Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):4018-4028. doi:10.7150/jca.20906 This issue Cite
Review
Roles of Smads Family and Alternative Splicing Variants of Smad4 in Different Cancers
Irfan Ullah1, Weichao Sun1, Liling Tang1 ![]() , Jianguo Feng2
, Jianguo Feng2 ![]()
1. Key Laboratory of Biorheological Science and Technology, Ministry of Education, College of Bioengineering, Chongqing University, Chongqing 400044, China
2. Department of Anesthesiology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan Province, China
Received 2017-5-7; Accepted 2018-8-20; Published 2018-10-16
Abstract
Transforming Growth Factor β (TGF-β) is one of the most common secretory proteins which are recognized by membrane receptors joined to transcription regulatory factor. TGF-β signals are transduced by the Smads family that regulate differentiation, proliferation, early growth, apoptosis, homeostasis, and tumor development. Functional study of TGF-β signaling pathway and Smads role is vital for certain diseases such as cancer. Alternative splicing produces a diverse range of protein isoforms with unique function and the ability to react differently with various pharmaceutical products. This review organizes to describe the general study of Smads family, the process of alternative splicing, the general aspect of alternative splicing of Smad4 in cancer and the possible use of spliceoforms for the diagnosis and therapeutic purpose. The main aim and objective of this article are to highlight some particular mechanisms involving in alternatives splicing of cancer and also to demonstrate new evidence about alternative splicing in different steps given cancer initiation and progression.
Keywords: Apoptosis, Alternative splicing, Cancer, Homeostasis, Proteins, Transforming Growth Factor β
Introduction
The word Smad is derived from the two orthologous protein “sma'' and “Mad” from Caenorhabditis elegans and Drosophila melanogaster, respectively [1]. It was first recognized in Drosophila melanogaster in the mid-1990 as a Mad protein. It mediates the signal of decapentaplegic (DPP), analogous to a mammalian Bone morphogenetic protein 2/4 (BMP2/4) which is a member of the TGF-β superfamily [2]. SMADs are intracellular proteins that mediate extracellular signal of TGF-β ligands to the nucleus and regulate gene transcription [3,4]. This pathway is called transforming growth factor beta signaling (TGF-β) pathway.
The TGF-β signaling pathway has been concerned with many biological activities including fibrosis, embryonic development, wound healing, tumor development, cell differentiation, apoptosis, homeostasis and immunity regulation [5]. The TGF-β superfamily contains a significant group of secreted peptides growth factors in metazoan, such as TGF-βs, inhibin, nodal, Activins, bone morphogenetic protein (BMP) and other correlated proteins [6]. TGF-β family, its receptors and signaling pathways play a critical role in the development or progression of several forms of neoplasia. The possibility of the loss of BMP, TGF-β signaling components and TGF-β-type II receptors is contributing to the development or progression of advanced meningiomas [7].
In the early stage of normal epithelial cells and tumorigenesis, TGF- beta is antiproliferative, but plays a role in tumor promoters at a later stage, assisting in metastatic progression through an autocrine TGF-β loop. The growth inhibition properties of TGF-β are paramount and demonstrated in normal and early tumor cells. This property is due to the suppression of c-Myc proto-oncogene or cyclin-dependent kinases (CDK) and even inhibition of CDK regulators such as p15, P21, and p27 [8]. TGF-β and other proteins that involved in TGF-β signaling pathways initially express in most of the cancers. However, during transfection of TGF-β into tumor cells, the tumor cells show resistance to the inhibitory effect of TGF-β and leading to cell proliferation, invasion and increases metastasis. The inactivation of some components of TGF-β signaling pathways such as TBRI, TBRII, and Smads contribute to metastasis, cell proliferation, and cancer development. TGF-β overexpression increases angiogenesis, metastasis, invasiveness and immune suppression. The overexpression of TGF-β has been detected in various tumors such as colon, breast, esophageal, gastric, Liver, lungs, pancreas, kidney, brain, prostate, malignant melanoma and certain blood disease. TGF-β increase epithelial cell motility and prompt metastasis development. It causes the transition of the epithelial cell into the Epithelial-mesenchymal cell (EMT). The EMT is vital for embryonic development, tissue restoration, and repair. The critical part of EMT is to lose the cell adherence and production of extracellular matrix which results in separation of the cells from one another and loss epithelial cell polarity [9]. The incorrect revival of EMT has been reported in human malignant tumors [10]. TGF-β may induce EMT through Smad-mediated or non-Smad signaling pathways [11]. The Smad-dependent pathway involves in TGF-β suppression function while Smad-independent pathway loses the tumor suppression function of TGF-β and essential for pro-oncogenic function. The several parts of TGF-β signal transduction pathway act as a tumor suppressor. However, their mutations cause some human diseases such as cancers [12-14]. The function of this pathway depends on the activation of cytoplasm mediators (TGFBRII, TGFBRI, and SMADs) into the nucleus, which regulates the gene expression. Smads gene family provides instruction for the formation of a particular protein that helps to normalize the activity of specific genes in cell growth and cell division (proliferation) [15]. Therefore, functional study of TGF-beta pathways and roles of Smads are vital for the development of new therapeutic approaches to cancer.
In this review, we discuss Smads family and its alternative splicing in a different type of cancers. Specifically, we focus on the alternative splicing of Smad4 in tumors and the possible use of spliceo-froms for the diagnosis and therapeutic purpose.
Smad family and its functions in TGF-beta signaling Pathway
There are three Smads proteins encoded in C. elegans, five in Drosophila and eight in human and mouse genome. Five of mammalian Smads act as a substrate for TGF-β family receptor. Five Smads (Smad1, 2, 3, 5 and 8) are commonly called receptor-regulated Smads (R-Smads); Smad 2 and Smad 3 serve as a substrate for TGF-β (Nodal and Activin) while Smads 1, 5 and 8 for BMP and anti-Mullerian [16]. All R-Smads are situated in the cytoplasm. The Smad4 is a general partner for all R-Smads and works as a shuttle between nucleus and cytoplasm. Thus it is called Co-Smad. The TGF-β pathway starts when TGF-β ligands bind to the receptors presenting on the surface of the cell membrane (TGFBRII and TGFBRI). These receptors activate a group of R-Smads. The R-Smads and Co-Smad make a complex and translocate into the nucleus where they interact with coactivator or corepressor act as a transcription factor [17]. Smad3 can bind with Smad-binding element while Smad2 cannot bind directly to DNA [18]. Smad4 gene is present on the long arm (q) of chromosome 18 at point 21.1 and covering the area from 46,810,610 to 46,860, 144 base pairs. The complete size of the gene is 49539 bases, located on the sense/coding / positive strand of DNA, and 3220 nucleotides transcribed to mRNA. This gene translates into a protein having 552 amino acids and 60439 Daltons molecular weight. Prior it was believed that it contains 11 exons, but the current mutational studies report one additional exon that is located upstream of exon and called exon 0 [19]. Smad4 is reported as a tumor suppressor gene and plays critical roles in the TGF-β signaling pathway. As it is deleted in most pancreatic cancer, therefore also called DPC4 (deleted in pancreatic cancer) [20]. The genetic imbalances in about 97% of pancreatic cancers, caused by point mutations, deletions amplifications, translocations, and inversions [21]. The key driver genes in pancreatic carcinogenesis are KRAS, CDKN2A, TP53, and SMAD4. Approximately 50% of the pancreatic carcinomas is due to Smad4 mutation, resulting in reduced proactive protein. Approximately 30% of the mutations occurred by homozygous deletion. Clinical studies have shown that SMAD4 can be used as a biomarker for pancreatic cancer (PC). A study of 90 patients with pancreatic cancer showed that (19%) of patients are due to Smad4 mutations. In another study, the SMAD4 gene is deactivated by a mutation in the MH2 domain by deletion homozygosity at (32%), (12%) and (20%) [22].
Smad4 inactivation is associated with different types of cancer. For example, wild-type Smad4 can inhibit tumorigenesis [23]. Germline mutation in Smad4 causes Juvenile polyposis [12,24]. It also plays an important role in the recruitment of specific transcriptional coregulator to the Smad transcriptional complex. Smad4 is mostly found as a homo-oligomer in the basal state [25].
There are two inhibitory SMADs (I-SMAD): SMAD6 and SMAD7. These I-SMADS turn off signaling pathway to abrogate TGF-β signal transduction rather than transducer acting as an antagonist. In BMP, TGF-β, and activin signaling, Smad7 occupies type I receptor to inhibit Smad phosphorylation. Mouse (mSmad), Xenopus (XSmad) preferentially inhibits different signaling pathways. mSmad prefers to inhibit TGF-beta, activin over BMP but XSmad preferentially inhibits BMP. Smad 7 plays a vital role in the negative feedback regulation of TGF-β signaling at receptor level [25]. Smad7 is present the nucleus, but due to TGF-β stimulation, it moved to the cytoplasm. The Smad6 preferentially inhibit BMP signaling pathway with a different mechanism. Smad6 competes with Smad4 instead of binding with TBRI. It makes Smad1-Smad6 complex instead of the Smad4-Smad1 complex to inhibit BMP signal transduction. The overexpression of Smad4 can save BMP signaling [26]. Smad6 can occupy TBRI and BMPR by imitating Smad7 to inhibit TGF-β and BMP signaling. Both Smad 6 and Smad7 expression increased in response to TGF-β, activin, and BMP. Smad7 transfection can block TGF-β mediated response in a mammalian cell. Smad7 mRNA injection can block TGF-β and activin signaling in Xenopus embryo. It is concluded that TGF-β induces Smad7 mRNA expression. Smad7 inhibits TGF-β mediated phosphorylation of Smad2 and Smad3. Smad7 acts as a participant in negative feedback to control TGF-β response. The different and diverse cellular function of TGF-β and interferon gamma (IFN-gamma) is unknown, but IFN-gamma induces the expression of Smad7 through Jak1 and Stat1. By this way, Smad7 inhibits the interaction of TBR1 and Smad3. TGF-β activates the transcription factors Smad2 and Smad3 while IFN-gamma activates the transcription factor Stat1[19]. Smad7 synthesis can also be caused by NF-kappa B/RelA transcription factor in response to the pro-inflammatory cytokines, interleukin -1 β(IL-1β) and TNF- α. In this way, Smad 7 inhibits TGF-beta signaling [27]. I-Smads is the regulator of TGF-β and BMP signaling by negative feedback. They may block the phosphorylation of R-Smads by making a stable complex with activated TBR1 or recruit ubiquitin E3 ligases like Smurf1/2 causing ubiquitination and deprivation of activated TBR1. Moreover, it can also inhibit TGF-beta and BMP signaling in nucleus due to interaction with transcriptional repressors, for example, Hoxc-8, CtBP, and histone deacetylases or disturbing the arrangement of TGF-β induce active Smad-DNA complex. Several human diseases like inflammatory diseases, tissue fibrosis, and carcinogenesis are linked with the deregulation of Smad 7 expression. Smad7 can be suggested as a therapeutic potential to treat these diseases such as inflammatory disease, carcinogenesis and tissue fibrosis [23,28].
A structural and functional feature of Smad signaling pathways and importance of Smad4. TGF-beta singling pathways start by binding TGF-beta/BMP ligands to a pair of membrane receptors (TBRII+TBRI/ BMPRII+BMPRI). These receptors phosphorylate R-Smads (2,3 and 1,5,8) to promote the establishment of the heterotetrameric receptor complex. The R-Smads interact with Co-Smads Smad4, which acts as a shuttle between nucleus and cytoplasm. The Samd4 and associated R-Smads translocate to the nucleus and bind to transcription factor to regulate gene expression.
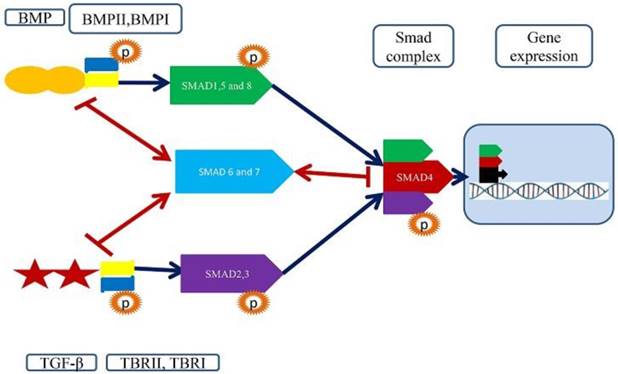
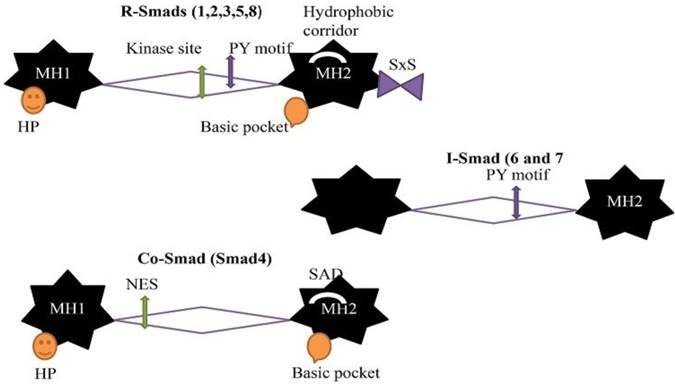
Structures of SMADs
The general stuff of all Smads is same but the few differences according to different categories. Due to these differences, they have different functions. There are two globular domains of Smads proteins connected by a region called linker region and containing five hundred amino acids in length [29]. The Mad Homology domain 1 (MH1) at N-terminal and Mad Homology domain 2 (MH 2) at C-terminal are conserved in all R-Smads and Co-Smad/Smad4 but not in I-Smads. The MH2 domain/ C-terminal are conserved in all Smads proteins, but the linker region is different in different subgroups. X-ray crystal structural and functional analysis determined that MH1 and MH2 domain provide interaction site for Smads proteins with other proteins and DNA. The MH1 domain binds with DNA while C-terminal/ MH2 domain mediates protein- protein interaction with many regulators and effectors protein, including TGF-beta receptor, particularly cytoplasmic anchor proteins, specific DNA binding cofactors, chromatin modifiers and nucleoporins [30,31]. The primary contact is formed between Smads (R-Smads and Co-Smad), and DNA is recognized by the β-hairpin structure at the MH1 domain and stabilized by a tightly bounded Zinc atom. The β-hairpin is conserved in all R-Smads and Co-Smad. There is a flexible segment between these two domains (MH1 andMH2 domain) having binding sites for Smad ubiquitination-related factor (Smurf), ubiquitin ligases, phosphorylation sites for various protein kinases in all R-Smads and nuclear export signal in Smad4. The linker region in R-Smads and I-Smads contain proline-tyrosine motif (PY) recognized by WW domain in Smurf ubiquitin ligases. The MH2 domain is one of the most adaptable protein interacting parts in signal transduction and highly conserved. R-Smads contain one of the core pockets at MH 2 domain for the interaction of activated Type I receptor and S-X-S motif for the interaction with Smad4. A hydrophobic patches/hydrophobic corridor is present at the surface of MH 2 domain providing a site for multiple interactions such as nucleoporins, cytoplasmic retention protein, and DNA binding cofactors. The surface of the MH2 domain in Smad4 contains SAD (Smad4 activation domain) that mediates the interaction with transcriptional activator and repressor. I-Smads have no MH1 domain [12].
The eight Smads proteins are listed in three different classes according to function. A linker region separates the two conserved globular domain (MH1 and MH2). The linker region is variable. Co-Smad and R-Smads contain β hairpin (Hp) at the MH1 domain for DNA binding while the I-Smads have no MH1 domain. The linker region of R-Smads and I-Smads contain PY motif for recognition of WW domain in Smurf ubiquitin ligases. The R-Smad linker region contains multiple phosphorylation sites for CDKs, MAPKs and protein kinase. The MH2 domain of R-Smads includes a basic pocket for receptor interaction, SXS for Co-Smad interaction and hydrophobic corridor for multiple interaction purposes. The linker region of Co-Smad contain nuclear export signal (NES), and MH2 domain contains Smad activation domain (SAD).
Structure of Smads (R-Smads, Co-Smad, and I-Smads)
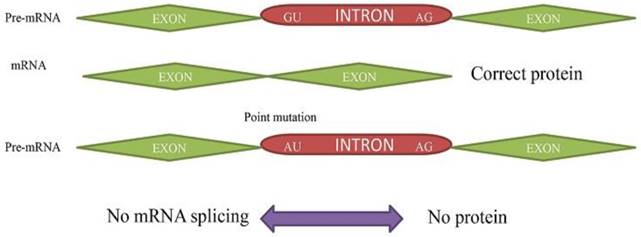
Alternative Splicing and its important roles in cancer
During the eukaryotic transcriptional process, the pre-mRNA undergoes splicing in which noncoding region is removed, and the exons rejoin to generate a mature mRNA. Alternative splicing is a fundamental mechanism to yield many diverse proteins, multiple transcriptions are able to create numerous mRNA transcripts. Approximately 95% of human polymorphic exon genes are subjected to alternative splicing [32]. Different types of alternative splicing exist, in which introns are detached, and exons are rejoined together, including mutually exclusive splicing, splicing alternative 3 'or 5' splice sites, and exon skipping, the most common is intron retention. Alternative splicing allows for significant amounts of protein diversity in humans with different functions in cellular processes, developmental processes, and disease states. The number of proteins encoding genes in mammals is about 20,000 to 35,000, but the proteins number is much higher due to alternative splicing [33]. Alternative splicing limits binding characteristics, protein stability, enzymatic activity, post-translational modifications and intracellular localization of various proteins. Many proteins have different domains with variable function can be used protein-protein interaction or enzymatic activity. The exons correspond to. Alternative splicing can mix the protein domains which affects the nature and function of proteins. The two main categories of alternative splicing are protein level and transcript level. Alternative splicing which produces abbreviated protein due to a frame shift and exclusion or inclusion of specific exons having different function such type of splicing is called protein level. In the transcript level, it produces different splice having different translation ability. Thus, it takes part a significant role in expanding protein diversity [34]. The current RNA sequencing data show that more than 95% of human genes yield at least two pre-mRNA and an average of seven mRNA isoforms from a single gene [35]. There are several methods for the detection of splice variants but computational prediction and microarray analysis is a most effective method. The microarray is the most popular, robust and accessible method for detection of splicing variants. RT-PCR and nested PCR can also be used for identification of gene isoform.
Alternative splicing events are linked with numerous human diseases like cardiovascular diseases including cancer, respiratory diseases, neurodegenerative diseases, and metabolic diseases. Multiple activities of tissues like the brain require many different proteins with dispersal function. The mRNA splicing can alter the protein composition leading to many neurological processes. Point mutations cause several human diseases such as ataxia-telangiectasia and neurofibromatosis. Half of the patients are suffering from the disease as a result of a mutation in pre-mRNA [36]. In some splicing disorders, the regulatory factor which is essential for splicing process become mutated and disturb splicing activity. The mutation of splicing factor altered pre-mRNA splicing. It can lead to serious disorders or mild disease [37]. According to the cell type, alternative splicing regulates developmental stage and disease stages. The detection of splice site is not exactly known, but some tissue-specific factor takes part in alternative splicing [38]. Approximately 50% human genetic disease including cancer is linked to the mutation of enhancer or silencer causing alternative exon formation. [39,40]. Notably, the aberrantly spliced variants are frequently found in cancer. It demonstrates that some isoforms of Tumor suppressor gene can play a fundamental role in cancer survival [41]. The accumulation of complex genetic and epigenetic mutation result cancer. The cancer cell starts irregular growth and metastases to the neighbor cells develop a malignant tumor. The splicing of cancer associated gene disturbs cell cycle, apoptosis and signal transduction pathways [42,43].
The genomic instability, aberrant alternative splicing, and sequence substitution frequently occur in cancer and results in invalid and dysfunctional proteins. The isoform produced in this process help in differentiation and survival of cancer cells. Alternative splicing has been determined in many types of cancers, and the most common case is mutually exclusive exon [42]. The splice variants of some tumor suppressor genes like BRCA1, Tensin homolog deleted on chromosome TEN (PTEN) and P53 is linked to cancer. The isoform of these tumor suppressor genes contributes to the advancement of diagnostic and therapeutic methods [44]. For example, the isoform of P53 protein produced through alternative splicing affects various biological process including dysregulation effect in tumorigenesis [45]. The genome-wide analysis studies allow us to observe the relationship between alternative splicing and tumorigenesis. The cancer marker can be identified in cancer research because it will make easy to determine the prognosis and therapy of cancer. Alternative splicing produces a miscellaneous variety of protein isoform having a unique function and the ability to respond differently with several medicinal products. Isoform-specific therapy is used to target individual protein isoform. For example, Etoricoxib, (COX-2 inhibitor) is used for the treatment of the inflammatory condition. Some small molecules can be used to target gene expression instead of isoform targeting. Some drugs have been designed to modify alternative splicing either targeting splicing factor (trans-acting elements) or splicing factor associated proteins. Several methods have been proposed for the use of traditional small molecule compounds based on oligonucleotides and RNAi-based gene therapy for drug development [46]. The alternative transcript which is produced by the splicing process can be conceded an excellent biomarker of cancer.
Primary Splicing Defect. The pre- mRNA splicing to mRNA without any mutation results in corrected protein. The (GU➔AU) point mutation in pre-mRNA results in no transcription to mRNA and protein.
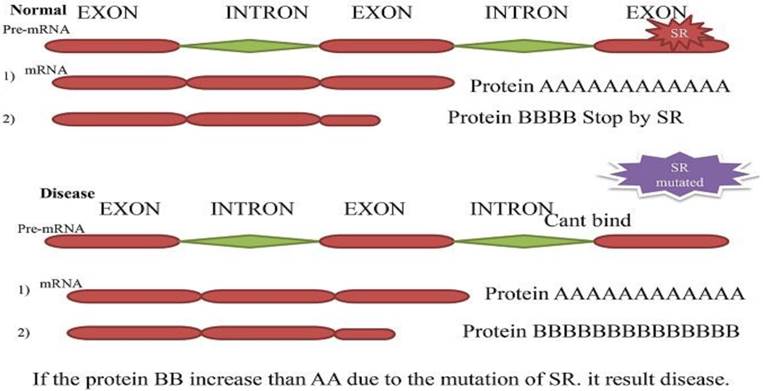
Secondary Splicing defect. The two-resulted mRNA and protein (A and B). In typical situation splicing factor blocks one splice site and second produce a significant amount of protein (A) than B. While in a diseased condition, the muted splicing factor cannot bind to pre-mRNA anymore, and both can produce protein. The protein (B) produces more than (A) and cause disease.
Roles of Smad4 in cancer
Inactivation of smad4 in cancers
Smad4 plays a key role in TGF-β signaling pathways and has been reported a tumor suppressor. The Smad4 expression is deregulated in several types of cancers [47]. The overexpression of Smad4 increases apoptosis and suppresses cell proliferation [48]. Smad4 is one of the key signal transducers of TGF-β which regulates cell function and pancreatic cancer development. The previous studies show that 55% of pancreatic cancers have inactivated Samd4 [19,49,50]. TGF-β signaling pathways are dysregulated in pancreatic cancer [51]. Approximately 90 percent of pancreatic carcinoma has Loss of Heterozygosity (LOH) at 18q chromosome. Recently, many researchers are trying to explore the function of Smad4 in the compound signal mechanism of TGF-β in cancer initiation and progression [52]. One of the best-characterized events in PC is a Smad4 loss. Smad4 is homozygously deleted in approximately 30%, 20% inactivated and 90% Allelic loss in all PC [53]. The mutation in Smad4 protein makes it more rapidly degradable [54]. The down regulations of Smad4 cause to disturb TGF-β induces apoptosis and cell cycle arrest. The restoration of Smad4 can reverse the invasive phenotype and can reduce proliferation rate in PC cells [55-57]. Smad4 restoration in many Smad4 null pancreatic tumor cell lines inhibited invasion and angiogenesis but did not affect proliferation [55]. Smad4 expressing PC Cells improve TGF-β mediated Epithelial-Mesenchymal Transition (EMT) with decreased expression of β catenin and E-cadherin and increased vimentin expression [58]. The loss of Smad4 increases the development of k-ras initiated neoplasms to the high-grade tumor. The k-ras oncogene activation and deactivation of tumor suppressor genes like SMAD4, p16, and p53 are the common events in pancreatic cancer [59]. The most efficient tumor suppressor is Smad4, but its function is highly different in pancreatic and Head and neck cancer. In head and neck cancer, the loss of Smad4 can promote and initiate tumor formation while in pancreatic cancer Smad4 loss can only develop metastases but can't initiate tumor formation [60]. Smad4 is also commonly inactivated in gastrointestinal carcinomas. Analysis of colon carcinoma shows that there are 47 proteins species depend on Smad4 expression. Smad4 takes part in the process of apoptosis, proliferation, and differentiation [61]. The mRNA expression of Smad4 in the metaplasia-dysplasia-adenocarcinoma gradually reduced. The promoter methylation has been found in 70% of primary adenocarcinoma. A quarter of adenocarcinoma samples showed abnormal isoform of a Smad4 protein having no change in gene sequence. The Smad4 locus is also affected by chromosomal rearrangement in esophageal cell lines. The antiproliferative response has been restored by the transient transfection of Smad4 cDNA.
Smad4 loss or knock down the result to induce tumorigenicity, angiogenesis, metastasis, invasion, migration, and resistance to 5FU. Smad4 loss results in the activation of Akt pathways that cause the upregulation of antiapoptotic protein Bcl-w, Bcl-2, and survivin. Smad4 loss induces resistance to 5FU with Akt pathway activation in CRC patients [62]. The absence of Smad4 also upregulates VEGF expression in CRC cell [63]. Overexpression of VEGF is directly related to drug resistance, increased vascular density, and development of metastasis [64]. The overexpression of Smad4 promoted tumor cell apoptosis, repressed VEGF-A and -C expression in vivo and in vitro, but had no effect on cell proliferation and migration [65].
Smad4 plays a fundamental role in the TGF-β signaling pathway. The loss of Smad4 occurs in 55% of PDAC [66]. It is downregulation or loss has been found at a malignant stage in various types of cancer. The loss of Smad4 also occurs in human head and neck squamous cell carcinoma (HNSCC) [67]. The Smad4 deletion causes the formation of Stomach, skin, and mammary gland cancer in mice. It affects both initiation and progression of tumorigenesis in these tissues [68-70]. The inactivation of SMAD4 in tumors emphasizes its role as a tumor suppressor gene. It has been reported that the loss of SMAD4 expression in vascular endothelial cells progress the invasion of ovarian cancer [71].
There are several causes of down-regulation of Smad4 containing a deletion, methylation, and protein modification [72]. The Smad4 inactivation may occur by the abolition of both alleles (homozygous deletion) or by one allele intragenic mutation together with the deletion of another allele (LOH) [73]. Approximately 50% of pancreatic cancer and 15% of colorectal cancer (CRC) reported muted Smad4 [74]. Recently, two mutations have been identified in the C-terminal domain of the Smad4, D351H (Asp351-His) and D537Y (Asp537- Tyr) in human CRC cell line [75]. Many missense mutations derived from a tumor which disturb the establishment of a heteromeric complex between R-Smads and Co- Smad (Smad2 and Samd4). The heteromeric complex translocates to the nucleus and binds with other transcriptional factors to regulate the gene expression [76]. The Smad4 protein is crucial for human physiology, and the mutation of Smad4 has been found in wide range of human cancer. C324Y mutation of Smad4 has been secluded from a nodal metastasis of papillary thyroid cancer. The C324Y mutant of Smad4 co-expressed with wild-type Smad4 increases homo-oligomerization of Smad4 with Receptor Smads and increases nuclear localization. The overexpression of Smad4 324Y shows reduction to the antiproliferative response of TGF-beta in FRTL-5 cell line. Smad4 C324Y plays a key role in thyroid cancer [77]. Smad4 is also mutated in 30% of biliary and colon cancers and some other cancer like lungs, ovarian, head, and neck, prostate, breast, esophageal, bladder, gastric, kidney and liver [78]. The role of Smad4 in tumor formation and embryonic development has been studied by mouse models. The mutations in Smad4 and APC (a colon tumor suppressor) have been observed in compound heterozygous mice. The mutation in both (Smad4 and APC) develop more malignant colon tumor as compared to only APC heterozygous mutation. It shows that Smad4 plays a pivotal role in the malignant development of colon tumor. The gastric polyposis and tumors can be developed in Smad4 heterozygous mice in old age [79].
Alternative splicing variants of smad4 in cancers
The smad4 act is a transporter between nucleus and cytoplasm. In unstimulated cells, the R-Smads and Co-Smad are bounded with microtubules, and TGF-β triggers these Smads to the nucleus [80]. These activated Smad complexes regulate transcription. The adjacent part of the MH2 domain in linker region take part in transcriptional activation and facilitated by the histone acetyltransferase coactivator (P300/CBP) [78,81]. Different isoforms of Smad4 have been identified with the deletion of the various exons in the linker region in different cancers. The six isoforms of Smad4 have been identified in Hacat cell. Any of the five exons in the linker region between the end of exon two and beginning of exon eight can be deleted. The deletion of exons in linker region is not a common property of all Smads. The Smad4 linker region is necessary for transcriptional activation, rather than the formation of transcription factor complexes. The linker region is also not needed for the association with activated Smad2. The human Smad4 exon 3 has a functional leucine-rich NES, which is compulsory for nuclear export. The exon 3 is critical but not sufficient. The important point is the boundary between exon 3 and four [82]. A portion of exon 7 and exon 6 adjacent to the MH2 domain (275-322 amino acids) is required for transcription [48,81]. The isoforms having deleted exon (∆4-7) is almost lacking the entire linker region is found in papillary thyroid carcinoma. These alterations of Smad4 may be involved in an early tumorigenesis [83].
Smad4 Isoforms with deletion of different exons in different cells
| Alternatively Spliced Variants | Mutation or deletion of exon | Normal cell/ cancer cells | Reference |
|---|---|---|---|
| Smad4 ∆E5 and ∆6, ∆4-6 | Linker region | Neuroblastomas and low level at normal tissue/ MDA-MB231 cells and Hacat cell line in both tumor cell and less in a normal cell. | [84,85] |
| Smad4 ∆6 and ∆4-7 | Entire linker region | Hacat cell line ∆4-7 also find in papillary thyroid carcinoma | |
| Smad4 ∆cod95-293, ∆117-373 | Internal exon-exon rearrangement | Frameshifts mutation with stop codon downstream | |
| Smad4 ∆190-240 | misses part of exon 4 and 5 | Found in Papillary thyroid carcinoma | |
| Samd4(∆-6, ∆5-6, ∆4-5-6, ∆4-5-6-7) | Linker region | Papillary thyroid carcinoma | [83] |
| Smad4 ∆3, ∆4, ∆5-6, ∆6, ∆4-6 and ∆4-7 | Hacat cell | [82] |
Alternative splicing plays a key role in gene regulation. The Splicing variance is involved in many cell and development processes such as axon guidance, apoptosis, sex determination and cell excitation. [86]. The abnormality in alternative splicing is linked with both inherited and acquired human genetic disorder. From last few years, it has been recognized that abnormal splicing of Some tumor suppressor gene causes tumorigenesis [87]. Some commonly reported alternative splicing in various human cancers like KLK12 [88], BRCA1 [89], PRMT2 and CDC25 phosphatases are found in breast cancer;; androgen receptor found in prostate cancer; TIMP1 and CD44 in colon cancer; calpain 3 in malignant melanoma; Bcl-xl and CD44 in lung cancer; KLF6 in liver cancer and LOXL4, GRB7 in ovarian cancer. The isoforms create during Alternative splicing (AS) may have continuous tumor formation or tumor inhibitor. That is why alternatively spliced variants is a potential biomarker for cancer diagnosis and used for treatment based on splice correction or chemotherapy to impair alternative splicing [90]. Alternative splicing is common in both normal gene regulatory process and certain disease including cancer. Genetic instability is encouraged due to DNA damage. The damaged DNA promotes genetic instability and harmful to the cell if left unrepaired [91]. There are two broad categories of genetic instability, microsatellite instability and chromosomal instability (CIN). Microsatellite instability involves defective DNA repair causing a change in DNA sequence while CIN is characterized by gain or loss of the chromosomal part. CIN is the causative force of tumorigenesis [92]. DNA damage is the essential component of human neoplasia and promotes genetic instability. Chromosomal mutations are predominantly found in human cancers [93]. The genetic instability increase due to Alternative splicing of DNA damage gene [94]. The transcript stability is affected by the introduction of Stop codons and leading mRNA degradation [95]. The Smad4 tumor suppressor function has been studied regarding of TGF-beta signaling. The mutation rate of the TGF-β signaling pathway is more than 80% especially highly mutated in pancreatic cancer. The TGF-beta signaling pathways component mutation is approximately 1, 4, 55 and 30% of TGFBR1, TGFBR2, Smad4 and P15 respectively. The restoration of Smad4 in the colon, breast, and pancreatic cancer can repair the TGF-beta mediated transcriptional activation, apoptosis, and growth inhibition.
Several isoforms of Smad4 have been found in various types of cancer. It shows that these isoforms compete against a cancerous cell or help in cancers development. Smad4 is vital in the TGF-β pathway and obligatory for transcriptional activation. Smad4 not only needed for nuclear translocation but also promote Smad2/Smad4 binding and FAST-1 complex with DNA to stimulate transcription [96]. Several parts of TGF-beta signaling pathways are called commonly mutated in various cancer, especially in the levels of Smads gene. Many epithelial tumors show missense, nonsense, point and frameshift mutations in Smad4 [97,98].
Pre-mRNA splicing is a complicated and comprehensive mechanism inside the nucleus and is a normal cause of cancer, resulting in the errors in the gene expression.
A transcription factor, cell signal transducer, and extracellular matrix are the most affected proteins. Currently, Antibodies against AS products is in clinical trials, but competitive RT-PCR is used as a simple diagnostic test. To understand the mechanism, function and role of the Alternative splicing in cancer may deliver a new direction for therapeutic information in future studies about cancer. In this review, we summarized alternative splicing of Smad4 gene in cancer.
Mechanism of Alternative splicing due to DNA damage. Mutation in splicing factor and DNA damage response related gene leads to alternative splicing.
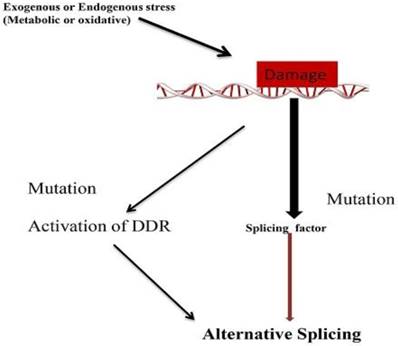
Conclusions
Alternative splicing of mRNA precursors is a ubiquitous machinery for the production of multiple transcripts with different functions from a genomic site in a mammalian cell. Aberrant splicing of tumor suppressor/oncogene gene has been found in many cancers to express a protein that promotes cell cycle or inhibit apoptosis. Therefore, it is critical to recognize the transcripts variants of a different oncogene and tumor suppressor gene. Smad4 is one of tumor suppressor gene and play a key mediator in the TGF-β signaling pathway. The splicing variants of Smad4 have been found in several cancers particularly pancreatic cancers. The imbalance of Smads signaling pathway can harm many cellular functions even lead to disease such as cancer. The identification of these transcripts is not only used as a biomarker but also prognostic and therapeutic purpose. The process of Alternative splicing is a possible target for gene therapy because alternative gene product is consistent with the activity in cancer. Therefore, a study on Smad4 isoforms and the mechanisms which regulate alternative splicing will provide a potential biomarker and therapeutic tool for cancer treatment. The research related with alternative splicing will be open a new window to solve the problem of tumorigenesis.
Acknowledgements
The work was funded by the Natural Science Foundation of China (No. 31670952, To Liling Tang), the Fundamental Research Funds for the Central Universities (No.2018CQDYSG0021, To Liling Tang) and Scientific Research Foundation for Doctors of the Affiliated Hospital of Southwest Medical University (To Jianguo Feng).
Conflict of Interest
The authors confirm that this article content has no conflict of interest.
References
1. Samanta D, Datta PK. Alterations in the Smad pathway in human cancers. Front Biosci. 2012;17:1281-93
2. Upadhyay A, Moss-Taylor L, Kim MJ, Ghosh AC, O'Connor MB. TGF-β Family Signaling in Drosophila. Cold Spring Harb Perspect Biol. 2017;9(9):a022152
3. Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science (80-). 2002;296(5573):1646-7
4. Wrana JL. Crossing Smads. Sci STKE. 2000;2000(23):re1
5. Jung B, Staudacher JJ, Beauchamp D. Transforming Growth Factor Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology. 2017;152(1):36-52
6. Tao S, Sampath K. Alternative splicing of SMADs in differentiation and tissue homeostasis. Development Growth and Differentiation. 2010;52:335-42
7. Johnson MD. Transforming Growth Factor Beta Family in the Pathogenesis of Meningiomas. World Neurosurg. 2017;104:113-9
8. Imamura T, Hikita A, Inoue Y. The roles of TGF-?? Signaling in carcinogenesis and breast cancer metastasis. Breast Cancer. 2012;19(2):118-24
9. Yagi K, Furuhashi M, Aoki H, Goto D, Kuwano H, Sugamura K. et al. c-myc is a downstream target of the Smad pathway. J Biol Chem. 2002;277(1):854-61
10. Zhang HT, Fei QY, Chen F, Qi QY, Zou W, Wang JC. et al. Mutational analysis of the transforming growth factor beta receptor type I gene in primary non-small cell lung cancer. Lung cancer. 2003;40(3):281-7
11. Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME. et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27-36
12. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes and Development. 2005;19:2783-810
13. Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295-309
14. de Caestecker MP, Piek E, Roberts a B. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst. 2000;92(17):1388-402
15. Luo K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017;9(1):a022137
16. Zhang H, Du L, Zhong Y, Flanders KC, Roberts JD. Transforming growth factor-beta stimulates Smad1/5 signaling in pulmonary artery smooth muscle cells and fibroblasts of the newborn mouse through ALK1. Am J Physiol - Lung Cell Mol Physiol. 2017;313(3):L615-L627
17. Ikushima H, Miyazono K. TGF-?? signal transduction spreading to a wider field: A broad variety of mechanisms for context-dependent effects of TGF-?? Cell and Tissue Research. 2012;347:37-49
18. ten Dijke P, Hill CS. New insights into TGF-b-Smad signalling. Trends Biochem Sci. 2004;29:265-73
19. Singh P, Wig JD, Srinivasan R. The Smad family and its role in pancreatic cancer. Indian J Cancer. 2011;48(3):351-60
20. Ormanns S, Haas M, Remold A, Kruger S, Holdenrieder S, Kirchner T. et al. The Impact of SMAD4 Loss on Outcome in Patients with Advanced Pancreatic Cancer Treated with Systemic Chemotherapy. Int J Mol Sci. 2017;18(5):1094
21. Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers (Basel). 2017;9(5):42
22. Hayashi H, Kohno T, Ueno H, Hiraoka N, Kondo S, Saito M. et al. Utility of assessing the number of mutated KRAS, CDKN2A, TP53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas. 2017;46(3):335-40
23. Cook T, Urrutia R. TIEG proteins join the Smads as TGF-β-regulated transcription factors that control pancreatic cell growth. Am J Physiol Gastrointest Liver Physiol. 2000;278(4):513-21
24. Calva D, Dahdaleh FS, Woodfield G, Weigel RJ, Carr JC, Chinnathambi S. et al. Discovery of SMAD4 promoters, transcription factor binding sites and deletions in juvenile polyposis patients. Nucleic Acids Res. 2011;39(13):5369-78
25. Macias MJ, Martin-Malpartida P, Massagué J. Structural determinants of Smad function in TGF-?? signaling. Trends in Biochemical Sciences. 2015;40:296-308
26. Miyazawa K, Miyazono K. Regulation of TGF-?? family signaling by inhibitory smads. Cold Spring Harb Perspect Biol. 2017;9(3):a022095
27. Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M. et al. A mechanism of suppression of TGF-b/SMAD signaling by NF-kB/RelA. Genes Dev. 2000;14(2):187-97
28. Yan X, Liu Z, Chen Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai). 2009;41(4):263-72
29. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685-700
30. Massague J. TGF[beta] signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616-30
31. Massagué J. TGF-β signaling in development and disease. FEBS Lett. 2012;586(14):1833
32. Sugnet CW, Kent WJ, Ares M, Haussler D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac Symp Biocomput. 2004;77:66-77
33. Rahmutulla B, Matsushita K, Nomura F. Alternative splicing of DNA damage response genes and gastrointestinal cancers. World J Gastroenterol. 2014;20(46):17305-13
34. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921
35. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413-5
36. Ars E, Serra E, García J, Kruyer H, Gaona A, Lázaro C. et al. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9(2):237-47
37. Meshorer E, Soreq H. Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends in Neurosciences. 2006;29:216-24
38. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457-63
39. Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6(5):386-98
40. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285-98
41. Skotheim RI, Nees M. Alternative splicing in cancer: Noise, functional, or systematic? International Journal of Biochemistry and Cell Biology. 2007;39:1432-49
42. Venables JP. Unbalanced alternative splicing and its significance in cancer. BioEssays. 2006;28:378-86
43. Brinkman BMN. Splice variants as cancer biomarkers. Clinical Biochemistry. 2004;37:584-94
44. Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, Matsuda S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochemical and Biophysical Research Communications. 2011;413:395-9
45. Wei J, Zaika E, Zaika A. P53 family: Role of protein isoforms in human cancer. Vol. 2012, Journal of Nucleic Acids. 2012;2012:687359
46. Le KQ, Prabhakar BS, Hong WJ, Li LC. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol Sin. 2015;36(10):1212-8
47. Waite KA, Eng C. From developmental disorder to heritable cancer: it's all in the BMP/TGF-beta family. Nat Rev Genet. 2003;4(10):763-73
48. Yang Z, Zhong L, Zhong S, Xian R, Yuan B. Adenovirus encoding Smad4 suppresses glioma cell proliferation and increases apoptosis through cell cycle arrest at G1 phase. Int Immunopharmacol. 2015;25(1):169-73
49. Van Heek T, Rader AE, Offerhaus GJA, McCarthy DM, Goggins M, Hruban RH. et al. K-ras, p53, and DPC4 (MAD4) alterations in fine-needle aspirates of the pancreas: A molecular panel correlates with and supplements cytologic diagnosis. Am J Clin Pathol. 2002;117(5):755-65
50. Hruban RH, Wilentz RE, Goggins M, Offerhaus GJ, Yeo CJ, Kern SE. Pathology of incipient pancreatic cancer. Ann Oncol. 1999;10(4):9-11
51. Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6(1):1-8
52. Xia X, Wu W, Huang C, Cen G, Jiang T, Cao J. et al. SMAD4 and its role in pancreatic cancer. Tumor Biology. 2014;36:111-9
53. Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Science. 2006:97 1-7
54. Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 2000;97(9):4820-5
55. Duda DG, Sunamura M, Lefter LP, Furukawa T, Yokoyama T, Yatsuoka T. et al. Restoration of SMAD4 by gene therapy reverses the invasive phenotype in pancreatic adenocarcinoma cells. Oncogene. 2003;22(44):6857-64
56. Yasutome M, Gunn J, Korc M. Restoration of Smad4 in BxPC3 pancreatic cancer cells attenuates proliferation without altering angiogenesis. Clin Exp Metastasis. 2005;22(6):461-73
57. Shen W, Tao GQ, Li DC, Zhu XG, Bai X, Cai B. Inhibition of pancreatic carcinoma cell growth in vitro by DPC4 gene transfection. World J Gastroenterol. 2008;14(40):6254-60
58. Zhao S, Venkatasubbarao K, Lazor JW, Sperry J, Jin C, Cao L. et al. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor beta-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res. 2008;68(11):4221-8
59. Liu F. SMAD4/DPC4 and Pancreatic Cancer Survival. Clin Cancer Res. 2001;7:3853-6
60. Malkoski SP, Wang XJ. Two sides of the story? Smad4 loss in pancreatic cancer versus head-and-neck cancer. FEBS Letters. 2012;586:1984-92
61. Stühler K, Köper K, Pfeiffer K, Tagariello A, Souquet M, Schwarte-Waldhoff I. et al. Differential proteome analysis of colon carcinoma cell line SW480 after reconstitution of the tumour suppressor Smad4. Anal Bioanal Chem. 2006;386(6):1603-12
62. Zhang B, Chen X, Bae S, Singh K, Washington MK, Datta PK. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br J Cancer. 2014;110(4):946-57
63. Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM. et al. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011;71(3):998-1008
64. Samuel S, Fan F, Dang LH, Xia L, Gaur P, Ellis LM. Intracrine vascular endothelial growth factor signaling in survival and chemoresistance of human colorectal cancer cells. Oncogene. 2011;30(10):1205-12
65. Li X, Li X, Lv X, Xiao J, Liu B, Zhang Y. Smad4 Inhibits VEGF-A and VEGF-C Expressions via Enhancing Smad3 Phosphorylation in Colon Cancer. Anat Rec. 2017;300(9):1560-1569
66. Leung L, Radulovich N, Zhu CQ, Wang D, To C, Ibrahimov E. et al. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLoS One. 2013;8(12):e84366
67. Nature Publishing Group. X, Brodie SG, Yang X, Im YH, Parks WT, Chen L, et al. Oncogene. Vol. 19, Oncogene. Scientific & Medical Division, Macmillan Press. 1987:1868-1874 p
68. Qiao W, Li AG, Owens P, Xu X, Wang XJ, Deng CX. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25(2):207-17
69. Yang L, Mao C, Teng Y, Li W, Zhang J, Cheng X. et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res. 2005;65(19):8671-8
70. Li W, Qiao W, Chen L, Xu X, Yang X, Li D. et al. Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development. 2003;130(24):6143-6153
71. Yang J, Wang Y, Zeng Z, Qiao L, Zhuang L, Gao Q, Ma D, Huang X. Smad4 deletion in blood vessel endothelial cells promotes ovarian cancer metastasis. International Journal of Oncology. 2017;50(5):1693-1700
72. Onwuegbusi BA, Aitchison A, Chin SF, Kranjac T, Mills I, Huang Y. et al. Impaired transforming growth factor beta signalling in Barrett's carcinogenesis due to frequent SMAD4 inactivation. Gut. 2006;55(6):764-74
73. Ralph HH, Offerhaus GJA, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-Suppressor genes in pancreatic cancer. J Hepatobiliary Pancreat Surg. 1998;5(4):1868-6982
74. Ahmed S, Bradshaw AD, Gera S, Dewan ZM, Xu R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. Journal of Clinical Medicine. 2017;6(1):5
75. Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS. et al. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci U S A. 2001;98(17):9719-23
76. Wu JW, Fairman R, Penry J, Shi Y. Formation of a Stable Heterodimer between Smad2 and Smad4. J Biol Chem. 2001;276(23):20688-94
77. D'Inzeo S, Nicolussi A, Donini CF, Zani M, Mancini P, Nardi F. et al. A novel human Smad4 mutation is involved in papillary thyroid carcinoma progression. Endocr Relat Cancer. 2012;19(1):39-55
78. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. New Engl J. 2000;342(18):13508
79. Weinstein M, Yang X, Deng CX. Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine and Growth Factor Reviews. 2000;11:49-58
80. Dong C, Li Z, Alvarez R, Feng XH, Goldschmidt-Clermont PJ. Microtubule binding to Smads may regulate TGF beta activity. Mol Cell. 2000;5(1):27-34
81. De Caestecker MP, Yahata T, Wang D, Parks WT, Huang S, Hill CS. et al. The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J Biol Chem. 2000;275(3):2115-22
82. Pierreux CE, Nicolás FJ, Hill CS. Transforming growth factor beta-independent shuttling of Smad4 between the cytoplasm and nucleus. Mol Cell Biol. 2000;20(23):9041-54
83. Lazzereschi D, Nardi F, Turco A, Ottini L, D'Amico C, Mariani-Costantini R. et al. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene. 2005;24(34):5344-54
84. de Winter JP, Roelen BA, ten Dijke P, van der Burg B, van den Eijnden-van Raaij AJ. DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-beta1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene. 1997;14(16):1891-9
85. Kageyama H, Seki N, Yamada S, Sakiyama S, Nakagawara A. DPC4 splice variants in neuroblastoma. Cancer Lett. 1998;122(1-2):187-93
86. Wang G-S, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8(10):749-61
87. Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119(Pt 13):2635-41
88. Talieri M, Devetzi M, Scorilas A, Pappa E, Tsapralis N, Missitzis I. et al. Human kallikrein-related peptidase 12 (KLK12) splice variants expression in breast cancer and their clinical impact. Tumour Biol. 2012;33(4):1075-84
89. Tammaro C, Raponi M, Wilson DI, Baralle D, Rappsilber J, Ryder U. et al. BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem Soc Trans. 2012;40(4):768-72
90. Tang JY, Lee JC, Hou MF, Wang CL, Chen CC, Huang HW. et al. Alternative splicing for diseases, cancers, drugs, and databases. The Scientific World Journal. 2013;2013:703568
91. Metcalf JL, Bradshaw PS, Komosa M, Greer SN, Stephen Meyn M, Ohh M. K63-Ubiquitylation of VHL by SOCS1 mediates DNA double-strand break repair. Oncogene. 2014;33:1055-65
92. Lee AJX, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA. et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71(5):1858-70
93. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623-7
94. Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A. et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002;3(3):248-54
95. Maquat LE, Carmichael GG. Quality Control of mRNA Function. Cell. 2017Apr30;104(2):173-6
96. Liu F, Pouponnot C, Massagué J. Dual role of the Smad4/DPC4 tumor suppressor in TGFbeta-inducible transcriptional complexes. Genes Dev. 1997;11(23):3157-3167
97. Miyaki M, Kuroki T. Role of Smad4 (DPC4) inactivation in human cancer. Vol. 306, Biochemical and Biophysical Research Communications. 2003;306:799-804
98. Iacobuzio-Donahue CA, Song J, Parmiagiani G, Yeo CJ, Hruban RH, Kern SE. Missense Mutations of MADH4 characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res. 2004;10(5):1597-1604
Author contact
![]() Corresponding authors: tanglilingedu.cn/ffengjianguocom
Corresponding authors: tanglilingedu.cn/ffengjianguocom