Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(10):1863-1869. doi:10.7150/jca.24950 This issue Cite
Research Paper
Feasibility of continuing crizotinib therapy after RECIST-PD in advanced non-small cell lung cancer patients with ALK/ROS-1 mutations
Jun Liu*, Shaohua Cui*, Feng Pan, Yiqian Ni, Hua Zhong, Liwen Xiong, Bo Jin, Tianqing Chu, Aiqin Gu, Liyan Jiang ![]()
Department of Pulmonary Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University, Shanghai, China
*These authors have contributed equally to this work
Received 2018-1-15; Accepted 2018-3-2; Published 2018-4-27
Abstract
Objectives: To study whether ongoing clinical benefits of continuing anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) inhibition are achieved by crizotinib treatment post progressive disease (PD) in advanced non-small cell lung cancer (NSCLC) patients harboring ALK/ROS1 mutations.
Materials and methods: Demographic and clinicopathologic parameters were collected from 38 patients who continued crizotinib therapy beyond Response Evaluation Criteria in Solid Tumors (RECIST)-defined PD and analyzed. After adjusting for potential confounding factors, factors influencing the time from RECIST-PD to crizotinib discontinuation (progress-free survival 2, PFS2) were analyzed.
Results: The median time from first dose treatment to RECIST-PD (PFS1) was 9.6 months (95% CI 5.6-13.6 months). The estimated median PFS2 was 5.9 months (95% CI 0.1-11.7 months). Six- and twelve-month crizotinib treatment probabilities after initial PD were 42.1% (95% CI 25.7-58.6%) and 21.1% (95% CI 7.5-34.6%), respectively. Patients who demonstrated RECIST-PD due to new lesions had a longer median PFS2 compared to patients who were attested to enlargement of original lesions (10.0 versus 2.4 months, p = 0.009). The median PFS2 was numerically longer among patients who received local therapy compared to those who did not receive local therapy, however the difference was not significant (9.9 versus 4.2 months, p = 0.094). Multivariable Cox regression analysis showed that only the progression pattern [new lesions versus enlargement of original lesions, HR = 0.329 (95% CI 0.138-0.782), p = 0.012] remained an independent prognostic factor of PFS2.
Conclusion: Continuation of crizotinib therapy after RECIST-PD in Chinese NSCLC patients with positive ALK/ROS1 mutations is feasible in clinical practice.
Keywords: Anaplastic lymphoma kinase, Crizotinib, C-ros oncogene 1, Non-small-cell lung cancer, RECIST, Treatment beyond disease progression
Introduction
Lung cancer has a high morbidity and mortality rate and is the main cause of global cancer-related deaths [1,2]. In recent years, molecular targeted therapies using tyrosine kinase inhibitors (TKIs) for non-small-cell lung cancer (NSCLC) patients harboring driver gene mutations have made a revolutionary impact on the care of this disease [3-5]. In recent studies, epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), and c-ros oncogene 1 (ROS1) have been identified as driver genes of NSCLC [3,5]. ALK gene rearrangements, leading to an in-frame fusion protein with oncogenic activity in vitro with the echinoderm microtubule- associated protein-like 4 (EML4) gene in NSCLC, were discovered in 2007 [6]. ALK rearrangements are identified in a minority of NSCLC cases, occurring in 2% to 7% of all NSCLC patients [7,8]. Additionally, ROS1 rearrangement was found in different types of malignant tumors, including NSCLC [9-11]. ROS1 gene rearrangements have recently been identified and are observed in 1% to 2% of all lung cancer patients [11,12].
Crizotinib is an ATP-competitive small- molecular TKI that targets ALK, ROS1, and c-MET [11,13]. Two randomized Phase III trials (PROFILE 1014 and 1007) have shown that crizotinib showed significant improvements in the objective response rate (ORR), progression-free survival (PFS), and quality of life when compared to cytotoxic chemotherapy in first-line or non-first line settings in ALK-positive advanced NSCLC patients [14,15]. The kinase domains of ALK and ROS1 share about 77% amino-acid identity within ATP-binding sites, indicating a high homology. In the PROFILE 1001 study [16], a total of 50 ROS1-rearranged patients were enrolled. The results indicated that the ORR was 72% (95% CI, 58 to 84) and that the median PFS was 19.2 months (95% CI, 14.4 to not reached), which was similar in clinical efficacy to the ALK-rearranged subgroup.
Response Evaluation Criteria in Solid Tumors (RECIST) is the gold standard and a common approach for tumor therapy evaluation, however it may not be the most appropriate criteria for terminating TKI treatment [17-19]. For example, two single-arm studies of crizotinib in advanced ALK-positive NSCLC, including a phase I trial (PROFILE 1001) and a phase II trial (PROFILE 1005), allowed continuation of crizotinib therapy beyond RECIST-defined progressive disease (PD) in patients who continued to obtain clinical benefits of the drug [20]. Ou et al. conducted a retrospective study in which patients were enrolled from the two clinical trials mentioned above. This study confirmed that continuing ALK inhibition with crizotinib after PD may provide survival benefits to patients with advanced ALK-positive NSCLC [18]. The limited number of patients described of ROS1 mutant may mainly account for the lack of reliable evidence related to continuing ROS1 inhibition with crizotinib after PD in ROS1-rearranged patients [12].
Without reliable evidence indicating that continuing ALK and ROS1 inhibition with crizotinib post RECIST-PD is beneficial for Chinese NSCLC patients, we conducted a retrospective study to investigate the effectiveness of continuing crizotinib treatment post RECIST-PD and determined the patient-specific characteristics associated with longer post PD treatment with crizotinib in these patients.
Patients and Methods
Patients
In this study, data on ALK- or ROS1- rearrangement patients with advanced NSCLC, who received oral crizotinib treatment between July 2013 and December 2016 at the Shanghai Chest Hospital, Shanghai Jiao Tong University (Shanghai, China) were retrospectively analyzed. Three weeks of treatment after initial PD was defined as the cut-off for dividing patients into a continuing or discontinuing treatment group (>3 versus ≤3 weeks, respectively). For most patients, this time frame was within one therapy cycle, and doctors generally decided whether to continue crizotinib treatment within this period of time [18]. Prior to initiation of therapy, all patients underwent baseline computed tomography (CT) of the thorax, enhanced magnetic resonance imaging (MRI) of the brain, electrocardiogram (ECG), and abdominal ultrasound.
This study was approved by the Ethics Committee of Shanghai Chest Hospital, Shanghai Jiao Tong University (Shanghai, China). Each patient signed formal informed consent before enrollment.
Molecular pathology testing
Tumor samples obtained by either surgical or diagnostic procedures were used for ALK and ROS1 mutation detection, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), and immunohistochemical (IHC) analysis were used for ALK-rearrangements detection. FISH was chosen as the testing method of ROS1-rearrangements, and IHC analysis was conducted using a monoclonal D5F3 antibody (Ventana Medical Systems, Tucson, AZ, USA) directed against ALK.
Treatment and response evaluation
All patients received crizotinib therapy 250 mg (twice daily) in 28-day cycles, with appropriate dosing adjustments as needed. To evaluate treatment response, CT scans were performed after the first cycle of crizotinib therapy, then subsequently after every 2 cycles. The tumor response was assessed by the RECIST, version 1.1 [19], including complete response (CR), partial response (PR), stable disease (SD), and PD. Additional locoregional therapy was conducted if necessary, depending on the patients' symptoms, and radiological information obtained.
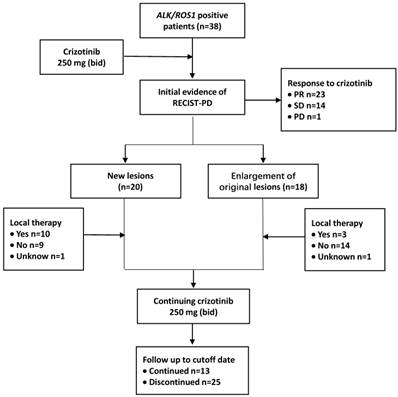
The time from the first dose of crizotinib treatment to the initial imaging evidence of RECIST-defined PD was defined as PFS1. After the disease progressed, crizotinib treatment was continued until unacceptable levels of toxicity were observed or when physician-determined absence of clinical benefits was achieved. The time between the initial imaging evidence of PD and terminating the usage of crizotinib was calculated and defined as PFS2. The cutoff follow-up time for this study was October 31, 2017 (Figure 1).
Treatment and follow-up process of patients. Abbreviations used: ALK: anaplastic lymphoma kinase; ROS1: c-ros oncogene 1; RECIST: Response Evaluation Criteria in Solid Tumors; PD: progressive disease; PR: partial response; SD: stable disease; bid: twice daily
Statistical analysis
The Kaplan-Meier survival method was used to estimate PFS1 and PFS2, whereas log-rank analysis was used to compare statistical significance of differences between patient groups. The Cox proportional hazards model analyses were used to identify the effect of different clinical features on PFS1 and PFS2. The hazard ratio (HR) between two subgroups was assessed using the Cox proportional hazard model with 95% Wald CIs. P < 0.05 was considered statistically significant. Statistical analyses were performed by SPSS® software, version 19.0 (SPSS Inc., Chicago, IL, USA).
Results
Patient characteristics
In this study, a total of 38 patients were enrolled. The demographic and clinicopathologic parameters of these patients are presented in Table 1. For all patients, the mean age was 53 years (range 34-73 years); 17 patients (44.7%) were male and 21 patients (55.3%) were female. Patients tended to be never or light smokers (smoking index <400) (31/38; 81.6%), and all patients had a confirmed adenocarcinoma histology. A total of 33 patients (86.8%) harbored ALK-rearrangements, while the remaining patients harbored ROS1-rearrangements. Nearly half of the patients (18/38; 47.4%) were therapy-naive, while 20 patients (52.6%) had received previous anticancer treatments as either a second- or third (or greater)-line therapy prior to crizotinib administration. The performance status of the Eastern Cooperative Oncology Group (ECOG) of 34 patients was 1 (89.5%), and most patients (31/38; 81.6%) suffered from one or two metastatic sites when receiving crizotinib therapy.
Demographic and clinicopathologic parameters.
| n | % | |
|---|---|---|
| Age, years | ||
| Mean | 53 | |
| Range | 34-73 | |
| Gender | ||
| Male | 17 | 44.7 |
| Female | 21 | 55.3 |
| ECOG performance status | ||
| 0 | 2 | 5.3 |
| 1 | 34 | 89.5 |
| 2 | 2 | 5.3 |
| No. of previous therapies | ||
| 0 | 18 | 47.4 |
| 1 | 15 | 39.5 |
| ≥2 | 5 | 13.2 |
| Radical surgery history | ||
| Yes | 8 | 21.1 |
| No | 30 | 78.9 |
| Histologic type | ||
| Adenocarcinoma | 38 | 100 |
| Non-adenocarcinoma | 0 | 0 |
| Smoking historya | ||
| Never smoker | 30 | 78.9 |
| Smoking index <400 | 1 | 2.6 |
| Smoking index ≥400 | 7 | 18.4 |
| Clinical stage | ||
| IIIB | 4 | 10.5 |
| IV | 34 | 89.5 |
| Gene detection | ||
| ALK | 33 | 86.8 |
| ROS1 | 5 | 13.2 |
| No. of metastatic sites when receiving initial crizotinib therapy | ||
| 0 | 4 | 10.5 |
| 1 | 18 | 47.4 |
| 2 | 13 | 34.2 |
| 3 | 3 | 7.9 |
a Smoking index was calculated as the number of cigarettes smoked per day, multiplied by the number of years of smoking.
Abbreviations used: ECOG: Eastern Cooperative Oncology Group; ALK: anaplastic lymphoma kinase; ROS1: c-ros oncogene 1
Response evaluation to initial crizotinib therapy
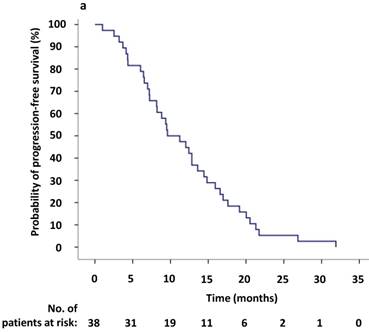
To evaluate the response to crizotinib in the 38 patients according to RECIST, 60.5% of patients (23/38) achieved PR, while 14 patients (36.8%) were evaluated as SD. One patient was assessed as PD because after one month of crizotinib treatment, a new lesion was determined at the lumbar. The ORR (CR and PR) was 60.5% (95% CI 44.2%-76.8%), and the median PFS1 was 9.6 months (95% CI 5.6-13.6 months) (Figure 2,3a). Moreover, the six- and twelve-month crizotinib treatment probabilities for 38 patients were 81.6% (95% CI 68.7-94.5) and 47.4% (95% CI 30.7-64.0), respectively.
The duration of crizotinib treatment in 38 ALK/ROS1-positive patients. Brown bars indicate PFS1, blue bars indicate PFS2.
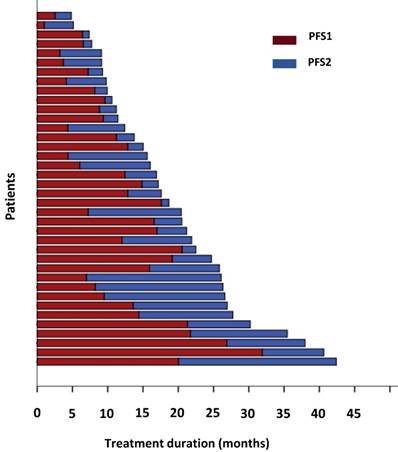
Univariate survival analysis showed that patients who assessed an ECOG performance status of ≤1 had a significant longer PFS1 to initial crizotinib treatment compared to patients who possessed an ECOG performance status of 2 (p = 0.001). The median PFS1 was numerically longer among patients who achieved PR (12.0 months, 95% CI 7.6-16.5 months) when compared to patients who assessed as an SD (8.2 months, 95% CI 4.0-12.3 months), however this difference was not significant. Multivariable Cox regression analysis indicated that only the ECOG performance status [2 versus ≤1, HR = 15.0 (95% CI 2.45-91.42), p = 0.006] was an independent predictor of PFS1. However, only two patients possessed an ECOG performance status of 2.
Characteristics of PD sites
All patients demonstrated RECIST-PD at the study cutoff date. Common disease progression sites included lung (16, 42.1%) and brain (13, 34.2%). In a total of 20 patients (52.6%), PD was confirmed due to the appearance of new lesions, while in the remaining patients (47.4%) enlargement of original lesions was observed. Thirteen patients (34.2%) received locoregional therapy at the time of initial PD. Seven patients with brain progression received palliative brain radiotherapy, and one patient received oral temozolomide instead. Three patients underwent bone radiotherapy, and 1 patient was treated with bone cement on lumbar vertebrae (Table 2).
Post-progression characteristics and clinical outcomes.
| n | % | |
|---|---|---|
| Disease progression site | ||
| Brain | 13 | 34.2 |
| Bone | 4 | 10.5 |
| Lung | 16 | 42.1 |
| Pleura | 2 | 5.3 |
| Subcutaneous nodule | 2 | 5.3 |
| Adrenal gland | 1 | 2.6 |
| Progression pattern | ||
| New lesions | 20 | 52.6 |
| Enlargement of original lesions | 18 | 47.4 |
| Local therapy when receiving PD | ||
| No | 23 | 60.5 |
| Yes | 13 | 34.2 |
| Brain radiation | 7 | 18.4 |
| Bone radiation | 3 | 7.9 |
| Subcutaneous nodule radiation | 1 | 2.6 |
| Bone cement | 1 | 2.6 |
| Temozolomide | 1 | 2.6 |
| Duration of PFS2 | ||
| 0 to <3 months | 13 | 34.2 |
| 3 to <6 months | 9 | 23.7 |
| 6 to <9 months | 3 | 7.9 |
| 9 to <12 months | 5 | 13.2 |
| ≥12 months | 8 | 21.1 |
Abbreviations used: PD: progressive disease; PFS: progression-free survival
Efficacy of continuing crizotinib therapy
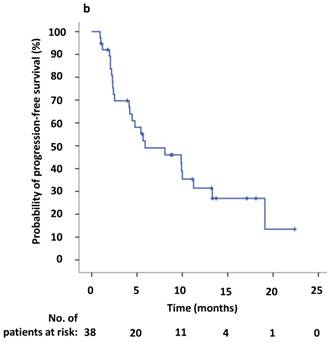
The estimated median PFS2 of the 38 patients was 5.9 months (95% CI 0.1-11.7 months) (Figure 2,3b). The six- and twelve-month crizotinib treatment probabilities after initial PD for 38 patients were 42.1% (95% CI 25.7 - 58.6%) and 21.1% (95% CI 7.5- 34.6%), respectively. Thirteen patients (34.2%) continued to receive crizotinib therapy at the cutoff date.
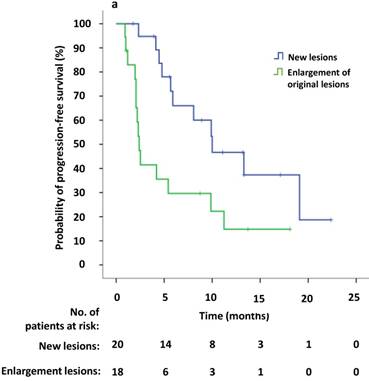
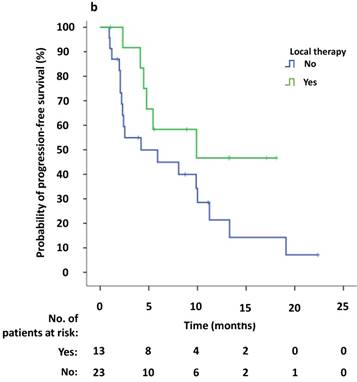
Univariate survival analysis revealed that the median PFS2 was significantly longer in patients that demonstrated new lesions at the time of PD compared to patients who were attested to enlargement of original lesions (10.0 versus 2.4 months, p = 0.009). The median PFS2 after continuing crizotinib treatment was numerically longer among patients receiving local therapy compared to those who did not receive local therapy, however this difference was not significant (9.9 versus 4.2 months, p = 0.094) (Figure 4a, b). Univariate Cox regression analyses showed that significant independent prognostic factors of PFS2 included new lesions [HR = 0.354 (95% CI 0.156-0.804), p = 0.013] and PFS1 [HR = 0.924 (95% CI 0.863-0.989), p = 0.022]. Moreover, multivariable Cox regression analysis showed that only progression pattern [new lesions versus enlargement of original lesions, HR = 0.329 (95% CI 0.138-0.782), p = 0.012] remained an independent prognostic factor of PFS2 (Table 3). At the study cutoff date, the median overall survival (OS) had not yet been reached.
(a) Kaplan-Meier survival analysis for progression-free survival 1 (PFS1) after initial crizotinib treatment prior to RECIST-defined progressive disease (PD). The median PFS1 was 9.6 months (95% CI, 5.6-13.6 months). (b) Kaplan-Meier survival analysis for progress-free survival 2 (PFS2) after continuing ALK/ROS1 inhibition with crizotinib beyond PD. The median PFS2 was 5.9 months (95% CI, 0.1-11.7 months). Tick marks represent censored observations.
(a) Kaplan-Meier survival analysis of PFS2 for continuous crizotinib treatment after identification of PD with new lesions and enlargement of original lesions (p = 0.009, log-rank test). (b) Kaplan-Meier survival analysis of PFS2 for continuous crizotinib treatment with or without local therapy (p = 0.094, log-rank test). Tick marks represent censored observations.
Multivariable Cox proportional hazards regression analyses to identify the relationship between clinical features and PFS2.
| Clinical features | HR a | 95% CI | P value |
|---|---|---|---|
| PFS1 (1-month increments) | 0.943 | 0.883-1.008 | 0.083 |
| Progression pattern (new lesions versus enlargement of original lesions) | 0.329 | 0.138-0.782 | 0.012 |
| Local therapy (yes versus no) | 0.513 | 0.198-1.328 | 0.169 |
aFor categorical variables, HR < 1 favors the first category and HR > 1 favors the second category; for continuous variables, for every unit increase in the variable, there was an increase (HR > 1) or decrease (HR < 1) in the risk of death.
Abbreviations used: HR: hazards ratio; CI: confidence interval; PFS: progression-free survival
Discussion
The present study, including 38 advanced NSCLC-harboring ALK or ROS1 mutations, revealed that crizotinib continuation after RECIST-PD was feasible in clinical practice. The median time between the initial imaging evidence of PD and terminating the usage of crizotinib (mPFS2) was nearly 6.0 months. Most patients developed disease progression despite an initial response to TKIs. In recent years, continuing molecularly targeted therapy beyond RECIST-PD by physicians has increased in frequency [18,21-24]. Moreover, several retrospective studies have suggested that molecularly targeted treatment beyond PD may be feasible and effective in patients with EGFR-positive mutations or ALK positive mutations [18,21,22]. Our findings are consistent with the outcomes of several previously published retrospective studies [18,24].
In a retrospective study [24], it was found that patients who received local therapy for disease progression may have longer periods of continuing crizotinib therapy (PFS2). We drew a similar conclusion using univariate survival analysis, however multivariable Cox regression analysis showed that receiving local therapy might not be an independent prognostic factor. Further analysis indicated that in 10 patients (10/13, 76.9%) who received local treatment, PD occurred due to the formation of new lesions, and in 3 patients (3/13, 23.1%) enlargement of original lesions was observed. Given the above findings, we concluded, based on multivariable Cox regression analysis, that new lesions rather than local therapy remained an independent prognostic factor of PFS2. Additional studies are of great necessity due to the limited number of patients enrolled.
At the study cutoff date, eight patients continued crizotinib therapy from the initial imaging evidence of PD for more than twelve months (8/38, 21.1%). Most of these patients (7/8) harbored ALK-rearrangements. In addition, five patients were therapy-naïve. When PD occurred, the disease progression site in four patients was the brain. In six patients, the formation of new lesions confirmed PD, while the other two patients demonstrated enlargement of originally identified lesions. Four patients received locoregional therapy at the time of initial PD. The PFS1 in these patients ranged from 7.0 to 21.7 months. Defining the demographic and clinicopathologic parameters of these patients was constrained by the limited number of patients.
The difference between ALK- and ROS1-positive subgroups treated with crizotinib was noticed by experts as follows: the ROS1-rearranged subgroup had longer responses [median PFS: 19.2 versus 9.7 months; median duration of response (DOR): 49.1 versus 64.5 weeks, respectively] [16,25]. In a retrospective study, it was concluded that the existence of ROS1 rearrangement was not only a predictive factor of crizotinib therapy, but also a prognostic marker for survival since patients with ROS1 rearrangement had a longer median survival (36.7 months) compared to other oncogene mutant populations, for instance EGFR mutation and ALK-positive NSCLC (25.3 and 23.9 months, respectively) [26]. In our study, we found that the median PFS1 was numerically longer in ROS1-mutant patients (13.6 months 95%, CI 11.9-15.3 months) compared to ALK-mutant patients (9.4 months 95%, CI 7.8-11.0 months), however this difference was not statistically significant (p = 0.432). A similar result was observed for the median PFS2 (9.9 months, 95% CI 0-21.4 versus 5.7 months 95%, CI 1.5-9.9, p = 0.495). Five patients with ROS1-positive NSCLC included in the study may attribute to the small number of patients described in ROS1-rearranged NSCLC [12], which apparently reduced accuracy and creditability of the outcomes.
Although most patients with ALK- and ROS1-rearranged NSCLC responded to crizotinib, tumor progression was inevitable after 1 to 2 years of oral treatment. Development of secondary genetic mutations in the primary oncogene (such as L1196M and G1269A in ALK kinase domain, and G2032R in the ROS1 kinase domain), as well as activation of alternative signaling pathways are known as common resistance mechanisms [27-29]. The exact resistance mechanism may sufficiently guide subsequent therapy. Ceritinib and alectinib are two well-known next-generation inhibitors [30-32]. Ceritinib, a highly potent oral ALK and ROS1 TKI, has proven efficacy against crizotinib-resistant ALK mutations, including L1196M, G1269A, I1171T, and S1206Y [30]. Alectinib is also a potent ALK inhibitor that can inhibit several crizotinib-resistant ALK mutations, including L1196M, G1269A, and F1174L). Alectinib is especially suitable for patients with central nervous system (CNS) metastases due to the penetrable features of the CNS [31, 32]. Recently, the latest National Comprehensive Cancer Network (NCCN) guideline (Version 1.2018, www.nccn.org) recommended continuing use of crizotinib or changing to next-generation ALK inhibitors, such as ceritinib, alectinib, and brigatinib for the treatment of ALK-positive patients with solitary lesions or asymptomatic progression to crizotinib. In this study, we found that patients would benefit from continuing crizotinib beyond PD. Therefore, it is necessary to compare next-generation inhibitors and crizotinib continuation treatment after RECIST-PD to investigate optimal treatment effects on patients who were initially receiving crizotinib therapy.
In conclusion, in this retrospective analysis, we demonstrated that ALK/ROS1-positive NSCLC patients received ongoing clinical benefits when continuing ALK and ROS1 inhibition using crizotinib post RECIST-PD. The demographic and clinicopathologic parameters that were described to be related to PFS2 may help physicians in their decision-making process. The main limitation of this study included a small number of patients enrolled on account of the low-level expression in ALK and ROS1-rearranged and discontinuing crizotinib therapy after RECIST-PD in a proportion of patients. Due to the limitations of this study, additional studies are warranted to investigate the underlying mechanisms of disease progression and to define the best therapeutic strategy to ameliorate prognosis of these patients.
Acknowledgements
This study was supported by the National Nature Science Foundation of China (Grant No. 81472175).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30
2. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Lynch TJ, Bell DW, Sordella R. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129-39
4. Paez JG, Jänne PA, Lee JC. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497-500
5. Ou SH, Bartlett CH, Mino-Kenudson M. et al. Crizotinib for the treatment of ALK rearranged non-small cell lung cancer: a success story to usher in the second decade of molecular targeted therapy in oncology. Oncologist. 2012;17:1351-75
6. Soda M, Choi YL, Enomoto M. et al. Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature. 2007;448:561-6
7. Gridelli C, Peters S, Sgambato A. et al. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40:300-6
8. Kwak EL, Bang YJ, Camidge DR. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693-703
9. Gu T-L, Deng X, Huang F. et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6:e15640
10. Birch AH, Arcand SL, Oros KK. et al. Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One. 2011;12:e28250
11. Bergethon K, Shaw AT, Ou SH. et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863-70
12. Takeuchi K, Soda M, Togashi Y. et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378-81
13. Ou SH, Kwak EL, Siwak-Tapp C. et al. Activity of Crizotinib (PF02341066), a Dual Mesenchymal-Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) Inhibitor, in Non-small Cell Lung Cancer Patient with De Novo MET Amplification. J Thorac Oncol. 2011;5:942-6
14. Shaw AT, Kim DW, Nakagawa K. et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385-94
15. Solomon BJ, Mok T, Kim DW. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167-77
16. Shaw AT, Ou SH, Bang YJ. et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963-71
17. Jackman D, Pao W, Riely GJ. et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357-60
18. Ou SH, Janne PA, Bartlett CH. et al. Clinical beneft of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol. 2014;25:415-22
19. Eisenhauer EA, Therasse P, Bogaerts J. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-47
20. Blackhall F, Ross Camidge D, Shaw AT. et al. Final results of the large-scale multinational trial PROFILE 1005: efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open. 2017;2(3):e000219
21. Lo PC, Dahlberg SE, Nishino M. et al. Delay of treatment change after objective progression on first-line erlotinib in epidermal growth factor receptor-mutant lung cancer. Cancer. 2015;121(15):2570-7
22. Nishie K, Kawaguchi T, Tamiya A. et al. Epidermal growth factor receptor tyrosine kinase inhibitors beyond progressive disease:a retrospective analysis for Japanese patients with activating EGFR mutations. J Thorac Oncol. 2012;7(11):1722-7
23. Park K, Yu CJ, Kim SW. et al. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients With Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer: The ASPIRATION Study. JAMA Oncol. 2016;2(3):305-12
24. Hong X, Chen Q, Ding L. et al. Clinical benefit of continuing crizotinib therapy after initial disease progression in Chinese patients with advanced ALK-rearranged non-small-cell lung cancer. Oncotarget. 2017;8(25):41631-40
25. Camidge DR, Bang YJ, Kwak EL. et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-9
26. Scheffler M, Schultheis A, Teixido C. et al. ROS1 rearrangements in lung adenocarcinoma: prognostic impact, therapeutic options and genetic variability. Oncotarget. 2015;6:10577-85
27. Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31(31):3987-96
28. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227-35
29. Awad MM, Katayama R, McTigue M. et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Eng J Med. 2013;368:2395-401
30. Shaw AT, Kim DW, Mehra R. et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189-97
31. Gadgeel SM, Gandhi L, Riely GJ. et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK- rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2study. Lancet Oncol. 2014;15:1119-28
32. Ou SH, Ahn JS, De Petris L. et al. Alectinib in crizotinib-refractory ALK- rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34:661-8
Author contact
![]() Corresponding author: Liyan Jiang, Department of Pulmonary Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241# Huai Hai (W.) Rd, Shanghai, China. Tel: +86 13916146759 Fax: +86 021-32260856 e-mail: Jiang_liyancom
Corresponding author: Liyan Jiang, Department of Pulmonary Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241# Huai Hai (W.) Rd, Shanghai, China. Tel: +86 13916146759 Fax: +86 021-32260856 e-mail: Jiang_liyancom