Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(9):1689-1697. doi:10.7150/jca.24137 This issue Cite
Research Paper
PIASy antagonizes Ras-driven NSCLC survival by promoting GATA2 SUMOylation
Bin Chen1,2*, Jie Luo1*, Yirui Zhou1*, Xu Xin1, Rong Cai3 ![]() , Chunhua Ling2
, Chunhua Ling2 ![]()
1. Department of Oncology, Shanghai Pulmonary Hospital, Shanghai Tongji University, Shanghai 200433, China;
2. Department of Respiratory Diseases, the First Affiliated Hospital of Soochow University, Suzhou 215000, China;
3. Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
*These authors contributed equally to this work
Received 2017-12-2; Accepted 2018-2-5; Published 2018-4-18
Abstract
GATA2 regulated transcriptional network has been validated requisite for RAS oncogene-driven non-small cell lung cancer (NSCLC). GATA2 has been reported as a SUMOylated protein. In endothelial cells, its transcriptional activity is attenuated by SUMO-2 conjugation, which is specifically catalyzed by its E3 ligase PIASy. In this study, we found a decreased expression of PIASy in RAS mutant NSCLC cell lines and specimens with RAS mutations. Forced expression of PIASy in NSCLC cells inhibits their viability in vitro, as well as tumorigenesis and growth in vivo. Mechanistically, we demonstrated overexpression of PIASy in A549 cells altered the regulated transcriptional network of GATA2, including proteasome, IL-1-signaling, and Rho-signaling pathways. Forced expression of PIASy resulted in the accumulated SUMOylation of GATA2, attenuating its transcriptional activity in A549 cells. These results collectively suggest that PIASy plays an antagonistic role in RAS-driven NSCLC survival, by enhancing the SUMOylation of GATA2 and inhibiting its transcriptional activity.
Keywords: PIASy, Ras, NSCLC, SUMOylaition, GATA2
Introduction
Lung cancer remains a major health challenge in the world. The 5-year survival rate is only about 15% for patients with lung cancer, despite improvements in the integrated application of surgery, radiotherapy, and chemotherapy (1,2). Histologically, non-small-cell lung cancer (NSCLC) accounts for 80% of the lung cancer (3). Among NSCLC patients, the most frequently mutated oncogenes are the RAS family of GTPases and various RTKs (receptor tyrosine kinases) (4). Even though tyrosine kinase inhibitors show efficacy in NSCLC patients with mutant RTKs, patients inevitably develop into therapeutic resistance (5-8). Hence, novel drugs are required to target RAS mutant NSCLCs.
In 2012, Kumar et al reported that loss of the transcription factor GATA2 (GATA binding protein 2), reduced the viability of NSCLC cells with RAS mutations (9). It is the first time reporting GATA2 regulated transcriptional network plays significant roles in survival of RAS-driven NSCLC cells. GATA2 is a member of GATA family of transcription factors that plays a critical role in hematopoietic and cardiovascular development (10,11). Mutation and desregulated expression of GATA2 are common in hematopoietic malignancy (12,13). In prostate cancer, GATA2 is an established pioneer factor for androgen receptor (AR)-regulated genes (14-16). Recently, Vidal et al reported that GATA2-IGF2 (insulin-like growth factor 2) axis confers aggressiveness in lethal prostate cancer and might serve as a compelling therapeutic target (17).
GATA2 is a sumoylated protein and PIASy (protein inhibitor of activated STATy) specifically enhances its SUMOylation in endothelial cells (18). SUMOylation has now been emerging as an important posttranslational modification (PTM) of target proteins possessing various biological functions (19-21). Conjugation of small ubiquitin-related modifier protein (SUMO) to its substrates is catalyzed by the activating (E1), conjugating (E2) and ligating (E3) enzymes (22-24). It is well known that E3 ligases are important regulators for protein SUMOylation (25). The largest group of E3 ligase is characterized by an SP-ring motif, including protein inhibitor of activated STAT (PIAS) family of proteins (26). PIAS family consists of five members, PIAS1, PIAS3, PIASxα, PIASxβ and PIASy (27). Among them, PIASy mediated SUMOylation of target proteins has been shown to be involved in a variety of cellular processes, such as chromosome segregation, apoptosis, autophagy, response to genotoxic stress, and epithelial-to-mesenchymal transition (EMT) (28-31). Although the relationship of PIASy with various cancers, has been in an extensive study, the biological role of PIASy in NSCLC tumorginesis and progression is not fully illustrated.
Since GATA2 is a target protein of PIASy, here we investigated whether PIASy is involved in the pathological process in NSCLC mediated by GATA2. Interestingly, we found that PIASy is downregulated in RAS mutant NSCLC cell lines as well as in 26 NSCLC specimens with RAS mutations. Furthermore, the forced expression of PIASy in NSCLC cells antagonized cell survival both in vitro and in vivo. Mechanistically, PIASy overexpression enhanced the SUMOylation of GATA2, attenuated its transcription activity and regulated transcriptional network in NSCLC cells. Thus, we revealed the role of PIASy, a E3 SUMO ligase, as a tumor suppressor in NSCLC, by altering GATA2 SUMOylation and its transcriptional activity.
Materials and methods
Ethical statement
This study was approved by the Ethics Committee of Shanghai Pulmonary Hospital, Shanghai Tongji University, and performed in strict accordance with the recommendations in the Guide for the Care and Use of Human Samples from Shanghai Pulmonary Hospital. We declare no ethical or conflicts of interest.
Cell culture
Human RAS mutant NSCLC cell lines A549, H460 and H1299 were all obtained from ATCC and incubated in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum. All cell lines were incubated in a 5% CO2, 95% air humidified atmosphere at 37°C.
Real-time PCR
Total RNA was isolated by Trizol kit (Invitrogen). RNA was treated with DNase I (Promega). Complementary DNA was synthesized using the cDNA synthesis kit (Takara) according to the manufacturer's instructions. Fluorescence real-time RT-PCR was performed with the double-stranded DNA dye SYBR Green PCR Core Reagents (Takara) using the ABI PRISM 7300 system (Perkin-Elmer). PCR was done in triplicate and standard deviations representing experimental errors were calculated. Pairs of PCR primers used to amplification of the target genes were as Table 1.
Primers for quantitative real-time PCR
| Gene name | Forward (5'-3') | Reverse (5'-3') |
|---|---|---|
| hPIASy | CGGGTGTCCCTCATCTGTC | TGGGCTTCTTCTCGTTCATC |
| hNrf1 | AGCCATAGCCCTTCTTCC | GTAGCCAACCGCACCTTC |
| hPSMA3 | TTGGCTACAACATTCCACTAAA | ATAACCGTATGAAACACCTGAT |
| hPSMA4 | TATTACAGTATCAGGAGCCA | ATGAAACACCAAAGGGAC |
| hPSMB8 | TAACCCTTACCTGCTTG | TGCCGACACTGAAATACG |
| hTRAF6 | GAGTTTGACCCACCCCTG | ACCTGCATCCCTTATTGA |
| hROCK1 | GGACCTCTCGGATTCTAC | ATTGCTCCTTATCTTGTT |
| hEPHA3 | AGTTGAATGCAAACATCACAAAGAAG | GAAACTCCTTTTTGATGTGTGCATTC |
| hCDC42 | GTCATAGCCTGTTTAAAGGTATTTTTACC | TAGGAGGCGGAGCTTGTAGTG |
Western blot analysis
Protein extracts were equally loaded onto 10-12% SDS-polyacrylamide gels, electrophoresed, and transferred to PVDF membranes. After blocking with 5% nonfat milk in PBS, the membranes were probed with antibodies against PIASy (Santa Cruz), Flag and β-actin (Sigma), followed by horseradish peroxidase-conjugated secondary antibodies. The signals were detected by chemiluminescent substrate kit (Millipore).
Immunohistochemistry
Paraffin-embedded tumor tissues and adjacent normal tissues from 26 cases of NSCLC patients were collected from Pulmonary Hospital of Shanghai Tongji University. The immunohistochemical analysis was performed on the 4 mm thick fraction mounted on charged slides and sectioned from each clinical sample. Then, each slide was deparaffinized in 60°C, followed by treatment with xylene and graded alcohol. After the antigen retrieval and being blocked with 5% bovine serum albumin, tissue slides were immunohistochemically stained by antibody against PIASy (Santa Cruz), then visualized by standard avidin-biotinylated peroxidase complex method. Hematoxylin was used for staining and morphologic images were observed with Olympus BX51 microscope.
Cell viability
Cells were plated in triplicate in 24-well plates. Seventy-two hours later, cell numbers were counted by haemacytometer.
Cell cycle analysis
To assess the distribution of nuclear DNA content, cells were collected, rinsed and fixed overnight in 75% cold ethanol at -20℃. Then, cells were treated with Tris-HCl buffer (pH 7.4) supplemented with 100 µg/mL RNase A and stained with 25 µg/mL propidium iodide (BD Biosciences). Cell cycle distribution was determined by flow cytometry (Becton Dickinson). Ten thousands cells were acquired and analyzed for DNA content. All data were collected, stored, and analyzed by ModFit software.
Stable cell line generation
The retroviruses containing wild-type PIASy or mutant PIASy were gifts from the lab of Prof. Jinke Cheng. They were constructed as previously described (31), infected cells to generate pBabe-PIASy-A549 cells or pBabe-PIASy-Mut-A549 cells after puromycin selection. These cell lines were cultured in DMEM with 10% FBS and 3μg/mL puromycin. All cell lines were incubated in a 5% CO2, 95% air humidified atmosphere at 37°C.
Animals and xenograft
Male BALB/c nude mice (6 weeks old, 5 mice per group) were subcutaneous injected into the left belly of the nude mice with the following tumor cells, 2×107 pBabe-A549, pBabe-PIASy-A549 or pBabe-PIASy-Mut-A549 cells. After 3 weeks, mice were killed, and tumors were weighed up.
Immunoprecipitation
293T cells (5*106) or A549 cells (2×107) pellets were lysed in 1ml of radioimmune precipitation assay buffer (50mM Tris-HCl (pH 7.4), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS and 450mM NaCl) containing 1mM phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin, 10 mg/ml aprotinin, 1.5mM pepstatin, 1mM Na3VO4 and 5mM N-ethylmaleimide. Cell lysis was performed for 30 min on ice, and DNA in the sample was sheared with a 22-gauge needle. After centrifugation at 20000 g for 10 min at 4°C, the supernatants were added to appropriate antibody coupled to 20 ml of protein G-Sepharose beads. The bead suspensions were rotated for 3 h at 4°C. Beads were then washed five times with radioimmune precipitation assay buffer. The immunoprecipitates were treated with 30 ml of 2% SDS treating solution containing 5% β-mercaptoethanol and analyzed by western blot.
ChIP-qPCR assay
pBabe-A549, pBabe-PIASy-A549 or pBabe-PIASy-Mut-A549 cells (with FLAG tag) were crosslinked with 1% formaldehyde, and sonicated. Solubilized chromatin was immunoprecipitated with anti-FLAG M2-agarose affinity gel (Sigma), washed, and then eluted. After crosslink reversal and proteinase K treatment, immunoprecipitated DNA was extracted with phenol-chloroform, and then precipitated with ethanol. The DNA fragments were further analyzed by qPCR. The specific primers used to amplify promoter regions were described as previously reported (9).
Statistical analysis
The values were expressed as means± s.d. The paired t test was used for statistical analysis between two groups. For clinical data, the p value was calculated by Pearson's chi-square (x2) test. Significant level was set at p < 0.05.
Results
PIASy expression was decreased in RAS mutant NSCLC cells and tumor tissues
To investigate the role of PIASy in lung cancer, we first examined its expression in NSCLC cell lines with RAS mutations, by using human normal bronchus epithelial cell BEAS-2B as the control. As shown in Figure 1A and 1B, PIASy mRNA and protein levels were both decreased in A549, H460 and H1299 cells. Along with this way, expression of PIASy, in lung cancer specimens from 26 NSCLC RAS (+) patients, and their adjacent normal tissues, were investigated by using Real-time PCR assay. As depicted in Figure 1C, PIASy mRNA in 20 out of 26 tumor tissues displayed more than 2-fold decrease as long as those in their paired adjacent normal tissues. Quantification result of Immunohistochemical staining for PIASy in tumor tissues from 26 NSCLC patients with RAS mutants, were summarized in Table 2. Figure 1D displayed the representative case of immunohistochemical staining results with anti-PIASy antibody, showing the apparently decreased expression of PIASy in NSCLC tumor tissues. Thereafter, whether the downregulation of PIASy in RAS mutant NSCLC cells and tumor tissues is causative in NSCLC tumorigenesis and progression need to be investigated.
PIASy expression was decreased in RAS mutant NSCLC cell lines and tumor tissues. (A) Decreased expression of PIASy mRNAs in three RAS-driven NSCLC cell lines, A549, H460 and H1299, was detected by Real-time PCR analyses, by using human normal bronchus epithelial cell line BEAS-2B as the control. *p<0.05, **p<0.01. The data are presented as means ± s.d. of three independent experiments. (B) Decreased expression of PIASy proteins in A549, H460 and H1299 cells, was detected by Western blot analyses, by using BEAS-2B cells as the control. (C) PIASy mRNA expression in lung cancer specimens from 26 NSCLC patients with RAS mutants, and their adjacent normal tissues, were investigated by using Real-time PCR. The mRNA level of PIASy in 20 out of 26 tumor tissues displayed more than 2 folds decrease than those in their paired adjacent normal tissues. (D) Immunochemical staining with anti-PIASy antibody was applied to test its expression in paired NSCLS tumor tissues and adjacent normal tissues. Representative case of immunohistochemical staining results is shown in original magnification: 10×and 40×.
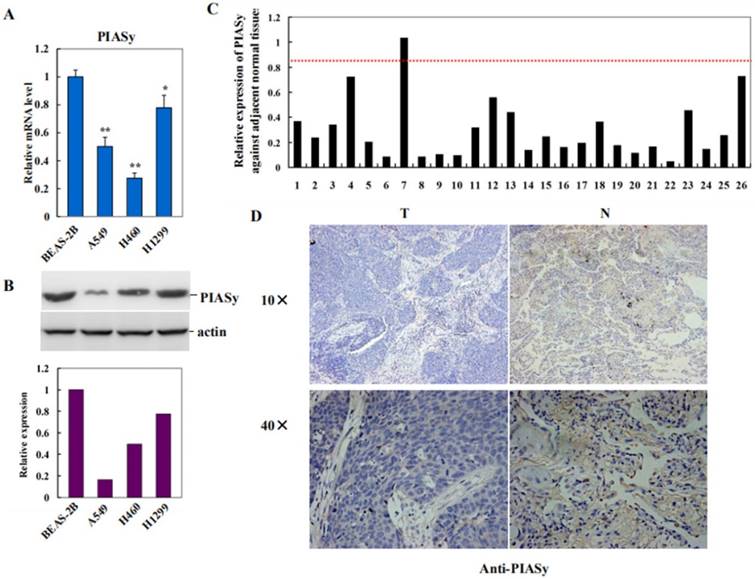
Immunohischemistry was performed to evaluate PIASy protein level in lung cancer specimens from 26 NSCLC patients with RAS mutants. The score of normal tissues was set as 3. The score of the expression level of PIASy ranges from 0 to 3 based on the intensity and percentage of immunoreactivity-positive cells.
| RAS mutants | PAISy | Total | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| + | 5 | 11 | 7 | 3 | 26 |
Forced expression of PIASy in RAS-oncogene drive NSCLC cells inhibited their viability in vitro
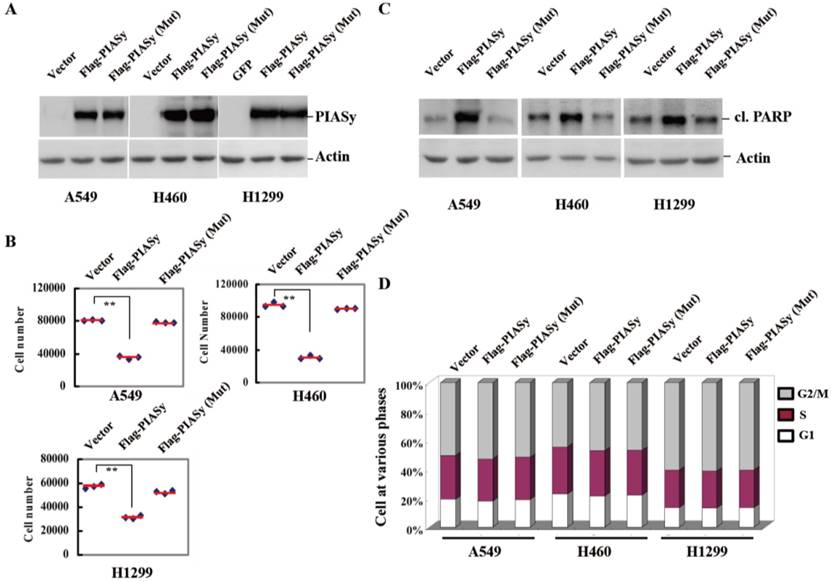
To see if the downregulation of PIASy in NSCLCs had pathological significance, we forced its expression into the NSCLC cells, and observed the effects of rescued expression of PIASy on tumor cell viability. Vector and PIASy E3 ligase catalytic site mutant proteins were also transfected into the cells at the same time as controls. As we can see from Figure 2A, anti-Flag tag antibody detected both wild-type and mutant PIASy protein expression in A549, H460 and H1299 cells. Overexpression of wild-type PIASy greatly reduced the viability of cells, while the mutant one did not (Figure 2B). In order to illustrate whether this reduced effect on cell viability is due to induced apoptosis or impaired proliferation, we examined both apoptotic markers and the cell cycle distribution in PIASy overexpressed cells. With wild-type PIASy overexpressed NSCLC cells, we observed elevated PARP cleavage, a marker of apoptotic cell death in comparison with mutant PIASy overexpressed ones, indicating the inhibition of PIASy on NSCLC cell viability depends on its catalytic activity (Figure 2C). Conversely, the cell cycle profiles of NSCLC cells shows no difference between wild-type and mutant PIASy overexpression groups (Figure 2D). Together, the data of Figure 2 showed that PIASy can inhibit RAS-driven NSCLC cell survival in vitro, which depends on its E3 ligase catalytic activity.
Forced expression of PIASy in RAS-oncogene driven NSCLC cell lines inhibited their viability in vitro. (A) Western blot analyses with anti-Flag antibody demonstrated the forced expression of PIASy Wild-type and PIASy mutant (Mut) protiens in A549, H460 and H1299 cells. (B) PIASy, PIASy (Mut) and control NSCLC (Vector) cells were plated in 24-well plates and cell numbers were counted 72 hr later. **p<0.01. The data are presented as means ± s.d. of three independent experiments. (C) Western blot analyses were performed on the cells above for cleaved PARP. (D) The cell cycle distribution of PIASy, PIASy (Mut) and control NSCLC cells was analyzed by flow cytometry and the percentages of cells in G1 phase, S phase and G2/M phase were quantified.
Overexpression of PIASy inhibits RAS-oncogene driven NSCLC progression in vivo
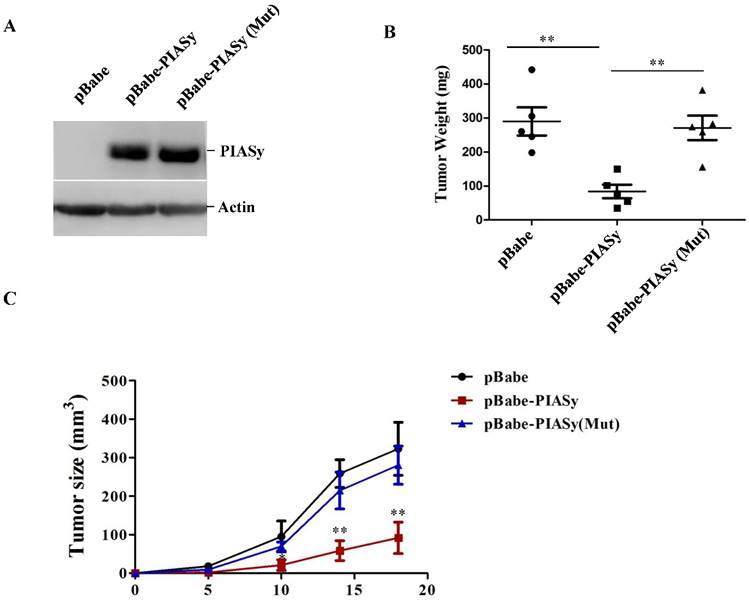
In order to examine the ability of PIASy's altering RAS mutant NSCLC viability in vivo, tumor xenograft models were used. We generated stably wild-type or mutant PIASy ovexpressed A549 cells (Figure 3A), and injected them subcutaneously in nude mice. As seen from Figure 3B and 3C, increased expression of PIASy in A549 cells significantly abrogated tumor formation in vivo, indicating the forced expression of wild-type PIASy markedly inhibited NSCLC tumorgenesis and growth. However, the tumour size in PIASy (Mut) A549 cells was similar to that in control cells, indicating that when PIASy loses its E3 SUMO ligase activity, it also loses the ability to inhibit NSCLC survival in vivo (Figure 3B and 3C).
Overexpression of PIASy inhibits RAS-oncogene driven NSCLC progression in vivo. (A)Western blot analyses with anti-Flag antibody demonstrated the establishment of stable cell lines overexpressing PIASy Wild-type and PIASy mutant (Mut) proteins in A549 cells. (B) 2×107 PIASy (n=5), PIASy (Mut) (n=5) and control (n=5) A549 cells were injected into the nude mice, and the tumor weight were measured at 3 weeks after injection. **p<0.01. (C)Growth curves of subcutaneous xenografts derived from pBabe, pBabe-PIASy, and pBabe-PIASy (Mut) A549 cells (n=5 in each group). *p<0.05, **p<0.01.
Overexpression of PIASy enhances GATA2 SUMOylation in A549 cells
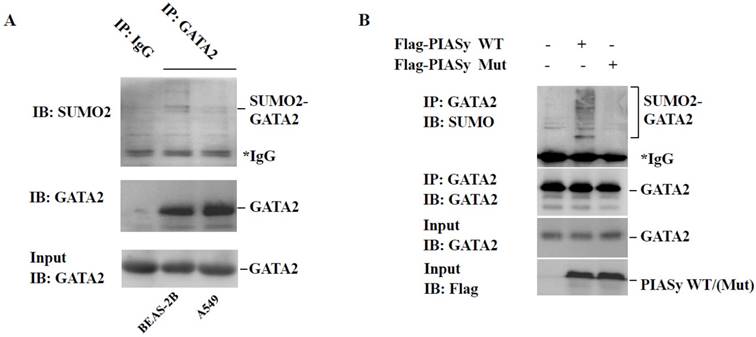
PIASy has been reported as a specific E3 ligase promoting GATA2 SUMOylation in endothelial cells (18). And GATA2 regulated transcriptional network is required for RAS muant NSCLC survival (9). For these two reasons, we speculated the role of PIASy in cell viability inhibition, may at least partially, plays via changing the SUMOylation status of GATA2. Firstly, immunoprecipitation analysis showed that in A549 cells, the modification of GATA2 by SUMO2 is lower, in comparison with that in BEAS-2B cells which is in RAS wild-type status (Figure 4A). More crucially, we found that overexpression of wild-type PIASy protein could enhance the SUMOylation of GATA2 in A549 cells, while the mutant one could not (Figure 4B), indicative of the regulation of PIASy on GATA2 SUMOylation in RAS mutant NSCLC cells.
Overexpression of PIASy enhances GATA2 SUMOylation in A549 cells. (A) Immunoprecipitation analysis showed endogenous GATA2 protein can be modified by SUMO2 in BEAS-2B and A549 cells. But in A549 cells, the modification of GATA2 by SUMO2 is attenuated, in comparison with that in BEAS-2B cells that is in RAS wildtype status. GATA2 was immunoprecipitated from cell lysates with anti-GATA2 antibody or non-specific IgG as a negative control (IP), and bound proteins were detected by immunoblotting with anti-SUMO2 or anti-GATA2. WCL were immunoblotted with anti-GATA2 (Input). (B) PIASy wild-type but not PIASy mutant (Mut) enhances GATA2 SUMOylation in A549 cells. GATA2 was immunoprecipitated from cell lysates with anti-GATA2 antibody (IP), and bound proteins were detected by immunoblotting with anti-SUMO2 or anti-GATA2. WCL were immunoblotted with anti-GATA2 or anti-Flag (Input).
SUMOylation of GATA2 by PIASy attenuates its transcriptional activity and regulated pathways
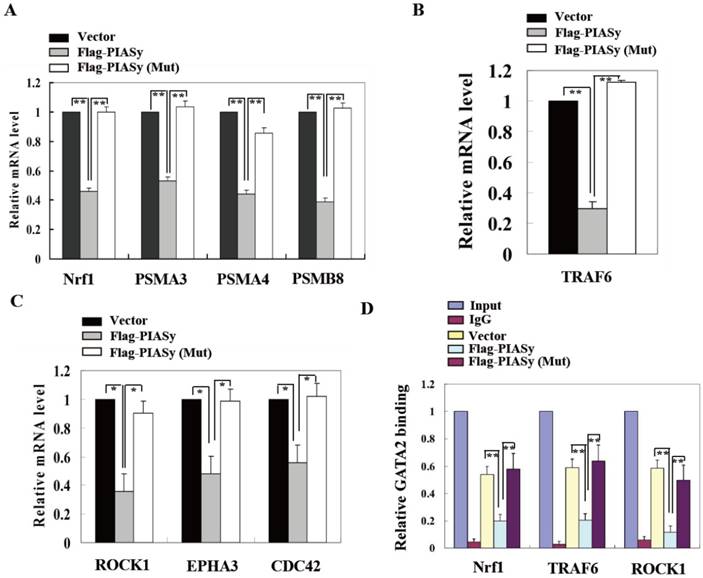
To further explain the mechanism of PIASy on A549 cell survival inhibition which is mediated by GATA2 SUMOylation, we explored the GATA2 regulated transcriptional network in wild-type and mutant PIASy overexpressed A549 cells. Gene expression involved in three GATA2 regulated pathways, including proteasome, IL-1, and Rho signallings, were all down-regulated in wild-type PIASy overexpressed A549 cells, in comparison with Vector and mutant PIASy overexpressed ones (Figure 5A, 5B and 5C). In addition, ChIP assay revealed that SUMOylation of GATA2 mediated by PIASy in A549 cells, greatly reduced its binding ability to the promoters of Nrf1, TRAF6 and ROCK1 genes (Figure 5D). Whereas, the mutant PIASy did not have the inhibitory effect on GATA2 transcriptional activity compared with the Vector control, with loss of its E3 ligase catalytic activity. These data collectively suggest that the PIASy inhibitory effects on RAS mutant A549 cell survival, was mediated, at least partially, by enhancing GATA2 SUMOylation and attenuating its transcriptional activity.
SUMOylation of GATA2 by PIASy attenuates its transcriptional activity and regulated pathways. PIASy wild-type but not PIASy mutant (Mut) attenuates GATA2 regulated transcriptional network composed of the proteasome (A), IL-1-signaling (B), and Rho signaling pathways (C) in A549 cells. *p<0.05, **p<0.01. The data are presented as means ± s.d. of three independent experiments. (D) Chromatin immunoprecipitation (ChIP) analysis showed that SUMOylation of GATA2 enhanced by PIASy decreased its binding ability to its target genes. **p<0.01.The data are presented as means ± s.d. of three independent experiments.
Discussion
The paradoxical role of PIASy in cancers has been reported by several studies (28-34). In ovarian and lung cancer cells, PIASy promotes epithelial-to-mesenchymal transition (EMT) and cancer metastasis via inhibiting SIRT1 transcription (33,34). Despite these two findings deciphering the pro-tumor effect of PIASy, in most conditions it acts as a tumor suppressor. In prostate cancer, PIASy acts as a transcriptional corepressor of AR and inhibits AR signalling (27). PIASy-mediated SUMOylation of TOPIIɑ inhibited mitotic chromosome segregation (28,31). In addition, PIASy has been reported able to provoke cell senescence and apoptosis by promoting p53 SUMOylation and activity (29). Kang et al reported PIASy reduces hypoxia-induced angiogenesis through enhancing HIF-1ɑ SUMOylation and degradation (30). In colon cancer, PIASy-mediated Tip60 SUMOylation promotes p53-induced autophagy (32). Therefore, the multiple functions of PIASy in tumorigenesis and progression chiefly depend on the different substrates targeted by PIASy in different cancer cells.
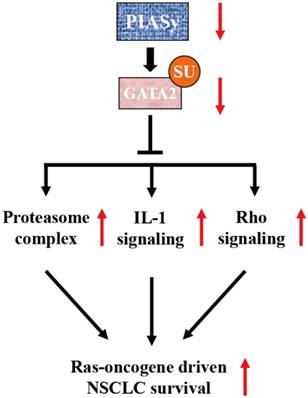
Previous study has identified PIASy as a specific E3 SUMO ligase for GATA2 in endothelial cells (18). It is a novel finding that GATA2 transcriptional network is requisite for RAS-driven NSCLC in recent years (9). Based on this finding, Shen et al utilized cationic lipid-assisted polymeric nanoparticles for systemic delivery of siRNA targeting GATA2 and found that suppression of GATA2 by NPsiGATA2 selectively inhibited proliferation of A549 cells harboring a KRAS mutation (35). In our study, the central achievement is the finding of SUMOylation-dependent regulation of GATA2 activity, mediated by PIASy in NSCLC cells. We found that due to the downregulation of PIASy in NSCLC cells, the SUMOylation level of GATA2 is decreased. SUMOylation defects on GATA2 abrogates its attenuation on GATA2 transcriptional activity, activating requisite transcriptional network for RAS mutant NSCLC cell survival, including proteasome, IL-1 and Rho signalling pathways, as illustrated in Figure 6.
Model of PIASy function in RAS-oncogene driven NSCLC. PIASy inhibited RAS-oncogene driven NSCLC cell survival by promoting GATA2 SUMOylation and downregulated its transcriptional network composed of the proteasome, IL-1-signaling, and Rho signaling pathways.
As a transcription factor investigated intensively in hematological and cardiovascular systems, GATA2 activity has been reported being modulated by several post-translational modifications. GATA2 is a short-lived protein that turns over rapidly through the ubiquitin-proteasome pathway (36). Nikajima et al identified Fbw7 a bona fide E3 ubquitin ligase for GATA2 in vivo (37). Hayakawa et al demonstrated acetylation provides multiple control points for the regulation of GATA2 function, like enhancing its DNA binding activity and transactivating potential (38). Phosphorylation of GATA2 via the MAP kinase constitutes a cytoplasmic link between GATA2 and growth factor receptors in hematopoietic progenitor cells (39). In adipocytes, insulin-dependent phosphorylation of serine 401 by PI3K/AKT impairs GATA2 translocation to the nucleus and its DNA binding activity (40). Recently, Katsumura et al defined a mechanism that oncogenic RAS-mediated amplification of GATA2 activity (41). Oncogenic RAS enhanced S192-dependent GATA2 phosphorylation, nuclear foci localization and transcriptional activation. Chun et al reported GATA2 is modified by SUMO2 in endothial cells by PIASy (18), but its SUMOylation site has not been identified yet. By using SUMOsp2.0 software, we found two potential SUMOylation sites (K222 and K375) in human GATA2 protein (Supplemental Figure 1). The further study to investigate whether GATA2 SUMOylation interacts with other forms of its PTMs, such as phosphorylation or ubiquination, during the pathological processes, is interesting. Moreover, the crosstalk between RAS oncogenic pathway and GATA2 SUMOylation pathway is also needed to be studied further.
Conclusions
In summary, we identified PIASy plays a key role in RAS-driven NSCLC cell survival. Forced expression of PIASy in RAS mutant NSCLCs cells inhibited cell survival in vitro and in vivo (Figure 2 and 3), which depends on its E3 ligase activity on GATA2 SUMOlyation (Figure 4). But the mechanism that causes PIASy downregulation in NSCLCs remains to be elucidated. The mechanism studies on regulation of PIASy expression and activity in cancers are quite rare, which might become an interesting field in the future. This study provides PIASy, a E3 SUMO liagase, as a novel and potential therapeutic target in RAS-driven NSCLCs.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This work was supported in part by the grants from Six talent peaks project of Jiangsu Province (WS-053), National Basic Research Program of China (973 Program) (2015CB910403), Natural Science Foundation of China (81572691).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-917
2. Ballatori E, Fatigoni S, Roila F. Treatment of lung cancer. New Engl J Med. 2009;361:2485 author reply 2486-7
3. Fischer B, Lassen U, Mortensen J. et al. Preoperative staging of lung cancer with combined PET-CT. New Engl J Med. 2009;361:32-9
4. Engelman JA, Chen L, Tan X. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6
5. Engelman JA, Cantley LC. A sweet new role for EGFR in cancer. Cancer Cell. 2008;13:375-6
6. Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cli Cancer Res: an official journal of the American Association for Cancer Research. 2008;14:2895-9
7. Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Gen De. 2008;18:73-9
8. Guix M, Faber AC, Wang SE. et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Biol Chem. 2008;118:2609-19
9. Kumar MS, Hancock D C, Molina-Arcas M. et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642-55
10. Tsai FY, Keller G, Kuo FC. et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221-6
11. Linnemann AK, O'Geen H, Keles S. et al. Genetic framework for GATA factor function in vascular biology. P Natl Acad Sci USA. 2011;108:13641-6
12. Shimamoto T, Ohyashiki JH, Ohyashiki K. et al. GATA-1, GATA-2, and stem cell leukemia gene expression in acute myeloid leukemia. Leukemia. 1994;8:1176-80
13. Nagai T, Harigae H, Ishihara H. et al. Transcription factor GATA-2 is expressed in erythroid, early myeloid, and CD34+ human leukemia-derived cell lines. Blood. 1994;84:1074-84
14. Wang Y, He X, Ngeow J. et al. GATA2 negatively regulates PTEN by preventing nuclear translocation of androgen receptor and by androgen-independent suppression of PTEN transcription in breast cancer. Hum Mol Genet. 2012;21:569-76
15. Chiang YT, Wang K, Fazli L. et al. GATA2 as a potential metastasis-driving gene in prostate cancer. Oncotarget. 2014;5:451-61
16. Wu D, Sunkel B, Chen Z. et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014;42:3607-22
17. Vidal SJ, Rodriguez-Bravo V, Quinn SA. et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell. 2015;27:223-39
18. Chun TH, Itoh H, Subramanian L. et al. Modification of GATA-2 transcriptional activity in endothelial cells by the SUMO E3 ligase PIASy. Circ Res. 2003;92:1201-08
19. Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1-12
20. Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947-56
21. Yeh ET. SUMOylation and De-SUMOylation: wrestling with life's processes. J Biol Chem. 2009;284:8223-7
22. Gill G. SUMO changes Sox for developmental diversity. Mol Cell. 2005;20:495-6
23. Shin JA, Choi ES, Kim HS. et al. SUMO modification is involved in the maintenance of heterochromatin stability in fission yeast. Mol Cell. 2005;19:817-28
24. Nayak A, Glockner-Pagel J, Vaeth M. et al. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J Biol Chem. 2009;284:10935-46
25. Johnson ES, Gupta AA. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 2001;106:735-44
26. Hochstrasser M. SP-RING for SUMO: new functions bloom for a ubiquitin-like protein. Cell. 2001;107:5-8
27. Gross M, Liu B, Tan J. et al. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene. 2001;20:3880-7
28. Azuma Y, Arnaoutov A, Anan T. et al. PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J. 2005;24:2172-82
29. Bischof O, Schwamborn K, Martin N. et al. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol Cell. 2006;22:783-94
30. Kang X, Li J, Zou Y. et al. PIASy stimulates HIF1alpha SUMOylation and negatively regulates HIF1alpha activity in response to hypoxia. Oncogene. 2010;29:5568-78
31. Ryu H, Furuta M, Kirkpatrick D. et al. PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J Cell Biol. 2010;191:783-94
32. Naidu SR, Lakhter AJ, Androphy EJ. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle. 2012;11:2717-28
33. Sun L, Li H, Chen J. et al. A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis. J Nat Cancer I. 2013;105:887-98
34. Sun L, Li H, Chen J. et al. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J Cell Sci. 2013;126:3939-47
35. Shen S, Mao CQ, Yang XZ. et al. Cationic lipid-assisted polymeric nanoparticle mediated GATA2 siRNA delivery for synthetic lethal therapy of KRAS mutant non-small-cell lung carcinoma. Mol Pharm. 2014;11:2612-22
36. Minegishi N, Suzuki N, Kawatani Y. et al. Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes Cells: devoted to molecular & cellular mechanisms. 2005;10:693-704
37. Nakajima T, Kitagawa K, Ohhata T. et al. Regulation of GATA-binding protein 2 levels via ubiquitin-dependent degradation by Fbw7: involvement of cyclin B-cyclin-dependent kinase 1-mediated phosphorylation of THR176 in GATA-binding protein 2. J Biol Chem. 2015;290:10368-81
38. Hayakawa F, Towatari M, Ozawa Y. et al. Functional regulation of GATA-2 by acetylation. J Leukocyte Biol. 2004;75:529-40
39. Towatari M, May GE, Marais R. et al. Regulation of GATA-2 phosphorylation by mitogen-activated protein kinase and interleukin-3. J Biol Chem. 1995;270:4101-7
40. Menghini R, Marchetti V, Cardellini M. et al. Phosphorylation of GATA2 by Akt increases adipose tissue differentiation and reduces adipose tissue-related inflammation: a novel pathway linking obesity to atherosclerosis. Circulation. 2005;111:1946-53
41. Katsumura KR, Yang C, Boyer M E. et al. Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA-2. EMBO R. 2014;15:938-47
Author contact
![]() Corresponding author: Rong Cai, Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China; rongcaiedu.cn. Chunhua Ling, Department of Respiratory Diseases, the First Affiliated Hospital of Soochow University, Suzhou 215000, China; linchunhua88com.
Corresponding author: Rong Cai, Department of Biochemistry & Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China; rongcaiedu.cn. Chunhua Ling, Department of Respiratory Diseases, the First Affiliated Hospital of Soochow University, Suzhou 215000, China; linchunhua88com.