Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(13):2492-2500. doi:10.7150/jca.19601 This issue Cite
Review
Chromosomal and Genomic Variations in Esophageal Squamous Cell Carcinoma: A Review of Technologies, Applications, and Prospections
Qi Song1, Dongxian Jiang1, Haixing Wang1, Jie Huang1, Yalan Liu1, Chen Xu1, Yingyong Hou1, 2 ![]()
1. Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai 200032, P. R. China;
2. Department of Pathology, School of Basic Medical Sciences & Zhongshan Hospital, Fudan University, Shanghai 200032, P. R. China.
Received 2017-2-10; Accepted 2017-5-30; Published 2017-8-2
Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignant tumors with poor prognosis worldwide. The poor prognosis is due to the advanced stage at the time of diagnosis and the limited clinical staging lacking significant molecular biomarkers to effectively stratify patients for treatment options. As cancer is a disease of genome instability and a resulting of accumulation of genetic alteration, mounting chromosomal and genomic technologies were developed and progressed rapidly which could be used for characterizing patients in genomics level. In this review, we summarized applications of multiple technologies and research progress at chromosomal and genomic level in ESCC.
Keywords: esophageal squamous cell carcinoma, chromosomal, genomic, technology, application.
Introduction
Esophageal cancer (EC) is the eighth most common cancer and the sixth most common cause of cancer-related death in the world, with an estimated 456,000 new cases and 400,000 deaths in 2012 [1]. However, according to the latest cancer statistics in China [2], EC is the third most commonly diagnosed cancer among men and the fifth among women. When concerning mortality, EC is the fourth leading cause of cancer death in both sexes. EC can be divided into esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC), which are two completely distinct subtypes from histopathological, epidemiologic and molecular aspects [3]. ESCC accounts for about 90% of cases of EC worldwide (Figure 1), and the 5-year survival rate for patients with ESCC, although has improved during the past decade, remains generally poor [4]. Many patients showed lymph node metastasis and tumor invasion into adjacent organs at the time of diagnosis and lacking effective chemotherapeutic approaches available to treat ESCC patients both contribute to the poor prognosis of ESCC [5]. Current clinical staging approaches are limited in their ability to effectively stratify patients for treatment options. The concept of precision medicine—coupling established clinical-pathological indexes with molecular profiling to create preventive, diagnostic, prognostic, and therapeutic strategies precisely tailored to each patient's requirements [6, 7]—was put forward in recent days. This concept gives all of us access to the personalized molecular information, bringing us closer to curing diseases.
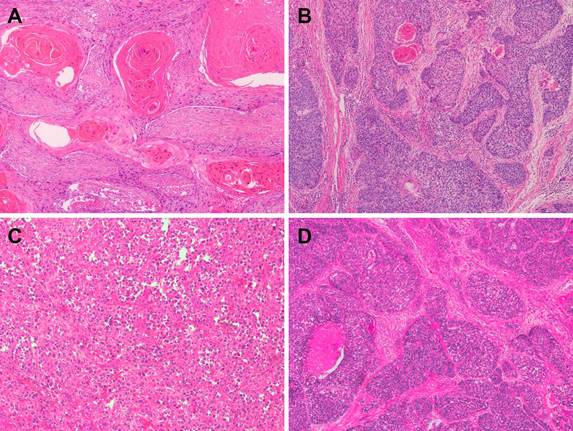
Representative microphotographs of sections from esophageal squamous cell carcinoma (ESCC). (A) Well-differentiated ESCC. (B) Moderately differentiated ESCC. (C) Poorly differentiated ESCC. (D) Basaloid squamous cell carcinoma.
In this review, we summarized a series of chromosomal and genomic technologies used in clinical and research field of ESCC in recent decades, helping us to understand the chromosomal and genomic characteristics and variations in ESCC and providing us a direct cognition to precision medicine concerning to molecular profiling level.
The most common used chromosomal and genomic technologies in ESCC
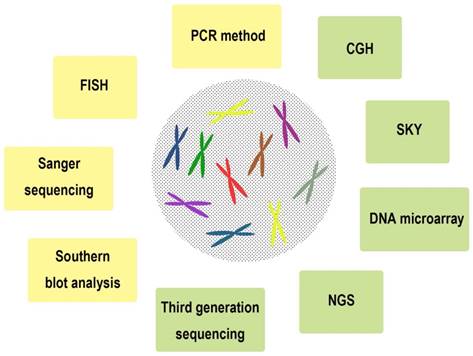
A series of chromosomal and genomic technologies were used in clinical and research field, including Southern blot analysis [8], Sanger sequencing [9], fluorescence in situ hybridization (FISH) [10], DNA microarray [11], PCR method [12], comparative genomic hybridization (CGH) [13], spectral karyotyping (SKY) [14], Next-generation sequencing (NGS) [15], Third generation sequencing [16], and so on (Figure 2, Table 1). We summarized the characteristics of these technologies with the purpose of understanding the chromosomal and genomic variations of ESCC in detail.
Representative chromosomal and genomic technologies used in clinical and research field of esophageal squamous cell carcinoma (ESCC).
The characteristics of a series of chromosomal and genomic technologies used in clinical and research field of ESCC
| Technology | The year of invention/ First proposed | Advantages | Limitations | Applications |
|---|---|---|---|---|
| Southern blot | 1975[8] | Semi-quantitative | Radiation; low-thoughput | Detection of the length or copy number of a specific DNA; identification of methylated sites |
| Sanger sequencing | 1977[9] | Highly exact; gold standard for genetic test | Expensive; low-thoughput; low sequencing speed; sequencing length limited in 400-900 bp; hard to manipulate | Detection of the specific variation of DNA fragment in clinical, e.g. gene mutation, deletion, insertion, and so on. |
| FISH | 1982[10] | In situ detection; sensitive; economy and safety; fast | Low resolution; low-thoughput; subjectivity; detection of the known loci | Detection of CNV, rearrangement, the number of chromosomes in clinical |
| DNA microarray | 1983 | High-thoughput; easy to manipulate | Design and manufacture | Gene expression profiling; comparative genomic hybridization; SNP detection |
| PCR method | 1983[12] | Fast; convenient; sensitive; economy | Detection of the known sequence; low quantity contamination may lead to misleading result; high cycle numbers lead to low fidelity | PCR-sequencing; PCR-SSCP; PCR-RFLP; gene expression; and so on |
| CGH | 1992 | In situ detection | Low-thoughput; low-sensitive; limited hybridization loci | Combined with DNA microarray (array based CGH, aCGH) to detect unbalanced chromosomal abnormalities |
| SKY | 1998[14] | High resolution | Limited in detecting complex rearrangements | Detection of structural rearrangements |
| NGS | 2000[15] | High-throughput; acquisition the sequence information of unknown sequence | Relative high cost; relative high work load; relative short reads; needed to be verified by repeated tests or Sanger sequencing | Whole genome sequencing; Targeted region deep sequencing |
| Third generation sequencing | ~2009[16] | Very long reads; fast; no need of pre-amplification; directly sequence the RNA and methylated DNA sequencing; relative easier data analysis compared with NGS | High cost; moderate throughput; dependence of enzymatic activity; low signal sensitivity | Single cell/molecular sequencing; whole genome sequencing; identify methylated sites; RNA-sequencing; detect rare mutations |
Southern blot analysis
The Southern blot named after its inventor is a method used for detection of a specific DNA sequence or identification methylated sites in samples. By the method of Southern blot, coamplification of genes, such as hst-1 and int-2 [17], MYEOV and CCND1 [18], was observed in ESCCs. Southern blot, as a conventional method to detect specific DNA sequence, has its limitations such as the harmful radiation generated from radioisotopes, which is needed for detection of DNA sequence. Nowadays, this method used as a validation tool to demonstrate the accuracy of other methods, such as quantitative polymerase chain reaction (qPCR) [19] and array-based comparative genomic hybridization (CGH )[20, 21].
Sanger sequencing
Sanger sequencing, as a most widely used direct DNA sequencing method developed in 1977, is based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase. When PCR method was developed in 1983, these two methods were perfectly combined, the sequencing always followed by the PCR amplification [22]. Recently, Sanger sequencing has been partly supplanted by Next generation sequencing (NGS), owning to the NGS's characteristics of high throughput, automated genome analyses. However, the Sanger method remains in wide use because of its accuracy, especially for validation of NGS results [23] as a classical method and smaller-scale projects in clinical.
FISH
FISH, used fluorescent probes that bind to those parts with a high degree of sequence complementarity, was widely used to detect the amplification [24], deletion [25], and gene rearrangement [26] of the targeted sequences on chromosome in situ, with the signal capturing by fluorescence microscopy. The biggest limitation of this technology is due to the subjectivity from different testers and the low resolution to detect the detailed variation.
DNA microarray
DNA microarrays offer a high-thoughput genomic approach to screen chromosomal alterations systematically and can be used to measure the alterations of large numbers of genes simultaneously or to genotype multiple regions of a genome for evaluating tumor heterogeneity [27]. However, the complexity of design and manufacture of DNA microarrays limited the use of this technology.
PCR method
The PCR method, is a widely-used technology for amplifying a particular DNA or cDNA sequence to generate thousands to millions of copies and acts as an important fundamental method for many applications, such as PCR-sequencing, PCR-restriction fragment length polymorphism (RFLP), PCR-single strand conformation polymorphism (SSCP), amplification refractory mutation system (ARMS) - PCR and so on. The major limitation of PCR is that the target sequence information is needed prior to primers' designation. Moreover, because PCR is a high sensitive technique, any form of contamination of the sample can produce misleading results [28].
PCR-sequencing
This combined technology is widely used in scientific research and molecular pathology examination for detecting single nucleotide polymorphism (SNP), mutation, and gene fusion in a particular district of a gene or more genes.
PCR-RFLP
PCR-RFLP is a technique that exploits variations in homologous DNA sequences. After the amplification of PCR, the DNA products are digested by restriction enzymes and the resulting restriction fragments are separated according to their lengths by gel electrophoresis. RFLP analysis was an important tool in genome mapping, localization of genes for genetic disorders and determination of risk for disease [12].
PCR- SSCP
SSCP is defined as conformational difference of single-stranded nucleotide sequences of identical length as induced by differences in the sequences under certain experimental conditions. This property allows sequences to be distinguished by means of gel electrophoresis, which separates fragments according to their different conformations [29].
ARMS-PCR
The basis of this technology is that oligonucleotides with a mismatched 3'-residue will not function as primers in the PCR under appropriate conditions [30]. ARMS-PCR is simple, rapid and reliable, allowing the direct analysis of any locus of interest provided sufficient sequence data.
Real-time qPCR
Real-time qPCR is a technique based on conventional PCR, monitoring the signal of amplification of a targeted DNA molecule during the PCR. Two common methods for the detection of real-time qPCR products are: (1) non-specific fluorescent dyes that intercalate with any double-stranded DNA, such as SYBR Green method, This method is a low cost and accurate way for detection of gene copy number alteration (CNA), including copy number amplification[31] and deletion [32]; (2) sequence-specific DNA probes consisting of oligonucleotides that are labelled with a fluorescent reporter which permits detection only after hybridization of the probe with its complementary sequence, such as TaqMan probe method. A series of TaqMan Assays were designed to detect SNP [33], CNA [34] and gene mutation. This method contains a specific DNA probe, which improves the specificity and sensitivity for detection, with a wider application, a higher accuracy than SYBR Green method, however, a higher cost.
Droplet digital PCR
The digital polymerase chain reaction simultaneously amplifies thousands of samples, each in a separate droplet within an emulsion. This can be used to quantitate mutant alleles or copy number of a specific gene in a DNA sample.
CGH
CGH is a powerful method that can survey the entire genome of tumor cells to detect DNA CNAs in one hybridization experiment [35], and has an improved resolution compared to the more traditional cytogenetic analysis techniques of FISH which are limited by the resolution of the microscope utilized. Nowadays, this technology is perfectly combined with DNA microarray to detect unbalanced chromosomal abnormalities.
SKY
SKY is used to simultaneously visualize all the pairs of chromosomes in an organism in different colors, detecting or defining genomic changes, such as chromosomal derivatives or chromosomal rearrangements [35]. However, it is limited in detecting complex rearrangements.
NGS
The high demand for low-cost sequencing has driven the development of NGS technologies that parallelize the sequencing process, producing thousands or millions of sequences concurrently [23, 36-38]. With the widely use of NGS, the genetic landscape of various diseases has been reported. However, without the bioinformatics, the results obtained from NGS costing serious funding can be worthless. Also, the sequence obtained from NGS need to be validated by other classical methods.
Third generation sequencing
Third generation sequencing is characterized by single molecular sequencing with no need of PCR amplification. This allows longer reads, faster sequencing speed, and more accuracy. Because of the direct sequencing of long sequence, it is easy to perform the data analysis without the need of joint of gene sequence. Moreover, RNA and methylated DNA sites can be directly sequenced.
Chromosomal variations in ESCC
Chromosomal variations are a series of missing, extra, or irregular portion of chromosomal DNA. It can be from an atypical number of chromosomes or a structural abnormality in one or more chromosomes or chromosomal segments.
Gains and deletions/losses of chromosomal arms
The most frequently detected chromosomal gains were found on 1p, 1q, 2p, 3q, 5p, 6p, 7p, 7q, 8q, 9q, 11q, 12p, 14q, 15q, 16p, 16q, 17q, 18p, 19q, 20p, 20q, 22q, and Xq, while the most frequently loss involved 3p, 3q, 4p, 4q, 5q, 6q, 7q, 9p, 13p, 13q, 18q and 19p [35, 39]. Gain in 12p is indicative of poor prognosis after esophagectomy[39]. However, it is also worth pointing that due to the limited technologies in that period, the results should be confirmed by further studies.
In recent years, high resolution array-based CGH (aCGH) has been applied to identify target oncogenes and tumor suppressor genes (TSGs) through defining recurrent gains and losses in various cancers. Studies on ESCC samples revealed that recurrent, high-level amplifications in 3q26.32-33, 3q27.1, 7p11, 7p22.3, 8p11.23, 8q21.11, 8q24.21, 11q13.2-11q13.3, 11q22, 12p12.1, 12q15-q21.1, 13q22.1, 14q11.2, 14q13.3, 18q11.2, and 19q13.11-q13.12, and homozygous deletions in 1p15.4, 2q22.1-22.2, 3p14.2, 4p16.1-p15.1, 4q34.3-q35.1, 5q12.1, 6p22.1, 9p21.3, 9p24.1, 13q14.2, 14q12, and 22q13.1 [13, 36, 40, 41]. Gain of 11q13.2 and loss of 7q34 and 18q21.1-q23 were associated with poor outcome [41].
Chromosomal structural variations
When the chromosome's structure is altered, several atypical forms appeared, such as deletions, duplications, translocations, inversions, insertions and so on. They often lead to an increased tendency to develop certain types of malignancies. Caixia Cheng, et al. [42] analyzed whole-genome sequencing (WGS) data from 31 ESCCs to predict somatic structural variations and determine copy number changes. They found deletions and translocations as the dominant SV types, and 16% of deletions were complex deletions. Chromothripsis, kataegis, and breakage-fusion-bridge (BFB) were identified as contributing to locally misarranged chromosomes that occurred in 55% of ESCCs.
Genetic variations in ESCC
Genetic variation is based on the variation in alleles of genes in a gene pool. Genetic variation will result in phenotypic variation if variation in the DNA sequence results in a difference in the order of amino acids in proteins coded by that DNA sequence. We focus on the gene CNAs and mutations in ESCC.
Gene mutations
Gene mutation is an important mechanism leading to the alteration or loss of the gene function. The most frequently mutated genes in ESCC, including TP53 [22], PIK3CA [43], BRCA2 [44], EGFR [45], NRF2 [46, 47], CDKN2A [48] were detected by several groups using traditional methods, such as PCR-sequencing or PCR-SSCP. A complete genetic landscape of ESCC remains incomplete, and it is likely that additional genes might also play a role in this disease and its progression. Recent advances in sequencing technology have overcome past limitation of scale and thousands of mutations can be identified in a single sample [49]. Chinese researchers conducted whole-genome sequencing or whole-exome sequencing on ESCC patients, hoping to define the mutational landscape of ESCC and providing an important molecular foundation for understanding esophageal tumors [23, 36-38, 50]. The mutations discovered by traditional methods were also detected by NGS, moreover, a number of novel mutated genes were firstly unmasked in ESCC. In table 2, we profiled the frequently mutated genes (≥5%) in two NGS studies. The most frequently mutated genes were TP53, TTN, MLL2, CDKN2A, PIK3CA, NOTCH1, NFE2L2, EP300, ADAM29, FAM135B and so on. And these genes were mostly involved in pathways containing epigenetic processes (MLL2, EP300, CREBBP, TET2); cell cycle (TP53, CCND1, CDKN2A, FBXW7); and the NOTCH (NOTCH1, NOTCH3), WNT (FAT1, YAP1, AJUBA) and receptor-tyrosine kinase phosphoinositide 3-kinase signaling pathways (PIK3CA, EGFR, ERBB2) [51].
Mutation profile of frequently mutated genes (≥5%) in two NGS studies
| Gene mutation rate | Authors | |
|---|---|---|
| De-Chen Lin, et al. [36] | Yi-Bo Gao, et al. [23] | |
| >50% | TP53 | TP53 |
| >30% | TTN | TTN |
| >15% | MLL2 | MLL2 |
| ≥10% | CSMD3, FAT1, GPR98, LRP1B, MUC16, PCLO, SYNE1, XIRP2 | CSMD3, DMD, EP300, FAT1, FAT3, MUC16, NOTCH1, RYR2, SYNE1 |
| ≥5% | ABCA13, ADCY8, ANK2, ARID2, CACNA1E, CEP350, COL11A1, CUBN, C10orf71, DMD, DNAH5, DNAH6, EP300, FAM135B, FAT2, FAT3, FSIP2, HMCN1, HYDIN, KDM6A, KLHL1, LRP2, MAP2, MLL3, MUC2, MUC4, MUC17, MYH4, MYH15, MYO3A, NFE2L2, NOTCH1, PBRM1, PIK3CA, PLEC, PPFIA2, PRDM9, PTEN, RB1, SI, SLCO5A1, SPHKAP, SYNE2, THSD7A, TRHDE, TRIO, UNC80, USH2A, VPS13D, ZFHX4, ZNF750 | ABCA13, ANK3, APOB, CACNA1E, CACNA1S, CDKN2A, CREBBP, CSMD1, CTNNA2, CTNND2, DNAH5, DNAH7, DNAH9, DNAH11, DNAH17, EYS, FAM135B, FAM171A1, FANCM, FAT2, FBN2, FBXW7, FSTL5, JUB, KDM6A, LAMA5, LRP1B, LRP2, MACF1, MLL3, MYH10, MYH15, NALCN, NAV3, NEB, NFE2L2, NOTCH3, NRXN1, PAPPA2, PCDH9, PCDH15, PCLO, PIK3CA, PKHD1, PKHD1L1, PTCH1, PXDNL, RB1, RIMS1, RP1, RYR3, SI, SLCO5A1, SLITRK3, SYT10, TRIM67, USH2A, XIRP2 |
Ling Zhang, et al. [38] discussed the mutation signature of ESCC. Signature A was characterized by C>G, C>T, and C>A mutations at TpCpX trinucleotides and was associated with mutations in the APOBEC family of cytidine deaminases. Signature B was characterized by an enrichment of C>T mutations at XpCpG trinucleotides because of an elevated rate of spontaneous 5-methyl-cytosine deamination. Genes involved in cell cycle and apoptosis regulation were mutated in 99% of cases and mutations in genes that regulate histone modification have been observed in about 63% of ESCCs [23].
To determine the deeper clinical implications of some significantly mutated genes, investigators focused on the prognostic values of genes with deleterious recurrent mutations. EP300 [23, 51], TET2 [51], FAM135B [37] mutations were associated poor survival respectively. However, NOTCH1 mutations had a better outcome than those individuals without deleterious mutations [50]. These results need further validation from multi-centers in future studies.
Gene CNAs
Gene amplifications
Gene amplification is one of the major causes leading to the proto-oncogene activation. As mentioned, high-level amplifications in 3q26 (P63), 3q26.32-33 (SOX2, PIK3CA), 3q27.1, 7p11.2 (EGFR), 7p22.3, 8p11.23 (FGFR1), 8q21.11, 8q24.21 (MYC), 11q13.2-11q13.3 (CCND1, FGF3/4/19, CTTN, CPT1A), 11q22, 12p12.1 (KRAS), 12q15-q21.1 (MDM2), 14q13.3 (NKX2-1), and 18q11.2 were detected (Figure 3) [36]. The amplified genes may be the key drivers giving rise to carcinogenesis.
High amplification of three genes assessed by fluorescent in situ hybridization (FISH) in esophageal squamous cell carcinoma (ESCC). (A) High fibroblast growth factor receptor (FGFR1) amplification. (B) High FGF4 amplification. (C) High FGF19 amplification.
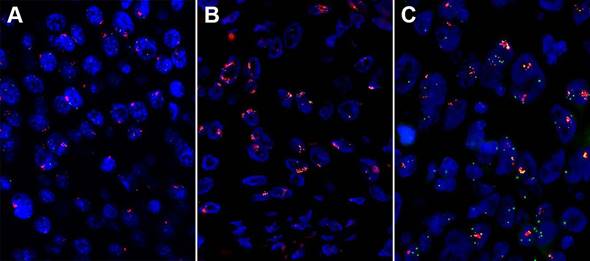
Wang, et al. [52] detected the CCND1 amplification on 100 ESCCs and 11 normal tissues using real-time qPCR and found that 41% of the patients had CCND1 amplification, which has a short survival time compared with the patients without CCND1 amplification. Our group's result had a consistence with them by FISH. The amplification rate of EGFR ranges from 7% to 15%, and it showed a correlation with poor prognosis in ESCC patients [24, 34, 53]. In ESCC, the 3q amplification peak includes only one annotated gene, SOX2 [54]. A copy number gain of SOX2 was observed in 6 of the 40 primary ESCCs (15%) [55]. De-Chen Lin, et al. [36] examined 59 tumors with aCGH and identified CCND1, EGFR, MYC, KRAS, FGFR1 were frequently amplified in ESCC. The amplification of FGFR1 was validated by FISH and high FGFR1 amplification is an independent poor prognostic factor in resected ESCC [56]. And our group's result showed that high FGFR1 amplification is a delayed poor prognostic factor in stage I and II patients (unpublished data).
Gene deletions/losses
Gene deletions/losses are not as common as amplification in ESCC. Relative high-level deletion in 2q22.1-22.2 (LRP1B), 9p21.3 (CDKN2A/B), 5q12.1 (PDE4D), 9p24.1 (PTPRD), and 3p14.2 (FHIT) were detected [36]. Madiniyet, et al. [25] examined 40 ESCC surgical specimens for TP53 gene deletion using FISH and TP53 gene deletion was significantly higher in poorly differentiated ESCC cases. The TP53 gene deletion rate was shown to be correlated with the level of differentiation and lymph node metastasis in ESCC. CDKN2A is a frequently deleted gene, with a loss rate of 14.3% (3/21) in ESCC patients [57]. Takehiko, et al. [32] investigated FBXW7 copy number aberrations in laser-microdissected 38 ESCC specimens using aCGH analysis. These evaluations found FBXW7 copy number loss rates of 44.7% (17/38) in the clinical samples.
Chromosomal and genomic variations in ESCC cell lines
Compared with ESCC samples, ESCC cell lines are pure cancer cells, playing important roles in the molecular mechanism research of ESCC. Studying the chromosomal and genomic variations of ESCC cells provide valuable insight for future studies using these cell lines as ESCC models.
KYSE 180 is an ESCC cell line. Loss of DNA copy number was observed at 4p, 5q, 6q, 9, 10p, 12p, 13, 14p, 15p, 18p, 18q, 20, 22, and Y. Chromosomal gains and translocations occurred at the entire or part of 1, 2p, 3, 4p, 5p, 5q, 6p, 7, 8, 10q, 11, 12q, 14q, 16, 17q, 19, and Xp. Seven derivative chromosomes (5, 8, 12, 14, 14, 14, and 17) presented complex translocations, each involving three or four chromosomes [58]. KYSE 410-4 is also an ESCC cell line. Chromosomal gains occurred at 2q, 3, 8, 17p, and X. Totally 16 structural arrangements were detected, including four derivative chromosomes. The rearrangement of the centromeric regions accounted for approximately 44% of all rearrangements [59]. Jianming Ying, et al. [60] profiled ten commonly used ESCC cell lines (EC1, EC18, EC109, HKESC1, HKESC2, HKESC3, SLMT1, KYSE70, KYSE410 and KYSE520) using aCGH for whole-genome DNA copy number alterations, finding that recurrent chromosomal gains were frequently detected on 3q26-27, 5p15-14, 8p12, 8p22-24, 11q13, 13q21-31, 18p11 and 20q11-13, with frequent losses also found on 8p23-22, 11q22, 14q32 and 18q11-23. Gao et al. [23] performed exome sequencing on 8 cell lines, including 7 from the KYSE series ESCC cell lines14 and 1 immortalized esophageal squamous epithelial cell line, Het-1A. Among the 8 cell lines, total mutations varied from 315 to 754.
These data provide significant, detailed information for appropriate uses of these ESCC cell lines for cytogenetic and molecular biological studies.
Current problems and future directions
As innovation and development of NGS have driven prices down and throughput up, projects have been transitioning from exome to whole-genome sequencing of tumor and matched germline samples, facilitating the discovery of new biology for ESCC. However, as data from different projects began to be collected and centralized, it became apparent that there are marked differences in how teams generate WGS data and analyze it. Benchmarking strategies are needed to be explored to standardize the sequencing method and data analysis [61].
Dissimilar to conventional biomarkers, big-data-based edge biomarker is a new concept to characterize disease features based on biomedical big data in a dynamical and network manner, which also provides alternative strategies to indicate disease status in single samples [62].
The carcinogenesis of ESCC is generally a multistep process reflecting cumulative chromosomal and genetic alterations. Moreover, multiple genetic variations may be involved in a single gene, such as CDKN2A, FBXW7. Plenty of researches had been done in ESCC, however, it is still lacking specific driver genes just as HER2 in breast cancer, KIT and PDGFRA in gastrointestinal stromal tumor, EGFR, EML4-ALK, ROS in lung cancer which can be used to diagnose the disease, stratify the patients, predict the prognosis or used as a therapeutic target. Big data era has arrived, researches on a large group of ESCC patients from multicenter urgently needed. Precision medicine based on genomic data could lead to new way for prevention, diagnosis and treatment of ESCC.
Acknowledgements
This work was supported by Shanghai Municipal Commission of Health and Family Planning, Key-developing disciplines (No.2015ZB0201).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer. 2015;136:E359-E86
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
3. Cancer Genome Atlas Research N, Analysis Working Group. Asan U, Agency BCC, Brigham, Women's H, Broad I, et al. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017
4. Rustgi AK, El-Serag HB. Esophageal carcinoma. The New England journal of medicine. 2014;371:2499-509
5. Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64:381-7
6. Mirnezami R, Nicholson J, Darzi A. Preparing for precision medicine. The New England journal of medicine. 2012;366:489-91
7. Collins FS, Varmus H. A new initiative on precision medicine. The New England journal of medicine. 2015;372:793-5
8. Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. Journal of molecular biology. 1975;98:503-17
9. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:5463-7
10. Langer-Safer PR, Levine M, Ward DC. Immunological method for mapping genes on Drosophila polytene chromosomes. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:4381-5
11. Hacia JG, Fan JB, Ryder O, Jin L, Edgemon K, Ghandour G. et al. Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays. Nature genetics. 1999;22:164-7
12. Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA. et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350-4
13. Shi ZZ, Shang L, Jiang YY, Hao JJ, Zhang Y, Zhang TT. et al. Consistent and differential genetic aberrations between esophageal dysplasia and squamous cell carcinoma detected by array comparative genomic hybridization. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:5867-78
14. Muller S, O'Brien PC, Ferguson-Smith MA, Wienberg J. Cross-species colour segmenting: a novel tool in human karyotype analysis. Cytometry. 1998;33:445-52
15. Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D. et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature biotechnology. 2000;18:630-4
16. Thompson JF, Steinmann KE. Single molecule sequencing with a HeliScope genetic analysis system. Current protocols in molecular biology. 2010 Chapter 7: Unit7 10
17. Tsuda T, Tahara E, Kajiyama G, Sakamoto H, Terada M, Sugimura T. High incidence of coamplification of hst-1 and int-2 genes in human esophageal carcinomas. Cancer research. 1989;49:5505-8
18. Janssen JW, Imoto I, Inoue J, Shimada Y, Ueda M, Imamura M. et al. MYEOV, a gene at 11q13, is coamplified with CCND1, but epigenetically inactivated in a subset of esophageal squamous cell carcinomas. Journal of human genetics. 2002;47:460-4
19. Law FB, Chen YW, Wong KY, Ying J, Tao Q, Langford C. et al. Identification of a novel tumor transforming gene GAEC1 at 7q22 which encodes a nuclear protein and is frequently amplified and overexpressed in esophageal squamous cell carcinoma. Oncogene. 2007;26:5877-88
20. Ishizuka T, Tanabe C, Sakamoto H, Aoyagi K, Maekawa M, Matsukura N. et al. Gene amplification profiling of esophageal squamous cell carcinomas by DNA array CGH. Biochemical and biophysical research communications. 2002;296:152-5
21. Kim DH, Muto M, Kuwahara Y, Nakanishi Y, Watanabe H, Aoyagi K. et al. Array-based comparative genomic hybridization of circulating esophageal tumor cells. Oncology reports. 2006;16:1053-9
22. Egashira A, Morita M, Kakeji Y, Sadanaga N, Oki E, Honbo T. et al. p53 gene mutations in esophageal squamous cell carcinoma and their relevance to etiology and pathogenesis: results in Japan and comparisons with other countries. Cancer science. 2007;98:1152-6
23. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM. et al. Genetic landscape of esophageal squamous cell carcinoma. Nature genetics. 2014;46:1097-102
24. Jiang D, Li X, Wang H, Shi Y, Xu C, Lu S. et al. The prognostic value of EGFR overexpression and amplification in Esophageal squamous cell Carcinoma. BMC cancer. 2015;15:377
25. Niyaz M, Turghun A, Ping ZH, Zhu Z, Sheyhedin I, Ren C. et al. TP53 gene deletion in esophageal cancer tissues of patients and its clinical significance. Molecular medicine reports. 2013;7:122-6
26. Hao JJ, Gong T, Zhang Y, Shi ZZ, Xu X, Dong JT. et al. Characterization of gene rearrangements resulted from genomic structural aberrations in human esophageal squamous cell carcinoma KYSE150 cells. Gene. 2013;513:196-201
27. Hu N, Wang C, Hu Y, Yang HH, Kong LH, Lu N. et al. Genome-wide loss of heterozygosity and copy number alteration in esophageal squamous cell carcinoma using the Affymetrix GeneChip Mapping 10 K array. BMC genomics. 2006;7:299
28. Garibyan L, Avashia N. Polymerase chain reaction. The Journal of investigative dermatology. 2013;133:e6
29. Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2766-70
30. Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N. et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic acids research. 1989;17:2503-16
31. Luo ML, Shen XM, Zhang Y, Wei F, Xu X, Cai Y. et al. Amplification and overexpression of CTTN (EMS1) contribute to the metastasis of esophageal squamous cell carcinoma by promoting cell migration and anoikis resistance. Cancer research. 2006;66:11690-9
32. Yokobori T, Mimori K, Iwatsuki M, Ishii H, Tanaka F, Sato T. et al. Copy number loss of FBXW7 is related to gene expression and poor prognosis in esophageal squamous cell carcinoma. International journal of oncology. 2012;41:253-9
33. Xu X, Chen G, Wu L, Liu L. Association of genetic polymorphisms in PTEN and additional gene-gene interaction with risk of esophageal squamous cell carcinoma in Chinese Han population. Diseases of the esophagus: official journal of the International Society for Diseases of the Esophagus / ISDE. 2015
34. Kato H, Arao T, Matsumoto K, Fujita Y, Kimura H, Hayashi H. et al. Gene amplification of EGFR, HER2, FGFR2 and MET in esophageal squamous cell carcinoma. International journal of oncology. 2013;42:1151-8
35. Yen CC, Chen YJ, Lu KH, Hsia JY, Chen JT, Hu CP. et al. Genotypic analysis of esophageal squamous cell carcinoma by molecular cytogenetics and real-time quantitative polymerase chain reaction. International journal of oncology. 2003;23:871-81
36. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X. et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nature genetics. 2014;46:467-73
37. Song Y, Li L, Ou Y, Gao Z, Li E, Li X. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91-5
38. Zhang L, Zhou Y, Cheng C, Cui H, Cheng L, Kong P. et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. American journal of human genetics. 2015;96:597-611
39. Kwong D, Lam A, Guan X, Law S, Tai A, Wong J. et al. Chromosomal aberrations in esophageal squamous cell carcinoma among chinese: gain of 12p predicts poor prognosis after surgery. Human pathology. 2004;35:309-16
40. Carneiro A, Isinger A, Karlsson A, Johansson J, Jonsson G, Bendahl PO. et al. Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC cancer. 2008;8:98
41. Shi ZZ, Liang JW, Zhan T, Wang BS, Lin DC, Liu SG. et al. Genomic alterations with impact on survival in esophageal squamous cell carcinoma identified by array comparative genomic hybridization. Genes, chromosomes & cancer. 2011;50:518-26
42. Cheng C, Zhou Y, Li H, Xiong T, Li S, Bi Y. et al. Whole-Genome Sequencing Reveals Diverse Models of Structural Variations in Esophageal Squamous Cell Carcinoma. American journal of human genetics. 2016;98:256-74
43. Hou J, Jiang D, Zhang J, Gavine PR, Xu S, Liu Y. et al. Frequency, characterization, and prognostic analysis of PIK3CA gene mutations in Chinese esophageal squamous cell carcinoma. Human pathology. 2014;45:352-8
44. Akbari MR, Malekzadeh R, Nasrollahzadeh D, Amanian D, Islami F, Li S. et al. Germline BRCA2 mutations and the risk of esophageal squamous cell carcinoma. Oncogene. 2008;27:1290-6
45. Liu QW, Fu JH, Luo KJ, Yang HX, Wang JY, Hu Y. et al. Identification of EGFR and KRAS mutations in Chinese patients with esophageal squamous cell carcinoma. Diseases of the esophagus: official journal of the International Society for Diseases of the Esophagus / ISDE. 2011;24:374-80
46. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY. et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. The Journal of pathology. 2010;220:446-51
47. Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K. et al. NRF2 Mutation Confers Malignant Potential and Resistance to Chemoradiation Therapy in Advanced Esophageal Squamous Cancer. Neoplasia. 2011;13:864-IN26
48. Hu N, Wang C, Su H, Li WJ, Emmert-Buck MR, Li G. et al. High frequency of CDKN2A alterations in esophageal squamous cell carcinoma from a high-risk Chinese population. Genes, chromosomes & cancer. 2004;39:205-16
49. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
50. Qin HD, Liao XY, Chen YB, Huang SY, Xue WQ, Li FF. et al. Genomic Characterization of Esophageal Squamous Cell Carcinoma Reveals Critical Genes Underlying Tumorigenesis and Poor Prognosis. American journal of human genetics. 2016;98:709-27
51. Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y. et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology. 2016;150:1171-82
52. Wang MT, Chen G, An SJ, Chen ZH, Huang ZM, Xiao P. et al. Prognostic significance of cyclinD1 amplification and the co-alteration of cyclinD1/pRb/ppRb in patients with esophageal squamous cell carcinoma. Diseases of the Esophagus. 2012;25:664-70
53. Sunpaweravong P, Sunpaweravong S, Puttawibul P, Mitarnun W, Zeng C, Baron AE. et al. Epidermal growth factor receptor and cyclin D1 are independently amplified and overexpressed in esophageal squamous cell carcinoma. Journal of cancer research and clinical oncology. 2005;131:111-9
54. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG. et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics. 2009;41:1238-42
55. Gen Y, Yasui K, Zen Y, Zen K, Dohi O, Endo M. et al. SOX2 identified as a target gene for the amplification at 3q26 that is frequently detected in esophageal squamous cell carcinoma. Cancer Genetics and Cytogenetics. 2010;202:82-93
56. Kim HS, Lee SE, Bae YS, Kim DJ, Lee CG, Hur J. et al. Fibroblast growth factor receptor 1 gene amplification is associated with poor survival in patients with resected esophageal squamous cell carcinoma. Oncotarget. 2015;6:2562-72
57. Smeds J, Berggren P, Ma X, Xu Z, Hemminki K, Kumar R. Genetic status of cell cycle regulators in squamous cell carcinoma of the oesophagus: the CDKN2A (p16(INK4a) and p14(ARF) ) and p53 genes are major targets for inactivation. Carcinogenesis. 2002;23:645-55
58. Wu YP, Yang YL, Yang GZ, Wang XY, Luo ML, Zhang Y. et al. Identification of chromosome aberrations in esophageal cancer cell line KYSE180 by multicolor fluorescence in situ hybridization. Cancer Genet Cytogenet. 2006;170:102-7
59. Yang Y, Chu J, Wu Y, Luo M, Xu X, Han Y. et al. Chromosome analysis of esophageal squamous cell carcinoma cell line KYSE 410-4 by repetitive multicolor fluorescence in situ hybridization. Journal of genetics and genomics = Yi chuan xue bao. 2008;35:11-6
60. Ying J, Shan L, Li J, Zhong L, Xue L, Zhao H. et al. Genome-wide screening for genetic alterations in esophageal cancer by aCGH identifies 11q13 amplification oncogenes associated with nodal metastasis. PloS one. 2012;7:e39797
61. Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E. et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nature communications. 2015;6:10001
62. Zeng T, Zhang W, Yu X, Liu X, Li M, Chen L. Big-data-based edge biomarkers: study on dynamical drug sensitivity and resistance in individuals. Briefings in bioinformatics. 2015
Author contact
![]() Corresponding author: Dr. Yingyong Hou, Department of Pathology, Zhongshan Hospital, Fudan University, 180 Fenglin Road, Xuhui District, Shanghai 200032, People's Republic of China. Phone: +8602164041990. Fax: +8602164038038. Email: houyingyongcom.
Corresponding author: Dr. Yingyong Hou, Department of Pathology, Zhongshan Hospital, Fudan University, 180 Fenglin Road, Xuhui District, Shanghai 200032, People's Republic of China. Phone: +8602164041990. Fax: +8602164038038. Email: houyingyongcom.