wnt, 73 SHH, 76 group 3, and 121 group 4 tumor samples as shown in the clinical file of this study. The SHH subgroup was further divided into eight samples with somatic TP53 mutations and 65 wildtype SHH samples. The remaining 553 samples were classified only by their histology or were not specified, and thus were discarded from the analysis.
Overview of putative driver genes by somatic mutation analysis
Multiple open source software packages in R available through platforms such as Bioconductor were used to determine the putative driver genes of the medulloblastoma samples from the COSMIC mutation data. SomInAClust, OncodriveFM, and OncodriveCLUST were used to analyze the somatic mutation data through mutation frequency, mutation pattern, functional impact, and other measures described by the authors of each package. Each package was used on the somatic mutation data for tumors from each medulloblastoma subgroup separately to determine candidate driver genes. In addition to these packages, all protein affecting mutations were classified by the “20/20” rule for the medulloblastoma subgroups. Candidate driver genes also included mutations that were classified by the “20/20” rule and were determined to affect at least three tumor samples for each embryonal brain tumor subgroup. The SHH wildtype, group 3, and group 4 medulloblastoma subgroups had these candidate drivers determined by the “20/20” rule alone. Eighty-four unique candidate driver genes were found in both primary histologies of the embryonal brain tumors, and the q values of candidate driver genes for each subgroup detected by each method is shown in the supplemental heatmap of all techniques (Supplementary Material: S4 File), as well as Fig. 6.
SomInAClust
SomInAClust is an R package that uses a mutation pattern and frequency based approach to scoring nonsynonymous mutations as tumor suppressor genes (TSGs) or oncogenes (OGs) [99]. The SomInAClust package used was updated to use the COSMIC v79 database MAF for its scoring protocol. The necessary TCGA equivalent MAF fields (Hugo_Symbol, Start Position, Variant_Classification, Tumor_Sample_Barcode, c_position_WU, amino_acid_change_WU) were constructed from the COSMIC master files of the medulloblastoma samples. As we were not working with TCGA data, the Tumor_Sample_Barcode field was simply the COSMIC ID_Tumour. ID_Tumour was used as it was the unique identifier for the tumor samples since each patient may have multiple tumor samples characterized in a study. Driver genes were selected on a q ≤ 0.05 cutoff and a TSG/OG score ≥ 20 as recommended by the authors [99].
OncodriveFM
OncodriveFM uses protein functional impact of the mutations using scores from three established methods (SIFT, PolyPhen2, and Mutation Assessor) to determine driver gene variants [100]. The accumulation of high functional impact variants across the tumor sample is the indicator for candidate driver genes by OncodriveFM. SIFT, PolyPhen2, and Mutation Assessor scores were assigned through ANNOVAR annotations of the VCF data. Missing SIFT, PolyPhen2, and Mutation Assessor scores were adjusted using an optimization from the authors: 1, 0, and, -2 were given to silent mutations and 0, 1, and 3.5 for nonsense or frameshift mutations respectively. Driver genes were significant only with a q ≤ 0.05 cutoff as recommended by the authors.
OncodriveCLUST
OncodriveCLUST identifies genes whose mutations are clustered in specific positions of the encoded protein against a baseline of synonymous/ coding silent mutations [101]. This clustering is especially common within oncogenic mutations [102]. The maftools package was used to implement the OncodriveCLUST algorithm in our somatic mutation data for medulloblastoma subgroups [103]. An FDR cutoff of q ≤ 0.1 with the minimum mutations set to 3 and the z-score p-value method was used to identify candidate driver genes by the OncodriveCLUST algorithm.
SomInAClust classification and the “20/20” rule role assignment
The driver genes found were assigned a subgroup level classification as either an oncogene (OG), tumor suppressor gene (TSG), or were not classified (NC). This classification of the driver genes resulted from the SomInAClust algorithm. Drivers determined by the other packages were classified by the “20/20” rule role assignment [102]. Genes were classified as oncogenes if > 20% of the recorded mutations in a gene are at recurrent positions and were activating. Similarly, genes were classified as tumor suppressors if > 20% of the recorded mutations were truncating or inactivating. The activating mutations for a gene were defined as missense mutations at the same amino acid or identical in-frame insertions or deletions. Inactivating mutations included nonsense mutations, frameshift insertions, frameshift deletions, splice-site mutations, or nonstop mutations. When a gene had > 20% oncogenic mutations but > 5% truncating mutations, it was classified as a tumor suppressor gene because well-studied oncogenes rarely harbor truncating or inactivating mutations. Calculations of the TSG and OG ratios were performed by coding in R. The “20/20” rule classification is shown in S2 Table for each candidate driver gene, and the amount for each type of mutation is displayed in S1 Table. It goes without saying that all of these classification results were also supplemented by individual study of the protein domains that were affected by the mutations as well as pathway analysis.
Pathway analysis for subgroups
To determine the significantly affected pathways, list-based gene set enrichment analysis (GSEA) was performed on the set of driver genes determined from the somatic analysis for each embryonal brain tumor subgroup. ToppGene and gProfiler were both used to implement the GSEA to determine enriched GO annotations as well as pathways [104,105]. For ToppGene, GO annotations and pathways included those that met the q ≤ 0.05 FDR Bonferroni cutoff [58,106]. The pathway data used consisted of BioCarta, REACTOME, KEGG, GO classifications, and other pathway databases provided by ToppGene. gProfiler was used to furnish similar KEGG pathway and GO annotations, with enriched pathways selected by the native g:SCS method when q ≤ 0.05. After the significant gene sets were determined by ToppGene and gProfiler, the percentage of patient tumors affected by each enriched gene set was calculated. This total gene set percentage was used to sort enriched gene sets by their impact on the tumor subgroups. Some of the significant gene sets were merged into broader classifications to illustrate trends in the data. The entire quantified enriched classification file with original classifications including all q values for each subgroup is in the Supplementary Material: S2 File.
Protein domain graphs
Protein domain graphs (PDGs) of the selected driver genes were drawn using the Lollipop plot mutation diagram generator in maftools [107]. This program uses Pfam API and UniProt REST API to build its PDGs. Mutations on the PDG are color-coded based on the cBioPortal variant classification of the mutation, and the height of the stem is a measure of the number of mutations at the amino acid position for a PDG.
Abbreviations
53BP1, p53-binding protein 1; AKT, protein kinase B; APC, adenomatous polyposis coli; AT/RT, Atypical teratoid/rhabdoid tumor; CK2, casein kinase 2; CMV, cytomegalovirus; CNS, central nervous system; COSMIC, Catalogue of Somatic Mutations in Cancer; CREBBP, CREB-binding protein; CSNK2B, casein kinase 2 beta subunit; CTNNB1, beta-catenin; CUL1, cullin 1; DG, driver gene; DNA, deoxyribonucleic acid; DYNC1H1, cytoplasmic dynein 1 heavy chain 1; EPHA7, ephrin type-A receptor A7; FBXW7, F-box/WD repeat-containing protein 7; FGFR2, fibroblast growth factor receptor 2; FGFR3, fibroblast growth factor receptor 3; Gli, GLI family zinc finger protein; GO, Gene Ontology; GSEA, Gene Set Enrichment Analysis; GSK-3β, glycogen synthase kinase 3 beta; H3K4, histone H3 lysine 4; HIVEP3, human immunodeficiency virus type 1 enhancer binding protein 3; IDH1, Isocitrate dehydrogenase 1; IFN-I, interferon type 1; JNK, c-Jun N-terminal kinase; KAT6A, lysine-specific acetyltransferase 6A; KDM6A, lysine-specific demethylase 6A; KMT2A, lysine-specific methyltransferase 2A; KMT2D, lysine-specific methyltransferase 2D; LC8, 8-kDa light chain; MAF, Mutation Annotation Format; MLL1, mixed-lineage leukemia 1; MLL3, mixed-lineage leukemia 3; MLL4, mixed-lineage leukemia 4; MSH2, MutS homolog 2; mTORC1, mammalian target of rapamycin complex 1; MUC16, mucin 16; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; OG, oncogene; PCDH9, protocadherin 9; PDG, protein domain graph; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; PNET, primitive neuroectodermal tumor; PTCH1, protein patched homolog 1; PTCHD4, patched domain-containing protein 4; RAD50, DNA double strand break repair protein; SCF, Skp/cullin/F-box containing complex; SHH, sonic hedgehog; SMARCA4, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4; SMO, smoothened, frizzled class receptor; SOX9, Sex-Determining Region Y Box 9; SUFU, suppressor of fused homolog; TCGA, The Cancer Genome Atlas; TNF-α, tumor necrosis factor Alpha; TNFR1, tumor necrosis factor receptor 1; TP53, cellular tumor antigen p53; TRAIL, TNF-alpha-related-apoptosis-inducing-ligand; TSG, tumor suppressor gene; VCF, variant call format; WHO, World Health Organization; Wnt, wingless-related integration site.
Supplementary Material
Supplementary Files S1_File, S1_Table, S2_Table, S3_File, S3_Table, S4_Table.
S2_File.
S4_File.
Acknowledgements
The authors wish to thank Dr. Kaixian Yu of the Department of Biostatistics, University of Texas M.D. Anderson Cancer Center for his critical review of the manuscript and constructive comments on methodology.
Author contributions
CJR participated in the study design, acquired and analyzed existing data from publically available sources, performed data analyses (including data processing, statistical calculations, programming, and pathway analysis) and interpretation, and drafted and revised the manuscript. MJB performed data analysis and interpretation and prepared and revised the manuscript. KS helped with data analysis and interpretation and revised the manuscript. MCR helped with scientific literature search and data analysis. QXAS conceived of the ideas, designed the study, directed the research, performed data analysis and interpretation, and drafted and revised the manuscript. All authors read and approved the final manuscript.
Funding information
This work was supported in part by grants from the Florida State University and an Endowed Chair Professorship in Cancer Research from anonymous donors to QXAS.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pollack IF. Pediatric brain tumors. Semin Surg Oncol. 1999;16:73-90
2. Brandão LA, Poussaint TY. Pediatric Brain Tumors. Neuroimaging Clin N Am. 2013;23:499-525
3. de Bont JM, Packer RJ, Michiels EM, den Boer ML, Pieters R. Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro Oncol. 2008;10:1040-1060
4. Lupo PJ, Nousome D, Okcu MF, Chintagumpala M, Scheurer ME. Maternal variation in EPHX1, a xenobiotic metabolism gene, is associated with childhood medulloblastoma: an exploratory case-parent triad study. Pediatr Hematol Oncol. 2012;29:679-685
5. Northcott PA, Korshunov A, Witt H. et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408-1414
6. Louis DN, Perry A, Reifenberger G. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803-820
7. Mir SE, Smits M, Biesmans D. et al. Trimethylation of H3K27 during human cerebellar development in relation to medulloblastoma. Oncotarget. 2017;8:78978-78988
8. Huang GH, Xu QF, Cui YH, Li N, Bian XW, Lv SQ. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016;107:583-589
9. Kijima N, Kanemura Y. Molecular Classification of Medulloblastoma. Neurol Med Chir (Tokyo). May. 2016
10. Sandén E, Dyberg C, Krona C. et al. Establishment and characterization of an orthotopic patient-derived Group 3 medulloblastoma model for preclinical drug evaluation. Sci Rep. 2017;7:46366
11. Iorgulescu JB, Van Ziffle J, Stevers M. et al. Deep sequencing of WNT-activated medulloblastomas reveals secondary SHH pathway activation. Acta Neuropathol. 2018;135:635-638
12. Morrissy AS, Garzia L, Shih DJH. et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529:351-357
13. Mcgranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. 2017.
14. Cavalli FMG, Remke M, Rampasek L. et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737-754.e6
15. Zhang J, Walsh MF, Wu G. et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. 2015;373:2336-2346
16. Trubicka J, Żemojtel T, Hecht J. et al. The germline variants in DNA repair genes in pediatric medulloblastoma: a challenge for current therapeutic strategies. BMC Cancer. 2017;17:239
17. Bourdeaut F, Miquel C, Di Rocco F. et al. Germline mutations in FGF receptors and medulloblastomas. Am J Med Genet Part A. 2013;161:382-385
18. Rausch T, Jones DTW, Zapatka M. et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59-71
19. Hahn H, Wicking C, Zaphiropoulous PG. et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841-851
20. Hamilton SR, Liu B, Parsons RE. et al. The Molecular Basis of Turcot's Syndrome. N Engl J Med. 1995;332:839-847
21. Taylor MD, Mainprize TG, Rutka JT, Becker L, Bayani J, Drake JM. Medulloblastoma in a child with Rubenstein-Taybi Syndrome: case report and review of the literature. Pediatr Neurosurg. 2001;35:235-238
22. Cordier S, Monfort C, Filippini G. et al. Parental exposure to polycyclic aromatic hydrocarbons and the risk of childhood brain tumors: The SEARCH International Childhood Brain Tumor Study. Am J Epidemiol. 2004;159:1109-1116
23. Barrington-Trimis JL, Nielsen SS, Preston-Martin S. et al. Parental smoking and risk of childhood brain tumors by functional polymorphisms in polycyclic aromatic hydrocarbon metabolism genes. PLoS One. 2013:8
24. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363-373
25. Stratton MR, Campbell PJ, Andrew F P. The cancer genome. Nature. 2009;458:719-724
26. Morrissy AS, Cavalli FMG, Remke M. et al. Spatial heterogeneity in medulloblastoma. Nat Genet. 2017;49:780-788
27. Hsia EYC, Gui Y, Zheng X. Regulation of Hedgehog signaling by ubiquitination. Front Biol (Beijing). 2015;10:203-220
28. Verdelho Machado M, Diehl A. Role of Hedgehog Signaling Pathway in NASH. Int J Mol Sci. 2016;17:857
29. Huangfu D, Anderson K V. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133:3-14
30. Taylor HC, Holwill MEJ. Axonemal dynein - a natural molecular motor. Nanotechnology. 1999;10:237-243
31. Raaijmakers JA, Tanenbaum ME, Medema RH. Systematic dissection of dynein regulators in mitosis. J Cell Biol. 2013;201:201-215
32. Lo KW-H, Kan H-M, Chan L-N. et al. The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J Biol Chem. 2005;280:8172-8179
33. Trostel S, Sackett DL, Fojo T. Oligomerization of p53 Precedes its Association with Dynein and Nuclear Accumulation. Cell Cycle. 2006;5:2253-2259
34. Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, Lauberth SM. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun. 2017;8:754
35. Cooks T, Pateras IS, Tarcic O. et al. Mutant p53 Prolongs NF-κB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell. 2013;23:634-646
36. Al-Fageeh M, Li Q, Dashwood WM, Myzak MC, Dashwood RH. Phosphorylation and ubiquitination of oncogenic mutants of beta-catenin containing substitutions at Asp32. Oncogene. 2004;23:4839-4846
37. Thompson M, Nejak-Bowen K, Monga SPS. Crosstalk of the Wnt Signaling Pathway. Targeting the Wnt Pathway in Cancer. New York, NY: Springer. 2011:51-80
38. Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161-168
39. Jiang JX, Sun CY, Tian S, Yu C, Chen MY, Zhang H. Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting ??-catenin for degradation in pancreatic cancer. Tumour Biol. 2016;37:13893-13902
40. Kam Y, Quaranta V. Cadherin-bound β-catenin feeds into the Wnt pathway upon adherens junctions dissociation: Evidence for an intersection between β-catenin pools. PLoS One. 2009:4
41. Daulat AM, Borg J-P. Wnt/Planar Cell Polarity Signaling: New Opportunities for Cancer Treatment. Trends in cancer. 2017;3:113-125
42. Nateri AS. The Ubiquitin Ligase SCFFbw7 Antagonizes Apoptotic JNK Signaling. Science (80- ). 2004;303:1374-1378
43. Vadlakonda L, Pasupuleti M, Pallu R. Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Front Oncol. 2013;3:85
44. Cizkova M, Cizeron-Clairac G, Vacher S. et al. Gene expression profiling reveals new aspects of PIK3CA mutation in eralpha-positive breast cancer: Major implication of the Wnt signaling pathway. McCubrey J, ed. PLoS One. 2010;5:e15647
45. Bhattacharya S, Eckner R, Grossman S. et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon- α. Nature. 1996;383:344-347
46. Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFNβ Enhanceosome Is Required for Synergistic Activation of Transcription. Mol Cell. 1998;1:277-287
47. Li W, Huang X, Tong H. et al. Comparison of the Regulation of β-Catenin Signaling by Type I, Type II and Type III Interferons in Hepatocellular Carcinoma Cells. Wong C-M, ed. PLoS One. 2012;7:e47040
48. Lee H, Park E, Kim Y, Park S. EphrinA5-EphA7 complex induces apoptotic cell death via TNFR1. Mol Cells. 2013;35:450-455
49. Litchfield DW. Protein kinase CK2 : structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1-15 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1223072/pdf/12396231.pdf. Accessed May 17, 2018
50. Voss H, Wirkner U, Jakobi R. Structure of the gene encoding human casein kinase II subunit beta. J Biol …. 1991:13706-13711 http://www.jbc.org/content/266/21/13706.short
51. Kristensen LP, Larsen MR, Højrup P, Issinger O-G, Guerra B. Phosphorylation of the regulatory β-subunit of protein kinase CK2 by checkpoint kinase Chk1: identification of the in vitro CK2β phosphorylation site. FEBS Lett. 2004;569:217-223
52. Yamane K, Kinsella TJ. CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 2005;65:4362-4367
53. Oukka M, Wein MN, Glimcher LH. Schnurri-3 (KRC) Interacts with c-Jun to Regulate the IL-2 Gene in T Cells. J Exp Med. 2004;199:15-24
54. Oukka M, Kim ST, Lugo G, Sun J, Wu LC, Glimcher LH. A mammalian homolog of Drosophila schnurri, KRC, regulates TNF receptor-driven responses and interacts with TRAF2. Mol Cell. 2002;9:121-131
55. Liu Q, Cheng Z, Luo L. et al. C-terminus of MUC16 activates Wnt signaling pathway through its interaction with β-catenin to promote tumorigenesis and metastasis. Oncotarget. 2016;7:36800-36813
56. Zhu P, Lv J, Yang Z. et al. Protocadherin 9 inhibits epithelial-mesenchymal transition and cell migration through activating GSK-3?? in hepatocellular carcinoma. Biochem Biophys Res Commun. 2014;452:567-574
57. Chung JH, Larsen AR, Chen E, Bunz F. A PTCH1 homolog transcriptionally activated by p53 suppresses hedgehog signaling. J Biol Chem. 2014;289:33020-33031
58. Northcott PA, Jones DTW, Kool M. et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818-834
59. Guo C, Chen LH, Huang Y. et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4:2144-2153
60. Northcott PA, Buchhalter I, Morrissy AS. et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311-317
61. Fernández LC, Torres M, Real FX. Somatic mosaicism: on the road to cancer. Nat Rev Cancer. 2015;16:43-55
62. Soto AM, Sonnenschein C. The somatic mutation theory of cancer: Growing problems with the paradigm? BioEssays. 2004;26:1097-1107
63. Taylor MD, Northcott PA, Korshunov A. et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465-472
64. Dubuc AM, Remke M, Korshunov A. et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013;125:373-384
65. Lin CY, Erkek S, Tong Y. et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530:57-62
66. Parsons DW, Roy A, Yang Y. et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2016;2:616-624
67. Jones DTW, Jäger N, Kool M. et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100-105
68. Pugh TJ, Weeraratne SD, Archer TC. et al. MEDULLOBLASTOMA EXOME SEQUENCING UNCOVERS SUBTYPE-SPECIFIC SOMATIC MUTATIONS. Nature. 2012;488:106-110
69. Robinson G, Parker M, Kranenburg TA. et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43-48
70. Subgroup specific structural variation across 1, 000 medulloblastoma genomes.
71. Remke M, Ramaswamy V, Peacock J. et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013;126:917-929
72. Parsons DW, Li M, Zhang X. et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435-439
73. Kool M, Koster J, Bunt J. et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008:3
74. Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29-46
75. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83-93
76. Vriend J, Ghavami S, Marzban H. The role of the ubiquitin proteasome system in cerebellar development and medulloblastoma. Mol Brain. 2015;8:64
77. Suryo Rahmanto A, Savov V, Brunner A. et al. FBW7 suppression leads to SOX9 stabilization and increased malignancy in medulloblastoma. EMBO J. 2016;35:2192-2212
78. Northcott PA, Jones DTW, Kool M. et al. Medulloblastomics: The end of the beginning. Nat Rev Cancer. 2012;12:818-834
79. Staub E. An Interferon Response Gene Expression Signature Is Activated in a Subset of Medulloblastomas. Transl Oncol. 2012;5:297-IN6
80. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836-848
81. Di Franco S, Turdo A, Todaro M, Stassi G. Role of Type I and II Interferons in Colorectal Cancer and Melanoma. Front Immunol. 2017;8:878
82. Bhatia S, Hirsch K, Baig NA. et al. Effects of altered ephrin-A5 and EphA4/EphA7 expression on tumor growth in a medulloblastoma mouse model. J Hematol Oncol. 2015:8
83. Petak I, Douglas L, Tillman DM, Vernes R, Houghton JA. Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis. Clin Cancer Res. 2000;6:4119-4127
84. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271
85. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168:37-57
86. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86
87. Shih R-H, Wang C-Y, Yang C-M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front Mol Neurosci. 2015;8:77
88. Pozniak PD, Darbinyan A, Khalili K. TNF-α/TNFR2 Regulatory Axis Stimulates EphB2-Mediated Neuroregeneration Via Activation of NF-κB. J Cell Physiol. 2016;231:1237-1248
89. Viatour P, Merville M-P, Bours V, Chariot A. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43-52
90. Zhu P, Lv J, Yang Z. et al. Protocadherin 9 inhibits epithelial-mesenchymal transition and cell migration through activating GSK-3β in hepatocellular carcinoma. Biochem Biophys Res Commun. 2014;452:567-574
91. Yang X, Chen MW, Terry S. et al. A human- and male-specific protocadherin that acts through the Wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005
92. Terry S, Queires L, Gil-Diez-de-Medina S. et al. Protocadherin-PC promotes androgen-independent prostate cancer cell growth. Prostate. 2006;66:1100-1113
93. Cavalli FMG, Remke M, Rampasek L. et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737-754.e6
94. Roussel MF, Stripay JL. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum. 2018;17:28-36
95. Forbes SA, Beare D, Gunasekaran P. et al. COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805-D811
96. R Core Team. R Core Team (2016). R: A language and environment for statistical computing. R Found Stat Comput Vienna, Austria URL http//wwwR-project.org/. 2016:R Foundation for Statistical Computing.
97. Yates A, Akanni W, Amode MR. et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710-D716
98. Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010:38
99. Van den Eynden J, Fierro AC, Verbeke LP, Marchal K. SomInaClust: detection of cancer genes based on somatic mutation patterns of inactivation and clustering. BMC Bioinformatics. 2015;16:125
100. Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012:40
101. Tamborero D, Lopez-Bigas N, Gonzalez-Perez A. Oncodrive-CIS: A Method to Reveal Likely Driver Genes Based on the Impact of Their Copy Number Changes on Expression. PLoS One. 2013:8
102. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546-1558
103. Mayakonda A, Koeffler HP. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv. 2016:052662
104. Reimand J, Kull M, Peterson H, Hansen J, Vilo J. G:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007:35
105. Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009:37
106. Robinson G, Parker M, Kranenburg TA. et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43-48
107. Jay JJ, Brouwer C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS One. 2016:11
Author contact
![]() Corresponding author: qxsangfsu.edu (QXS)
Corresponding author: qxsangfsu.edu (QXS)