Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(6):913-923. doi:10.7150/jca.17301 This issue Cite
Research Paper
Super Aqueous Solubility of Albendazole in β-Cyclodextrin for Parenteral Application in Cancer therapy
Krishna Pillai ![]() , Javed Akhter, David Lawson Morris
, Javed Akhter, David Lawson Morris
Department of Surgery, University of New South Wales, St. George Hospital, Kogarah, NSW, Australia
Received 2016-8-22; Accepted 2016-11-10; Published 2017-3-12
Abstract
Poor aqueous solubility of anticancer drug, albendazole (ABZ), prevents parenteral application. Here, we demonstrate how to increase the aqueous solubility of ABZ to 6- 8 mg/ml using sulfobutylether - β-cyclodextrin (SBE-β-CD) or Hydroxypropyl- β-cyclodextrin (HP- β-CD) by manipulation of complexation parameters such as the physical state of ABZ (ionized in acetic acid), the concentration of ionised ABZ, agitation time and temperature. Solubility was first examined with suspension of excess ABZ powder in cyclodextrin (CD) solutions at pH (2.3, 4.0 & 7.0), subsequently with excess ionised ABZ [ABZ] at pH. 2.3 with the determination of optimal quantity of [ABZ] use for maximal complexation. Complexation time, temperature effect, stability of formulation, with in vitro and in vivo cytotoxicity of [ABZ]-SBE-β-CD was assessed. Suspended ABZ formulation at pH 2.3 showed maximum solubilisation of 2.29 & 1.72 mg/ml, whilst excess addition of [ABZ] showed poor complexation (1.26 & 1.20 mg/ml) in SBE-β-CD & HP- β-CD, respectively. The addition of 8.0 mg/ml and 7.0 mg/ml of [ABZ] to 40% CD solutions at 25ºC showed maximum complexation with SBE-β-CD & HP- β-CD, respectively, at three days, with 2 weeks stability. [ABZ] complexed with SBE-β-CD showed potent cytotoxicity (in vitro & in vivo) in ovarian tumour cells. Hence, the current method may be used for solubilising ABZ for parenteral use.
Keywords: Albendazole, aqueous, solubility, cyclodextrin, complexation, cytotoxicity.
Introduction
Albendazole (ABZ) is an antihelminth that targets parasitic nematodes and cestodes in both human and animals [1-3]. Recently the molecule has been successfully tested for anticancer properties against a wide range of cancers such as ovarian, breast, prostate and a number of other cancers in both in vitro and in vivo models [4-6]. However, ABZ by virtue of its molecular structure is sparingly soluble in water (0.2µg/ml) although it is highly permeable through biological membranes [7]. ABZ is quite soluble in organic solvents (log P = 3.5), however the inherent toxicity found in such solvents is a disadvantage [8, 9] when preparing for intravenous (IV) or intra-peritoneal (IP) formulations.
For clinical application of ABZ in cancer using IP or IV routes, a much higher aqueous solubility of ABZ may be required usually 5 - 10 mg/ml (http://www.fda.gov/Drugs/default.htm) since low concentration of drugs in solution would require very large volumes to be used in order to reach the required therapeutic dose and thereby make the application impractical. Hence, numerous formulations have been carried out with ABZ [4, 10, 11], to enhance its aqueous solubility. Most recently a group has investigated the use of a number of different cyclodextrins (CDs) to increase ABZ aqueous solubility and reached a maximum of 1.52 mg/ml in a 40 % solution of Meth- β- CD (Me-β- CD) [12].
Cyclodextrins are polymeric glucopyranose units that form a truncated inverted cone with a central hydrophobic cavity and an external rim of hydroxyl groups [13]. They have been successfully used for the solubilisation of a number of hydrophobic therapeutic molecules [14-16]. The hydrophobic molecule (guest) is said to be attracted to the central cavity with stabilisation by the formation of weak hydrogen bonds, hydrophobic interactions and Van der Waals forces [17] resulting in enthalpy changes [18]. Three types of CDs exist, and their classification depends on the number of glucose units present, 6, 7 & 8 referred to as α, β & γ. Hence the size of the central pocket also varies, with γ being the largest and able to accommodate bigger guest molecules [19]. Modification by substitution on the hydroxyl group of the outer rim has produced a number of different CDs amongst which is the sulphobutylether-β-CD (SBE-β-CD) and the hydroxypropyl-β-CD (HP--β-CD). These two CDs have been found to exert minimal renal toxicity and hence more acceptable for clinical application [20, 21].
Since ABZ is a stable molecule at acidic pH [22] and ionisable [23], we set out to investigate whether a better complexation and a higher aqueous solubility of ABZ can be achieved with SBE-β-CD and HP-β-CD when ABZ is ionised in solution at lower pH. Further we also investigated how the concentration of ionised guest molecule in solution would affect complexation and solubilisation of ABZ, time duration for maximum ABZ-CD complexation, temperature effect (25 & 37ºC) on complexation, stability of complexed ABZ-CD at ambient temperature, along with in vitro and in vivo cytotoxicity evaluation of the formulation using only SBE-β-CD since it showed a higher ABZ solubility compared to HP-β-CD. With optimization of complexation parameters such as the physical state of ABZ (ionised in acetic acid) ,concentration of ionised ABZ used with the different percentages of cyclodextrins, complexation time and temperature, we were able to achieve a very much higher solubility of ABZ in both the SBE-β-CD and HP-β-CD as compared to other studies that have been cited earlier.
Materials and Methods
Reagents
Albendazole (ABZ) (Mw =265 g mol -1) and hydroxypropyl-β cyclodextrin (HP-β-CD, Mw = 1375 g mol -1) was purchased from Sigma Aldrich, Australia; Sulfobutylether -β-cyclodextrin (SBE-β-CD, Mw = 2241 g mol -1) was supplied by CYDEX Pharmaceuticals, Ter Lenexa, KS, USA, whilst all other reagents were of analytical grade supplied by Sigma Aldrich Chemicals. For solubilising the cyclodextrins, distilled water was used.
Standard Curves for ABZ-β-CD
Standard curve for the ABZ-CDs was prepared in 5% CD solution in distilled water acidified with glacial acetic acid (3ml acetic acid in 47 ml distilled water, pH = 2.38)). A 0.1 ml glacial acetic acid containing 1 mg of ABZ was introduced into 0.9ml of 5% CD solution and allowed to complex in a shaker overnight, at room temperature (25ºC). The resulting complex was adjusted to pH 7.0 using 0.1 M sodium hydroxide (no precipitate formed indicating full complexation with CD) dilution factor was taken into consideration and then serially diluted to prepare standards containing 1, 5, 10, 20, 40 & 80 µg/ml. The concentration of ABZ at µg/ml was measured using spectrophotometer (TECAN Infinite M200 PRO) at 230 nm and a standard curve was generated.
Phase solubility curves
Powdered ABZ in suspension at different pH (solid/liquid phase)
Cyclodextrins were prepared in varying concentrations ranging from 5% - 40% in aqueous media at different pH (2.3, 4.0 and 7.0) adjusted with glacial acetic acid. To 2 ml of the cyclodextrin solution in a 10 ml vortex tube was added excess ABZ in powder form as reported by Higuchi and Connors [24]. The tubes were then placed on an agitator for a period of 120 hours at 25ºC. The controls only contained distilled water and ABZ, with pH adjusted. After 120 hrs of agitation, those tubes containing cyclodextrins that were in acidic media were neutralised using 0.1 M sodium hydroxide. All tubes were then centrifuged at 3600 rpm, after which the supernatant were collected and filtered through 44 micron membrane filters. The filtered supernatant was then analysed for complexed ABZ with spectrophotometer (TECAN Infinite M200 PRO) at 230 nm.
ABZ solubilised in Acetic acid with aqueous cyclodextrin (pH.2.3) (with excess ABZ addition)
β-Cyclodextrins were prepared in acidified distilled water (pH.2.3), at different percentage (5, 10, 20, 30 & 40). The acidified aqueous media was prepared by adding 3 ml of glacial acetic acid to 47 ml of distilled water. To 0.8 ml of cyclodextrin was added excess ABZ (15 mg in 0.2 ml of acetic acid). The tubes containing, the various % of cyclodextrins were then placed in an agitator for a period of 120 hours at ambient temperature (25ºC). The tubes were then neutralised with 0.1 M sodium hydroxide, centrifuged at 3600 rpm, filtered with a 44 micron membrane and then analysed for ABZ content.
Solubilised ABZ in Acetic acid with aqueous β-Cyclodextrins (pH.2.3) - Increasing quantities of ABZ added to separate tubes containing specific percentage of cyclodextrins to study the effect of super saturation
As before, β-Cyclodextrins were prepared at various concentrations (5%-40%), in acidified aqueous media (pH 2.3). To a 0.8 ml of 40% cyclodextrin solution was added 0.2 ml of ionised ABZ in acetic acid containing (1,2,3,4,5,6,7,8,9,10,12, 14 mg) in separate tubes and then the tubes were placed on a shaker for 72 hours. A similar procedure was carried out for 30, 20 and 10 % CD) At the end of 72 hours, the reaction mixture was adjusted to neutral pH, centrifuged at 3600 rpm and then filtered with 44 micron membrane filter. The filtrate was then analysed for complexed ABZ, as before. Controls only contained solubilised ABZ in water (pH.2.3).
Ideal phase solubility curve using predetermined ionised ABZ to be added
The CDs were prepared in varying % (0-40) in aqueous acid media (pH 2.3) with the addition of predetermined quantities of ionised ABZ in acetic acid to the respective CD solutions. The solutions were then agitated at 25ºC for three days, after which the pH was adjusted, centrifuged, filtered and analysed for ABZ. The binding constant (K) and complexation efficiency (CE) was calculated as shown below:
Binding constant (K) 1:1 = slope / So (1- slope),
where So = intrinsic solubility of albendazole in aqueous media (in the absence of cyclodextrin). Slope is derived from the phase solubility plot.
Complexation efficiency (CE) = slope/ 1- slopeºC
Effect of incubation time on complexation efficiency
β-Cyclodextrins were prepared from 5-40% in acid media as before (ph. 2.3) and 0.8 ml of CD was treated to 0.2 ml of acetic acid containing various quantities of ionised ABZ (maximum quantities of ionised ABZ that has been determined earlier for each % of CD). For SBE-β-CD, the 40% solution was treated with 8.0 mg of ABZ in 200 µl of acetic acid, 30% to 7 mg, 20% to 5 mg and 10 % to 3 mg of ABZ. For the HP-β-CD, the ABZ added were 7, 6, 4 & 3 for 40, 30, 20 & 10 % CDs, respectively. For each day there were 5 tubes (0, 10, 20, 30 and 40% of CDs with addition of solubilised ABZ) (5 sets were prepared, one for each day). The tubes were then placed on an agitator for a period of 5 days, samples were analysed for ABZ complexation at days 1, 2, 3, 4 and 5.
Effect of temperature on complexation of solubilised ABZ in cyclodextrins
β-Cyclodextrins were prepared ranging from 0 - 40% at pH. 2.3 and treated to ionised ABZ as in the previous experiment. The tubes were then agitated at room temperature (25ºC) or at 37ºC in a water bath. At 72 hours, the tubes were neutralised, centrifuged and filtered before analysing the ABZ content.
Stability study of β-Cyclodextrins- ABZ complex at 25ºC
To a 0.8 ml of 20% SBE-β-CD solution in acid media (pH.2.3) was added 0.2 ml of acetic acid 5.0 mg of ABZ and agitated over 3 days in a shaker, after which, the solution was neutralised using 0.1 M sodium hydroxide, filtered with 44micron filter and then analysed for ABZ content (day 0). Suitable quantity of solutions was prepared and 100 µl under sterile conditions in triplicates were used for stability studies for each time point. The solution was stored at 25ºC and analysed on alternate days over 14 days. A similar arrangement was made for 40% CD but with the addition of 8.0 mg of ABZ. The procedure was the same for HP-β-CD, however, 4 & 7 mg/ml ABZ was used for complexation in 20% and 40% CD solutions.
In vitro cytotoxicity evaluation of ABZ-SBE- β-Cyclodextrin on ovarian tumour cell line OVCAR-3 and human ovarian epithelial cells (HOSE)
Ovcar -3 cells (5000/well) were seeded into a 96 well plate and grown in RPMI following standard procedures over 3 days. The cells were then treated with varying quantities (0, 1 X10 -7, 2.5 X 10 -7, 5 X 10 -7 & 1 X 10 -6) of sterile complexed ABZ-SBE-β CD that was prepared in RPMI. A similar procedure was adopted for ABZ that was solubilised in absolute ethanol (<1%). After 72 hours, the cells were fixed in 10% Trichloroacetic acid (TCA) and subjected to sulfohordomine assay using standard methods [25]. Optical density (OD) of solubilised dye was measured at 570 nm. A similar procedure was carried out for HOSE cells.
In vivo cytotoxicity evaluation of ABZ-SBE- β-Cyclodextrin with ovarian tumour - OVCAR-3
Animal study was conducted with animal ethics approval from University of New South Wales. After one week acclimatization, twenty nude balb C mice were randomised into 4 groups of 5 mice/group and they were each injected intraperitoneally with 3 X 10 -6 ovcar-3 cells in matrigel using 25 gauge needle. Under sterile conditions. Two weeks later, treatment began with IP injection of ABZ-SBE- β-CD of 50 mg/kg in phosphate buffer saline (200µl solution volume) in group 1. Controls only received SBE- β-CD in group 2. For comparison, group 3 received a suspension of ABZ in HPMC (Hydroxypropylmethylcellulose) at a dose 75 mg/kg in 200 µl solution), whilst group 4 only received HPMC in PBS (control). All animals were treated every day.
The animals were monitored daily and weight and circumference of abdomen were recorded every alternate day. At termination of the experiment on the 33rd day, the animals were euthanized following recommended protocol; their ascities volume and the weight of the tumours recovered were recorded.
Statistical analysis
All data were presented as the mean ± SEM and comparison of mean values between two groups were carried out using Student t-test with 95% confidence limit denoted as p=<0.05. Graph pad prism software 5.0 version were used for statistical analysis and drawing of the graphs (version 5.0, Graph pad software, La Jolla, CA, USA)
Results
Standard Curve for measuring ABZ complexed with β- Cyclodextrins (β- CD)
The standard curve that was generated was linear within the range of 0-40 µg/ml. (Figure 1A & B), after which it was nonlinear (not shown). Hence, in the rest of the experimental investigation a range of dilutions (0, 5, 10, 20, 30, and 40) µg/ml were used to generate the standard curve to measure the complexed ABZ in the CDs.
A & B show standard curves for ABZ in CDs in the range of 0 - 40 µg/ml that has been generated using complexed ABZ in a 5% solution of the CDs. C & D show phase solubility curves for the ABZ (solid suspension) when they were treated with β-CDs in solution at different pH. At pH 2.3, ABZ was most complexed as shown by the gradient of solubility curves; CD = cyclodextrin.
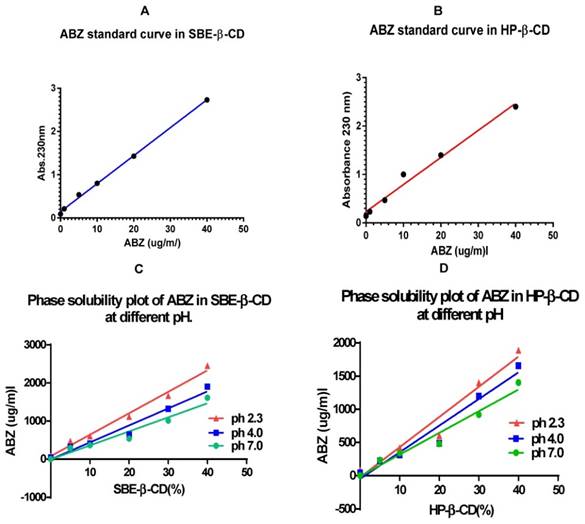
Phase solubility in cyclodextrin with suspended ABZ at different pH
The best complexation of ABZ was achieved at pH 2.3 for all the percentages of both the CDs investigated (Figure 1C & D). In particular 2292 µg/ml complexed in a 40% solution of SBE- β-CD and a slightly lower amount of 1729 µg/ml in HP -β-CD, with a difference of 24% (Table 1). At pH of 4.0, the difference in complexed ABZ in a 40 % solution between the two CDs was only 14%, whilst at pH 7.0 it was also 14%.
Complexation of suspension of ABZ powder in CD solution at various pHs
| SBE-β- CD (sulphobutylether β-CD) | HP-β-CD (Hydroxypropyl- β-CD) | |||||
|---|---|---|---|---|---|---|
| CD (%) | pH 7.0 ABZ (µg/ml) | pH 4.0 ABZ (µg/ml) | pH 2.3 ABZ (µg/ml) | pH 7.0 ABZ (µg/ml) | pH 4.0 ABZ (µg/ml) | pH 2.3 ABZ (µg/ml) |
| 0 | < 1 | 41±8 | 82±14 | < 1 | 41±7 | 82 ±10 |
| 5 | 113 ±12 | 162 ±31 | 310 ±29 | 72 ±5 | 62 ±4 | 70 ±2 |
| 10 | 205 ±22 | 252 ±35 | 440 ±36 | 178 ±12 | 151 ±7 | 259 ±8 |
| 20 | 379 ±29 | 486 ±52 | 951 ±48 | 321 ±18 | 326 ±14 | 440 ±21 |
| 30 | 850 ±28 | 1160 ±61 | 1503 ±46 | 759 ±36 | 1040 ±32 | 1240±38 |
| 40 | 1450 ±46 | 1740 ±56 | 2292 ±62 | 1243 ±43 | 1496 ±43 | 1729±41 |
Table 1 shows the amount of ABZ (µg/ml) that has been complexed and hence solubilised by SBE-β-CD and HP- β- CD at various pHs when suspension of excess ABZ powder was agitated for 5 days at 25ºC in 2 ml of cyclodextrin of varying percentages. The highest solubility was observed at pH. 2.3 for both the CDs; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin
Comparing the slopes of the graphs (Y values, Table 2), the relative complexation at pH 2.3 was 1.53 times that at pH 7.0 for SBE-β-CD whilst it was a little smaller with a Y value of 1.39 for HP-β-CD. The Y vales for both the CDs at pH 4.0 were quite similar (1.21 vs. 1.23).
Comparison of slope values of phase solubility curves of suspension of solid ABZ in CDs (5 -40%) at various pH.
| SBE - β- CD | HP- β- CD | |||
|---|---|---|---|---|
| pH | Slope | Y | Slope | Y |
| 2.3 | 56.36±3.187 | 1.53 | 45.53±4.722 | 1.39 |
| 4.0 | 44.66± 4.271 | 1.21 | 40.15±4.543 | 1.23 |
| 7.0 | 36.87± 4.064 | 1.0 | 32.69±3.148 | 1.0 |
Table 2 shows the slope values of the phase solubility curves at different pH and their Y values (normalization to values at pH. 7.0). Y = slope at pH (x) / slope at pH 7.0; (x) = pH 2.3, 4.0 & 7.0; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin.
Complexation of ionised ABZ (solubilised in glacial acetic acid) with β cyclodextrin (0 - 40%) over 5 days at 25ºC, when excess (15 mg/ml) of ABZ was added
From the phase solubility curves (Fig 2A & B) it is apparent that when excess quantities of solubilised ABZ was added, the loading of ABZ in CDs were rather low, for SBE -β -CD, it was around 1260µg/ml with 40% CD solution, whilst for HP- β -CD it was 1200 µg/ml. On a comparative basis to the addition of powdered ABZ to CDs at pH 2.3 in a 40% solution, there is a relative drop in complexation, for SBE-β-CD of 45% and 30% for HP-β-CD. This may indicate that super saturation of solubilised ABZ in acetic acid may actually interfere with the complexation process, commonly referred to as stearic hindrance and overcrowding in a reaction media.
A & B show phase solubility curve for CDs when excess (15 mg) of solubilised (ionised) ABZ was added to cyclodextrin solution. The maximum complexed ABZ with SBE-β-CD was 1260 µg/ml whilst with HP- β-CD was 1200 µg/ml in a 40% CD solution at pH 2.3; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin. C & D show that for each % of CDs, a maximum quantity of solubilised ABZ that can be added to get the best complexation, exceeding the maximum, results in decline in complexation. E & F is a phase solubility curve for CDs that has been generated with agitation over a period of 1- 5 days, maximum complexation was shown on the third day.
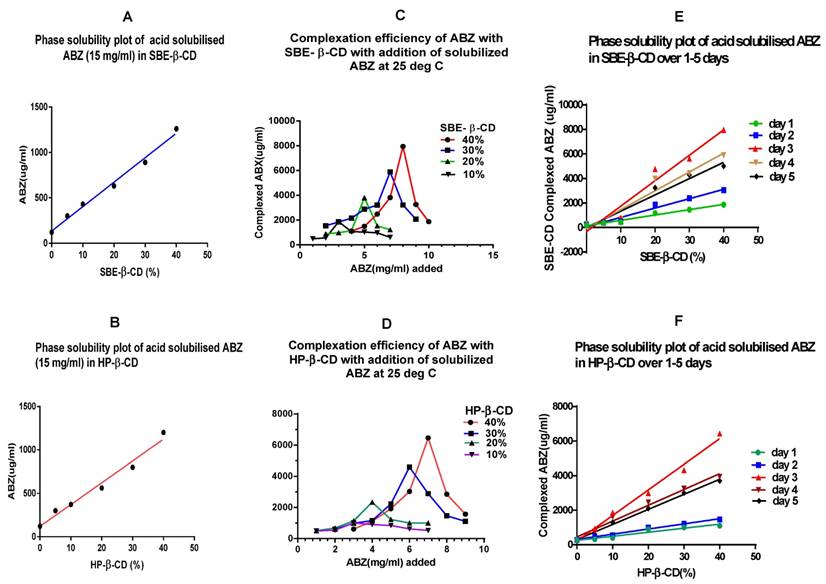
Complexation efficiency of β -cyclodextrin (10 - 40%) when small incremental quantities of ionised ABZ was added and agitated at 25ºC for 3 days
When added with incremental quantities of ABZ to the different percentages of CDs in separate tubes, it appears that maximum complexation can only be reached with addition of ionised ABZ that was compatible for the percentage of CD used. Addition of excess ionised ABZ reduced the complexation process. In the case of 40% CDs, for SBE- β- CD, after addition of 8.0 mg of ionised ABZ, further additions only results in a lower complexation, similar is the case with the other percentage of CDs used in this study. The same is observed for the HP- β -CDs. Hence, in our system, over loading only reduced the quantity of ionised ABZ from complexing with the CDs (Figure 2C & D). The quantities that can be added to different percentages of CDs to get the best complexation are given as Z value in table 3.
Maximum quantity of ionised ABZ addition to different percentages of CDs for best complexation and solubilisation of ABZ.
| SBE-β-CD | HP-β-CD | |||
|---|---|---|---|---|
| CD % | Z (mg/ml) | L (µg/ml) | Z (mg/ml) | L (µg/ml) |
| 10 | 3 | 1869±86 | 3 | 1010±61 |
| 20 | 5 | 3800±112 | 4 | 2342±82 |
| 30 | 7 | 5889±120 | 6 | 4600±101 |
| 40 | 8 | 7940±140 | 7 | 6460±110 |
Table 3 shows the maximum amount of ionised ABZ that can be added before inhibition of complexation occurs. Z = maximum concentration (mg/ml) of ABZ that can be added after which inhibition of complexation occurs; L = maximum amount (µg/ml of ABZ) that was complexed to the CD at 25ºC; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin.
Time profile study of complexation of ionised ABZ with β- CDs (5-40%)
Time profile study indicates that the maximum loading (complexation) of ionised ABZ with the CDs takes place at day 3, with agitation at 25ºC, after which further agitation only tend to reduce the complexation process (Fig. 2E & F). In this system, over loading with the solute, the ionised ABZ, was prevented by adding the predetermined quantity of ionised ABZ that the CD solution would complex (Table 3). The reduction of complexation after three days of agitation at 25ºC may be due to the rigorous agitation that may enhance the disruption of complexed ABZ, since only weak non covalent bonds hold the ABZ in the cavity.
Further examining the comparative slope values (Y) (Table 4), complexation is maximal for both the CDs at 3 days, the Y values being 4.76 and 6.29 for SBE -β-CD and HP -β-CD, respectively, after which it declines. Hence, in the current system, three days agitation at pH 2.3 is the best for maximal complexation.
Slope values of phase solubility curves at different days
| SBE-β- CD | HP- β- CD | |||
|---|---|---|---|---|
| Day (x) | Slope | Y | Slope | Y |
| 1 | 43.4 ±3.143 | 1.0 | 23.5 ±3.325 | 1.0 |
| 2 | 76.23 ± 6.7 | 1.7 | 29.87 ±1.782 | 1.27 |
| 3 | 207.3 ± 21.82 | 4.76 | 148.2 ±7.499 | 6.29 |
| 4 | 153.5 ±19.55 | 3.54 | 91.71 ±6.164 | 3.87 |
| 5 | 132.6 ± 15.5 | 3.0 | 87.36 ± 3.335 | 3.72 |
Table 4 shows the slope values of the phase solubility curves with normalisation of slope value to day 1. Y = (slope at x day/ slope at day 1) and this indicates a comparative complexation to day 1; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin
Phase solubility of ionised ABZ (solubilised in acetic acid) with aqueous β- CD at pH 2.3, 25ºC with three days agitation
A plot of phase solubility of ionised ABZ (solubilised in acetic acid) with SBE-β-CD (5-40%) indicates that in a 40% solution, 8 mg/ml of ABZ can be complexed at 3 days/25ºC (Fig 3A). Figure 3B shows a phase solubility plot for HP-β-CD indicating that in a 40% solution, 6.4 mg/ml of ABZ can be complexed.
A & B show an ideal phase solubility plot for both the CDs with maximal complexation, with the correct amount of acid solubilised- ionised ABZ added for each percentage (%) of CDs. C & D are phase solubility plots for the two CDs at 25 and 37ºC, much higher solubility was seen at 25ºC compared to 37ºC. E & F show the stability curves for the CDs tested over 14 days indicating that the complexation was stable when stored at 25ºC; CD=cyclodextrin
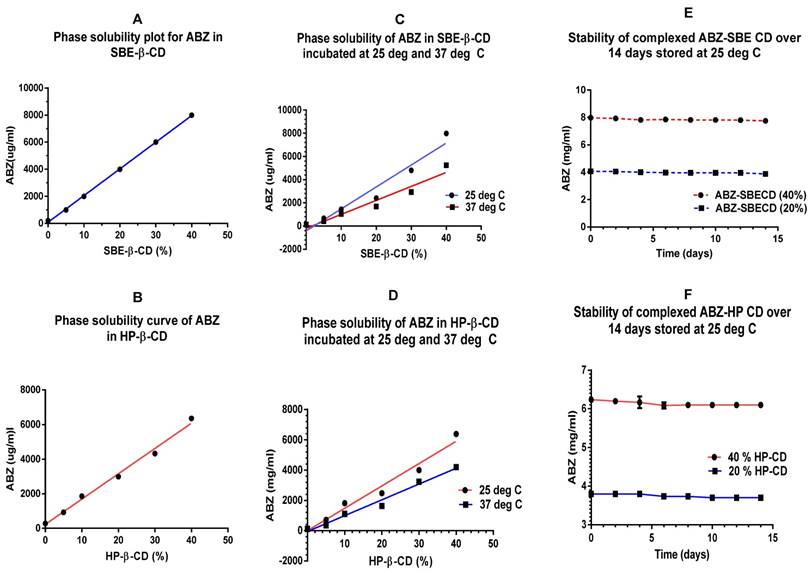
The binding constant (K) was calculated to be 48751M-1 for ABZ with SBE-β-CD and the complexation efficiency (CE) was 0.034126, indicating that 1 in every 28 SBE-β-CD molecule in solution forms a water soluble complex with ABZ. On a comparative basis, the K value for ABZ with HP-β-CD was 20571 M -1 and the CE was 0.0144, indicating that 1 in every 69 HP-β-CD molecules in solution complexes with ABZ.
Phase solubility of ionised ABZ in CDs at two different temperatures over 3 days
Complexation of ionised ABZ with CDs at 25ºC indicated that it is a more suitable temperature to work at, compared to 37ºC. Based on the slope of the graphs, for SBE-β -CD, complexation with ionised ABZ at 37ºC diminished by 36 % whilst for HP- β- CD it diminished by 29%. This may be expected since higher temperatures may have effect on the weaker bonds holding the ABZ in the hydrophobic cavity of the CDs (Figure 3 C & D)
Stability studies complexed ABZ with CDs at 25ºC over two weeks
Stability studies conducted over 14 days at 25ºC, under aseptic conditions, shows that the preparation of ionised ABZ in CDs (20 and 40%) were stable (Figure 3E & F). For long term stability studies, further investigation needs to be conducted. Earlier studies has shown that storage at 4ºC causes slight precipitation and this may be due to the solubility effect of solutions at low temperatures.
In vitro toxicity studies using complexed ABZ -SBE-β-CD on ovarian cancer cell (OVCAR 3 and Human ovarian surface epithelial cells (HOSE)
The in vitro evaluation of cytotoxic potency of ABZ in SBE- β-CD showed that it has similar cytotoxicity as that of ABZ in ethanol (Fig. 4A), indicating that the complexed ABZ is just as active as that of uncomplexed ABZ molecules. Similar evaluation with human ovarian epithelial cells (HOSE) indicated minimal cytotoxic effect that suggests that adverse reactions that may be encountered with ABZ treatment may also be minimal (Figure 4B).
A shows comparative cytotoxicity of ABZ in SBE- β-CD with that of ABZ in ethanol whilst B shows a similar comparative cytotoxicity in normal HOSE cells. C and D show the variation of abdominal circumference and body weight during the treatment period (14 - 36 days). The weight and abdominal circumference was stable to 32 days. E shows the ascites volume measured at day 36 (at euthanasia) in the control versus treated groups for the ABZ-SBE-β-CD as opposed to ABZ suspension in HPMC. ABZ- SBE- β-CD treated groups shows substantial ascities volume control (p = 0.0135). Similar comparison of weight of the tumours recovered at day 36. F shows that neither ABZ- SBE- β-CD nor ABZ in HPMC had any significant effect. HOSE= Human ovarian surface epithelial; SBE-β-CD = sulfobutylether-β- cyclodextrin; HP-β-CD = Hydroxypropyl -β-cyclodextrin; HPMC= Hydroxypropylmethylcellulose.
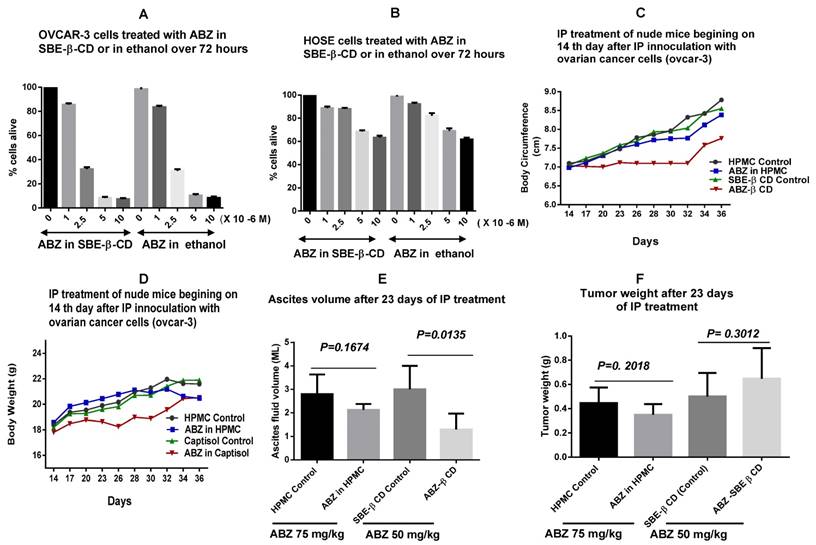
In vivo cytotoxicity evaluation using complexed ABZ -SBE-β-CD on ovarian cancer cell line (OVCAR-3) IP in nude mice
Figure 4C shows that the abdominal circumference remained almost constant to 32 days with treatment commencing on day 14, using ABZ- SBE -β-CD as compared to control with just SBE -β-CD. There was not much of a difference between control and drug treated groups with HPMC. In the case of body weight, the SBE-β-CD treated group had almost a constant weight to 30 days after which it began to increase, there was not much of a difference between the other groups, and they showed a gradual increase over the time period monitored (Figure 4D). Figure 4E shows that the ascites volume in the ABZ-SBE- β-CD treated was considerably reduced (>50%) compared to the control (p= 0.0135) whilst in the case of the HPMC groups, there was a smaller difference between the control and the treated groups but not statistically significant (p= 0.1674). Hence, only ABZ-SBE- β-CD was effective in controlling ascites volume in this study.
When the tumour weight was measured at the end of the treatment period of 36 days, ABZ- SBE -β-CD group was the highest compared to all the groups and it was 30% higher compared SBE -β-CD control (Fig. 4F), however, not statistically significant (p=0.3012). On the other hand in the HPMC groups, the ABZ-HPMC group tumour weight was reduced by 21 % compared to its control, but not statistically significant (p= 0.2018). Hence, neither ABZ-SBE -β-CD nor ABZ in HPMC had any effect on tumour weight.
Discussion
Several studies have been conducted to enhance the aqueous solubility of ABZ using cyclodextrins [26-28] with recent reported solubilities ranging from 0.79 - 1.52 mg/ml using different β - CDs [12]. For intravenous and intraperitoneal use, a higher solubility of ABZ is required. Although in our early study with ionised ABZ resulted in 2.14 mg/ml solubility in a 25 % of HP-β-CD solution, this study did not examine parameters that would maximize complexation [29]. Hence, in the current investigation we set out determine parameters that would ultimately affect complexation and hence use an optimised system to derive a super concentrated solution of aqueous ABZ. To do this we chose to investigate the aqueous solubility of ABZ, by complexation with SBE-β-CD since it has been identified to have minimal renal toxicity [20, 21]. As a comparator molecule we chose HP-β-CD since it also has a good safety profile in biological systems [30].
Using solubilised ABZ prepared in 5% CDs, a standard curve was generated that measured accurately a range of ABZ from 0 - 40 µg/ml. In the initial investigation using excess powdered suspension of ABZ with different percentages of both SBE-β-CD and HP- β-CD at three different pH values, indicated that at the lower pH of 2.3, complexation was the highest for both the CDs (2292µg/ml and 1729 µg/ml respectively). On a comparative basis, the solubility of ABZ in SBE-β-CD increased by a factor of 1.53 at pH 2.3 compared to that at pH 7.0. Similarly for the HP-β-CD, it was 1.39. A similar comparison at pH 4.0, showed an enhancement of solubility factor of 1.21 for SBE-β-CD and 1.23 for HP-β-CD. This observation may indicate that ionisation of ABZ plays a crucial role in enabling a more efficient complexation. Hence, this observation prompted us to use solubilised ABZ in acetic acid in subsequent investigation. However, excess addition of acid solubilised ABZ (15 mg/ml) to the different % (5 -40%) of CDs showed that complexation was extremely poor, indeed lower than what was observed using suspension of solid ABZ at pH 2.3. For SBE-β-CD, it was 1.260 mg/ml vs. 2.292 mg/ml (drop of 45%) and for HP- β-CD it was 1.2 mg/ml vs. 1.729 mg/ml (a drop of 30%). Subsequent investigations with addition of different quantities of solubilised ABZ to the various percentages of CDs showed that for a 40% solution of SBE-β-CD, maximum addition of 8.0 mg/ml complexed readily whilst an increase thereof showed a reduction in complexation. A 30% solution was compatible with 7.0 mg/ml of ABZ, whilst a 20% with 5.0 mg/ml. In the case of HP-β-CD a 40% solution was compatible with 7.0 mg/ml, 30% with 6.0mg/ml and 20% with 4.0 mg/ml, excess additions only inhibited the complexation process. Hence, this observation may indicate that overcrowding (stearic hindrance) at the entrance of the hydrophobic cavity of the CDs may in fact hinder the process of complexation. Stearic hindrance may not play a part in the powdered ABZ suspension formulation, since only minute quantities of ABZ was solubilised at low pH, i.e.310 µg/ml at pH 2.3 and therefore over loading of ABZ may not affect the system. With this predetermined quantities of solubilised ABZ to be added for maximum complexation, we proceeded to develop an ideal (best) phase solubility curve and then investigated the effect of other parameters such as the time duration for complexation and effect of temperature on complexation efficency.
Phase solubility curves for both the CDs indicated that the K value was K= 48751 M-1 for SBE-β-CD and 20571 M-1 for HP- β-CD indicating that 1 in 29 and 1 in 69 of the respective solubilised CDs interacted and complexed with ABZ. Compared to the K of 32160 M-1 for SBE-β-CD reported in a more recently [12], our K value was higher. This may be expected since they conducted their experiment using powdered excess ABZ in CD media with neutral pH. Their ABZ solubilities observed for a 40 % solution of HP- β-CD was 0.79 mg/ml as compared to 6.4 mg/ml in our investigation that is almost 8 fold higher. Their reported maximal solubility in a 40 % SBE-β-CD was 1.17 mg/ml as compared to our system where 8.0 mg/ml of ABZ was solubilised which is again about 6.8 fold higher. This high complexation may be attributed to the enhanced ability of interaction of two solubilised molecules as well as the low pH which encouraged the ionisation of ABZ molecule [23].
Investigation on the time duration for maximum complexation process to occur indicated that it took place at three days at 25ºC using predetermined quantity of solubilised ABZ with each percentage of CDs. Complexation (Y value) increased to 4.46 times at day 3, compared to day 1 and then declined to 3.0 at day 5 for SBE-β-CD whilst for HP-β-CD, the complexation process accelerated at day 3 to 6.29 times (Y value) that of day 1 and then after declined to 3.72 at day 5. Hence, the decline in complexation after three days may suggest that the weak intermolecular forces tethering ABZ in the hydrophobic cavity may be disrupted with agitation.
Complexation process may be temperature dependant and hence we investigated the process over 25ºC and at 37ºC. The results indicate that complexation was better at a lower temperature of 25ºC, for both the CDs. This is contrary to that reported by a researcher who has proposed that complexation is better at higher temperatures [31] although the molecule investigated and the complexation system was quite different. A rise in temperature may provide heat energy for the disruption of the weak hydrogen bonds that bind the ABZ in the hydrophobic cavity, hence the current observation [32].Temperatures below 25ºC may prove to be an advantage although the time of agitation (complexation time) may have to be increased. In our system a temperature of 25ºC seems to work rather well since the process can be completed within 3 days.
Finally, the stability of the complexed CD-ABZ over two weeks at room temperature (25ºC) was investigated. Since the formulation was kept under sterile condition and light excluded, the complex was stable over this period. ABZ is sensitive to light [33.34]. Complexation with CDs is known to stabilise the host molecules since it is encapsulated within the CDs [35]. Earlier work with storage at 4ºC only caused slight precipitation. We did not investigate lipolysation with reconstitution in saline or phosphate buffer before use, but this method could prove to be useful for storage and transport.
Cytotoxicity of the formulation was tested using both in vitro and in vivo models. We only chose to use the ABZ- SBE-β-CD formulation, since this was the formulation that we wished to use in clinical studies. The in vitro study showed that it was just as efficient as ABZ dissolved in ethanol (< 1%) indicating that complexed ABZ was just as active as the free form dissolved in ethanol. Further cytotoxic evaluation with HOSE (Human ovarian surface epithelial) cells, indicated that they were minimally affected suggesting that adverse reactions may also be minimal. More promising results emerged with nude mice studies showing that the body weight and body circumference was controlled by the ABZ-SBE-β-CD formulation, it was more effective (50% difference between treated and control, p = 0.0135) compared to a formulation of ABZ with hydroxypropylmethylcellulose (HPMC) that we had developed in earlier studies (unpublished data) for a period of 18 day. In fact in the HPMC groups (treated vs. control), the difference in ascites volume was small and non- significant (p = 0.1674) Ovcar-3 tumours produce mucinous ascites intraperitoneally that normally causes abdominal distension with gain in body weight owing to fluid accumulation [36]. Hence, the difference between the two formulations may be attributed to the nature of delivery of ABZ, in the SBE-β-CD formulation it is fully solubilised, whilst in the HPMC, ABZ was suspended. Hence, the full solubilisation of ABZ may provide an enhanced transfer of the drug into the tumour compared to the suspended formulation. The ascites volume was also considerably reduced when using the SBE-β-CD as compared to HPMC further indicating the superiority of fully solubilised ABZ.
When the tumour weight was evaluated, there was no difference between the ABZ- SBE-β-CD treated group and the control (p= 0.3012). Similarly, for the HMPC groups, the difference between the treated and controls was non-significant (p= 0.2018). Animals were treated every day with 50 mg/kg in the ABZ-SBE-β-CD group and this meant that the animals were exposed to high levels of ABZ whilst in the HPMC -ABZ group it was 75 mg/kg in suspension. Suspension formulation of hydrophobic drugs has relatively poor drug availability to the cells owing to poor solubility [29]. On the other hand, ABZ has a certain toxicity profile [37] and treatment at high dosage may have induced the animals to develop an efficient way of reducing the toxicity. Overloading with drugs into biological system has been shown to result in the development of efficient excretory pathways [38, 39]. Further, in cancer therapy, there are several studies indicating that treatment over a longer period of time induces the tumour cells to develop resistant pathways to overcome the effect of the treatment [39]. In this study using ABZ-SBE-β-CD at 50mg/kg with daily treatment regime for 18 days may have induced the tumours to develop pathways to overcome the effect of high dosage of the drugs [40]. The abdominal circumference and the weight gain (ascities fluid accumulation) measurements indicates that after 32 days (32-14 = 18 days of treatment), the tumour began to accelerate its growth. Long term treatment has been shown to cause resistance to the several anti-tumour drugs [39, 41, 42]. In the present study, one the one hand, the ABZ-HPMC suspension formulation had poor drug availability to affect the tumours whilst in the ABZ-SBE-β-CD, ABZ was present at supra dose which may have induced resistance to the treatment. Hence, in future in vivo studies, treatment on alternate days will be investigated with a lower dosage.
Therefore, the current formulation may be used for IP or IV delivery with judicious use of treatment regime. The current method of complexation may be conveniently carried out in phosphate buffer solution, which we have accomplished (unpublished data), with the use of minute amount of acetic acid to solubilise the ABZ such that after neutralisation only trace amount of acetate and sodium ions were added to the buffer, allowing easy and safe use of the formulation. Further the formulation can be diluted to the required concentration without any precipitate being formed. Although the present study only involves increasing the aqueous solubility of ABZ by complexation with cyclodextrins, a similar approach may be used to increase the aqueous solubility of other ionisable hydrophobic drugs.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Horton J. Albendazole: a review of anthelmintic efficacy and safety in humans. Parasitology. 2000;121(Suppl):S113-132
2. Jagota SC. Albendazole, a broad-spectrum anthelmintic, in the treatment of intestinal nematode and cestode infection: a multicenter study in 480 patients. Clin Ther. 1986;8:226-231
3. Vercruysse J, Behnke JM, Albonico M, Ame SM, Angebault C, Bethony JM. et al. Assessment of the anthelmintic efficacy of albendazole in school children in seven countries where soil-transmitted helminths are endemic. PLoS Negl Trop Dis. 2011;5:e948
4. Pourgholami MH, Wangoo KT, Morris DL. Albendazole-cyclodextrin complex: enhanced cytotoxicity in ovarian cancer cells. Anticancer Res. 2008;28:2775-2779
5. Pourgholami MH, Woon L, Almajd R, Akhter J, Bowery P, Morris DL. In vitro and in vivo suppression of growth of hepatocellular carcinoma cells by albendazole. Cancer Lett. 2001;165:43-49
6. Pourgholami MH, Yan Cai Z, Lu Y, Wang L, Morris DL. Albendazole: a potent inhibitor of vascular endothelial growth factor and malignant ascites formation in OVCAR-3 tumor-bearing nude mice. Clin Cancer Res. 2006;12:1928-1935
7. Meena AK, Sharma K, Kandaswamy M, Rajagopal S, Mullangi R. Formulation development of an albendazole self-emulsifying drug delivery system (SEDDS) with enhanced systemic exposure. Acta Pharm. 2012;62:563-580
8. Mottu F, Laurent A, Rufenacht DA, Doelker E. Organic solvents for pharmaceutical parenterals and embolic liquids: a review of toxicity data. PDA J Pharm Sci Technol. 2000;54:456-469
9. Laurent A, Mottu F, Chapot R, Zhang JQ, Jordan O, Rufenacht DA. et al. Cardiovascular effects of selected water-miscible solvents for pharmaceutical injections and embolization materials: a comparative hemodynamic study using a sheep model. PDA J Pharm Sci Technol. 2007;61:64-74
10. Palomares-Alonso F, Gonzalez CR, Bernad-Bernad MJ, Montiel MD, Hernandez GP, Gonzalez-Hernandez I. et al. Two novel ternary albendazole-cyclodextrin-polymer systems: dissolution, bioavailability and efficacy against Taenia crassiceps cysts. Acta Trop. 2010;113:56-60
11. Joudieh S, Bon P, Martel B, Skiba M, Lahiani-Skiba M. Cyclodextrin polymers as efficient solubilizers of albendazole: complexation and physico-chemical characterization. J Nanosci Nanotechnol. 2009;9:132-140
12. Pradines B, Gallard JF, Iorga BI, Gueutin C, Loiseau PM, Ponchel G. et al. Investigation of the complexation of albendazole with cyclodextrins for the design of new antiparasitic formulations. Carbohydr Res. 2014;398:50-55
13. Vecsernyes M, Fenyvesi F, Bacskay I, Deli MA, Szente L, Fenyvesi E. Cyclodextrins, Blood-Brain Barrier, and Treatment of Neurological Diseases. Arch Med Res. 2014;45:711-729
14. Rudrangi SR, Bhomia R, Trivedi V, Vine GJ, Mitchell JC, Alexander BD. et al. SR. Influence of the preparation method on the physicochemical properties of indomethacin and methyl-beta-cyclodextrin complexes. Int J Pharm. 2015;479:381-390
15. Chi L, Liu R, Guo T, Wang M, Liao Z, Wu L. et al. Dramatic improvement of the solubility of pseudolaric acid B by cyclodextrin complexation: preparation, characterization and validation. Int J Pharm. 2015;479:349-356
16. Ai F, Ma Y, Wang J, Li Y. Preparation, Physicochemical Characterization and I n - vitro Dissolution Studies of Diosmin-cyclodextrin Inclusion Complexes. Iran J Pharm Res. 2014;13:1115-1123
17. Rekharsky MV, Inoue Y. Complexation Thermodynamics of Cyclodextrins. Chem Rev. 1998;98:1875-1918
18. Fujiwara H AH, Murata S, Sasaki Y. Solvation effect on reactions in solution. II. Entropy changes in the inclusion complex formation of alpha-cyclodextrin with alcohols as studied by the titration calorimetry. Bull Chem Soc Jpn. 1987;60:3891-3894
19. Suarez-Cerda J, Nunez GA, Espinoza-Gomez H, Flores-Lopez LZ. A comparative study of the effect of alpha-, beta-, and gamma-cyclodextrins as stabilizing agents in the synthesis of silver nanoparticles using a green chemistry method. Mater Sci Eng C Mater Biol Appl. 2014;43:21-26
20. Luke DR, Tomaszewski K, Damle B, Schlamm HT. Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci. 2010;99:3291-3301
21. Hafner V, Czock D, Burhenne J, Riedel KD, Bommer J, Mikus G. et al. Pharmacokinetics of sulfobutylether-beta-cyclodextrin and voriconazole in patients with end-stage renal failure during treatment with two hemodialysis systems and hemodiafiltration. Antimicrob Agents Chemother. 2010;54:2596-2602
22. Ganapaty NVVJMRaS. Development and Vlidation of Stability indicating RP-HPLC method for Simultaneous Estimation of Pyrantel pamoate and Albendazole in Bulk and tablet dosage form. Int J of Phar Science. 2015;5:1220-1225
23. Jung H, Medina L, Garcia L, Fuentes I, Moreno-Esparza R. Absorption studies of albendazole and some physicochemical properties of the drug and its metabolite albendazole sulphoxide. J Pharm Pharmacol. 1998;50:43-48
24. Higuchi T CK. Phase solubility techniques. Adv Anal Chem Inst. 1965;4:117-212
25. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112-1116
26. Garcia A, Leonardi D, Salazar MO, Lamas MC. Modified beta-cyclodextrin inclusion complex to improve the physicochemical properties of albendazole. complete in vitro evaluation and characterization. PLoS One. 2014;9:e88234
27. Castillo JA, Palomo-Canales J, Garcia JJ, Lastres JL, Bolas F, Torrado JJ. Preparation and characterization of albendazole beta-cyclodextrin complexes. Drug Dev Ind Pharm. 1999;25:1241-1248
28. Diaz D, Bernad Bernad MJ, Gracia Mora J, Escobar Llanos CM. Complexation and solubility behavior of albendazole with some cyclodextrins. Pharm Dev Technol. 1998;3:395-403
29. Ehteda A, Galettis P, Chu SWL, Pillai K, Morris DL. Complexation of albendazole with hydroxypropyl-β-cyclodextrin significantly improves its pharmacokinetic profile, cell cytotoxicity and antitumor efficacy in nude mice. Anticancerr Research. 2012;32:3659-3666
30. Gould S, Scott RC. 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): a toxicology review. Food Chem Toxicol. 2005;43:1451-1459
31. Chiang PC, Shi Y, Cui Y. Temperature Dependence of the Complexation Mechanism of Celecoxib and Hydroxyl-beta-cyclodextrin in Aqueous Solution. Pharmaceutics. 2014;6:467-480
32. Tang XC, Pikal MJ, Taylor LS. The effect of temperature on hydrogen bonding in crystalline and amorphous phases in dihydropyrine calcium channel blockers. Pharm Res. 2002;19:484-490
33. Al-Kurdi Z, Al-Jallad T, Badwan A, Jaber AM. High performance liquid chromatography method for determination of methyl-5-benzoyl-2-benzimidazole carbamate (mebendazole) and its main degradation product in pharmaceutical dosage forms. Talanta. 1999;50:1089-1097
34. Del Brutto OH, Sotelo J, Roman GC. Therapy for neurocysticercosis: a reappraisal. Clin Infect Dis. 1993;17:730-735
35. Otzen DE, Knudsen BR, Aachmann F, Larsen KL, Wimmer R. Structural basis for cyclodextrins' suppression of human growth hormone aggregation. Protein Sci. 2002;11:1779-1787
36. Peterson NC. Behavioral, clinical, and physiologic analysis of mice used for ascites monoclonal antibody production. Comp Med. 2000;50:516-526
37. Dayan AD. Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta Trop. 2003;86:141-159
38. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-726
39. Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214-226
40. De Souza R, Zahedi P, Badame RM, Allen C, Piquette-Miller M. Chemotherapy dosing schedule influences drug resistance development in ovarian cancer. Mol Cancer Ther. 2011;10:1289-1299
41. Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat Rev. 2012;38:890-903
42. Wang H, Vo T, Hajar A, Li S, Chen X, Parissenti AM, Brindley DN, Wang Z. Multiple mechanisms underlying acquired resistance to taxanes in selected docetaxel-resistant MCF-7 breast cancer cells. BMC Cancer. 2014;14:37
Author contact
![]() Corresponding author: Krishna Pillai, Department of Surgery, University of New South Wales, St. George Hospital, Kogarah, NSW, Australia Email: z3179288edu.au
Corresponding author: Krishna Pillai, Department of Surgery, University of New South Wales, St. George Hospital, Kogarah, NSW, Australia Email: z3179288edu.au