Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(3):323-331. doi:10.7150/jca.17482 This issue Cite
Research Paper
Immunohistochemical and molecular characterizations in urothelial carcinoma of bladder in patients less than 45 years
Veronika Weyerer1, Roland Schneckenpointner2, Thomas Filbeck3, Maximilian Burger4, Ferdinand Hofstaedter5, Peter J. Wild6, Samson W. Fine7, Peter A. Humphrey8, Louis P. Dehner9, Mahul B. Amin10, Josef Rüschoff11, Carsten Boltze12, Andrea Tannapfel13, Ellen Zwarthoff14, Antonio Lopez-Beltran15, Rodolfo Montironi16, Cord Langner17, Robert Stoehr1, Arndt Hartmann1, Johannes Giedl1 ![]()
1. Institute of Pathology, University Hospital Erlangen, Friedrich-Alexander University Erlangen-Nürnberg, Erlangen, Germany;
2. Department of Internal Medicine II, University Medical Center Regensburg, Regensburg, Germany;
3. Department of Urology, University Hospital Erlangen, Friedrich-Alexander University Erlangen-Nürnberg, Erlangen, Germany;
4. St Josef Medical Center, Department of Urology, University of Regensburg, Regensburg, Germany;
5. Tumor Center Regensburg, University of Regensburg, Regensburg, Germany.
6. Department of Pathology and Molecular Pathology, University Hospital Zurich, Zurich 8091, Switzerland.
7. Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, USA;
8. Department of Pathology, Yale University School of Medicine, New Haven, CT, USA;
9. Division of Anatomic and Molecular Pathology, Department of Pathology and Immunology, St. Louis Children's Hospital, Washington University Medical Center, St. Louis, MO, USA;
10. Department of Pathology & Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
11. Pathology Nordhessen, Kassel, Germany;
12. Institute of Pathology, SRH-Klinikum, Gera, Germany;
13. Institute for Pathology, Ruhr-University, Bochum, Germany.
14. Department of Pathology, Erasmus Medical Centre, Rotterdam, the Netherlands.
15. Department of Surgery, Cordoba University Medical School, Cordoba, Spain.
16. Section of Pathological Anatomy, Polytechnic University of the Marche Region, School of Medicine, United Hospitals, Ancona, Italy.
17. Institute of Pathology, Medical University of Graz, Graz, Austria.
Received 2016-9-6; Accepted 2016-11-10; Published 2017-2-5
Abstract
Bladder tumours in early-onset patients are rare and seem to exhibit unique clinicopathological features. Only few studies have investigated somatic alterations in this specific age of onset group and evidence is accumulating of a distinct molecular behaviour of early-onset bladder tumours. We collected the largest cohort of early-onset tumours of patients 45 years old or younger and aimed to test genomic alterations typically found in bladder cancer. Tumours of 118 early-onset patients were compared with a consecutive group of 113 cases. Immunohistochemistry of TP53, CK20 and Ki-67 was carried out. Molecular analysis was conducted to test for loss of heterozygosity of chromosome 9 and 17, as well as TP53 and FGFR3 mutations. Fisher´s exact and chi-squared test were appropriately used. No differences in grade/stage characteristics were observed. Overexpressed TP53 was differentially distributed between the two groups. TP53 nuclear accumulation was significantly more frequent in early-onset papillomas, PUNLMPs and pTa low-grade tumours compared to the consecutive cohort (p=0.005). Moreover, chromosome 9 deletions (29.5% vs. 44.6%) and FGFR3 mutations (34.5% vs. 63.7%) were less often detected in early-onset patients (p=0.05 and p<0.0001). By comparing the largest cohort of early-onset bladder cancer patients with an unselected group, we demonstrated that the typical molecular features are not independent of age at diagnosis. Our study supports the hypothesis of a distinct biological behaviour in early-onset tumours.
Keywords: Early-onset, Bladder cancer, FGFR3, TP53 positivity, Mutation analysis.
Introduction
Urothelial bladder cancer (UBC) usually occurs late in life, with a mean age at diagnosis of approximately 67 years [1]. UBC is a heterogeneous disease, both clinically and molecularly. Low-grade non-muscle invasive tumours are associated with activating mutations of FGFR3 and PIK3CA and a lower risk of progression, whereas TP53 and RB pathway alterations play a key role in the development of high-grade non- and muscle-invasive UBC [2-5]. Additionally, total or partial loss of chromosome 9 is found in almost half of the tumours of all stages and grades [6, 7].
Worldwide estimates of cancer incidences for 2012 indicate that the proportion of UBC in patients younger than 45 years is at about 3.4% [8]. Because of these low incidence rates and since no uniform threshold is used for young patients, the natural history is still under debate. However evidence is accumulating, that especially tumours of patients of the first two decades of life are more frequently low-grade/-stage with a favourable prognosis [9]. Besides distinct clinicopathological features, very few studies of paediatric and adolescent patients reported, that molecular alterations in paediatric and adolescent patients were much rarer compared to typical UBC samples [10-14]. These unexpected results gave evidence, that tumours of this age of onset group may represent a distinct biological entity. However, only small sample-sized studies were reported and thus these findings have still to be validated.
The aim of our study was to investigate specific molecular alterations of UBC in the largest cohort to date of early-onset patients, defined herein for the study as patients aged 45 or younger. Immunohistochemical analysis was performed to detect CK20, Ki-67 and TP53 expression levels. In addition, we investigated the frequencies of loss of heterozygosity (LOH) of chromosome 9 and 17. Moreover, molecular analysis revealed mutations in the FGFR3 and TP53 gene. All obtained results were compared with a consecutive, unselected cohort of patients.
Materials and methods
Tumour samples
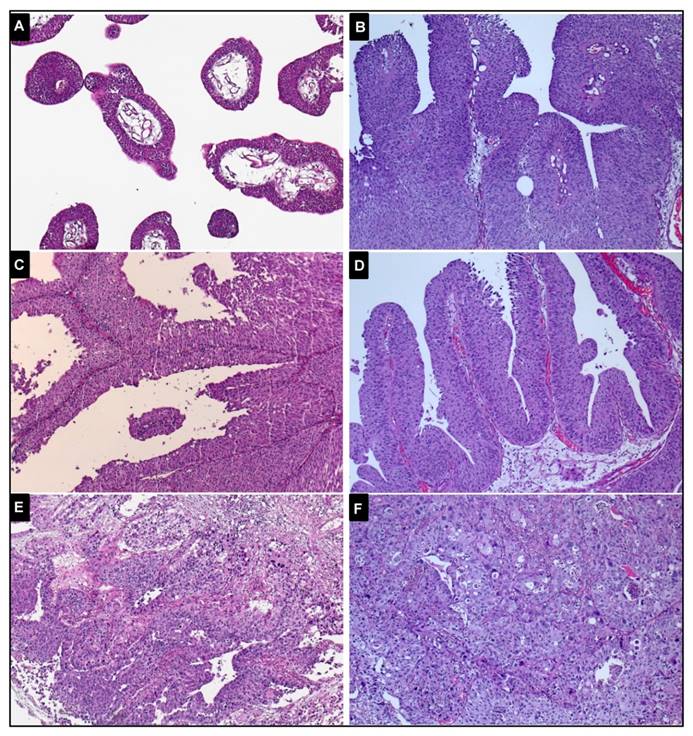
118 patients aged 45 or younger were identified from 1991 until 2001 from the files of the contributing Institutes of Pathology. 113 consecutive cases were included from the period between 1996 and 2002. For each case, formalin-fixed paraffin-embedded primary UBC and normal tissues were available and reclassified according to the 2016 WHO classification by two experienced uropathologists (A.H., J.G.) [15]. Figure 1 demonstrates representative images of the histopathological specimens of early-onset patients. Eight urothelial papillomas and ten samples of papillary urothelial neoplasia of low malignant potential (PUNLMP) were included. Due to similar molecular features reported, they were grouped together with pTa low-grade tumours [16]. Table 1 shows the characteristics of the two cohorts. Institutional Review Board approval (University Hospital Erlangen) was obtained for molecular analysis on archival material.
Representative images of histopathological specimens of the early-onset tumours (magnification: x100) A, papilloma, no analysed mutation or deletion was identified. B, pTa low-grade tumour, a FGFR3 mutation (p.S249C) was detected. C, pTa low-grade tumour, none of the analysed alterations was observed. D, pTa high-grade tumour, a mutation of the FGFR3 mutation (p.S249C) and LOH of chromosome 9 were observed. E, pT1 high-grade tumour, not mutated. F, pT2 high-grade tumour, a TP53 mutation in exon 5 was identified.
Clinicopathological characteristics of the two different cohorts.
| Characteristics | Early-onset group | Consecutive group | Group comparison | ||
|---|---|---|---|---|---|
| (n=118) | (n=113) | p-value | |||
| Age distribution, yr | |||||
| Minimum/maximum | 17/45 | 48/87 | |||
| Mean ± SD | 37.6 ± 6.5 | 69.8 ± 8.4 | |||
| Median age | 39 | 70 | |||
| Age categories, yr | |||||
| ≤ 20 | 3 | - | |||
| 21-25 | 4 | - | |||
| 26-30 | 8 | - | |||
| 31-35 | 23 | - | |||
| 36-40 | 32 | - | |||
| 41-45 | 48 | - | |||
| >45 | - | 113 | |||
| Gender, No. | |||||
| Male | 92 | (79.3) | 89 | (78.8) | p=1.0 |
| Female | 24 | (20.7) | 24 | (21.2) | |
| Not available | 2 | 0 | |||
| Stage/grade distribution I, No. | |||||
| Urothelial papilloma | 7 | 0 | |||
| Inverted urothelial papilloma | 0 | 1 | |||
| PUNLMP | 0 | 10 | |||
| pTa low-grade | 56 | 62 | |||
| pTa high-grade | 20 | 9 | |||
| pT1 low-grade | 4 | 1 | |||
| pT1 high-grade | 12 | 23 | |||
| pT2 high-grade | 8 | 7 | |||
| pT3 high-grade | 8 | 0 | |||
| pT4 high-grade | 1 | 0 | |||
| Not available | 2 | 0 | |||
| Stage/grade distribution II, No. | |||||
| Inverted/ Urothelial papilloma & PUNLMP & pTa low-grade | 63 | (54.3) | 73 | (64.6) | p=0.08 |
| pTa high-grade & pT1 | 36 | (31.0) | 33 | (29.2) | |
| ≥ pT2 | 17 | (14.7) | 7 | (6.2) | |
| Not available | 2 | 0 | |||
SD, standard deviation; PUNLMP, papillary urothelial neoplasm of low malignant potential.
Immunohistochemistry (IHC)
IHC of TP53, CK20 and Ki-67 were performed using whole tumour sections and an avidin-biotin peroxidase method with a 3,3'-diaminobenzidine chromatogen. According to the manufacturer's instructions, IHC was carried out in a NEXES immunostainer (Ventana, Tucson, AZ, USA). Anti-TP53 (mouse monoclonal IgG, clone Bp53-12 (sc-263) [Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA], dilution 1:1000), anti-CK20 (mouse monoclonal IgG2a, clone IT-Ks20.8 (61026) [Progen Biotechnik GmbH, Heidelberg, Germany]; dilution 1:10) and anti-Ki-67 (mouse monoclonal IgG1, clone MIB1 (M7240) [Dako, Glostrup, Denmark], dilution 1:50) were used as primary antibodies. The slides were evaluated by one surgical pathologist (A.H.). Nuclear staining reaction for TP53 was scored from 0% to 100% in 10% increments. The intensity of the TP53 staining was recorded as negative, weak or strong. Considering a cut-off level at 10% of positive cells, three different categories were defined: ≤10% negative, >10% weak or >10% strong. Anti-Ki-67-immunostaining was scored from 0% to 5% in 1% increments and from 5% to 100% in 5% increments and cut-off level for increased proliferation was defined at 10%. Staining of CK20 as a urothelial associated marker was defined as either normal (cytoplasmic staining pattern of the superficial cells only) or aberrant (negative or more than 10% of the urothelial cells stained) according to Harnden et al. [17].
Microdissection and DNA isolation
Tumour and normal DNA was isolated as described previously [18]. An experienced pathologist (A.H.) marked areas with highest tumour cell density on a representative H&E-stained section. 5-μm tissue sections were deparaffinised and tumour and non-malignant tissue were separately microdissected in sterile tubes by using sterile needles. Due to separation, tumour cells with a purity of at least 80% were obtained. DNA isolation was performed using the High Pure PCR Template Preparation Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions.
Detection of LOH
For detection of LOH two microsatellite markers at chromosomal region 9p21 (D9S304, D9S1751), two markers at 9q (D9S303, D9S747) and one marker at 17p13.1 (p53Alu) were used as described previously [19]. The Polymerase chain reaction (PCR) amplification was performed in a MJ Research Thermocycler (PTC100, MJ Research, Watertown, MA, USA). By using 6.7% polyacrylamide/50% urea gel electrophoresis (1h, 1500V, 55°C) PCR products were analysed in a SequiGen sequencing gel chamber (BioRad, Hercules, CA, USA) followed by silver nitrate staining [20]. In informative cases LOH was defined as a decrease in signal intensity of the tumour sample allele to at least 50% relative to the matched normal DNA allele. All cases of LOH were confirmed at least once. Silver nitrate gels were evaluated independently by three different investigators (J.G., R.Sch., R.S.).
FGFR3 and TP53 mutation analysis
SNaPshot method was used to analyse FGFR3 mutations, as described previously [21]. Three regions of the FGFR3 gene comprising the mutations were amplified simultaneously in a multiplex PCR reaction. Eight SNaPshot primers were used to detect nine mutations. An automatic sequencer (ABI Prism 310) analysed the extended primers. Direct sequencing of exons 5-9 of the TP53 gene was assessed as described previously [22]. Identified mutations were verified in a second sequencing run to rule out polymerase errors.
Statistical analysis
Two-sided Fisher´s exact and chi-squared test were appropriately used to assess differences in the distributions of tested markers between both groups. P-values less than 0.05 were interpreted as statistically significant. All analyses were performed using R version 3.2.5 (www.r-project.org).
Results
Immunohistochemical analysis of CK20, Ki-67 and TP53
Table 2 summarizes the results of the IHC of CK20, Ki-67 and TP53. Neither the staining pattern of Ki-67, nor the expression of the differentiation marker CK20 showed a significant difference between the two groups. When we differentiated the aberrant staining of CK20 in negative or >10% of stained cells, we observed a significantly higher number of negative staining among the early-onset compared to the consecutive cases (p=0.03).
Expression of CK20, Ki-67 and TP53 in the two different groups.
| Early-onset group | Consecutive group | Group comparison | |||
|---|---|---|---|---|---|
| IHC marker | n | (%) | n | (%) | p-value |
| Ki-67 IHC | |||||
| ≤10% | 63 | (58.9) | 55 | (49.1) | p=0.19 |
| >10% | 44 | (41.1) | 57 | (50.9) | |
| Not available | 11 | 1 | |||
| CK20 IHC | |||||
| Normal | 37 | (37.4) | 40 | (35.4) | p=0.88 |
| Aberrant | 62 | (62.6) | 73 | (64.6) | |
| Negative | 13 | (13.1) | 5 | (4.4) | |
| >10% of stained cells | 49 | (49.5) | 68 | (60.2) | |
| Not available | 19 | 0 | |||
| TP53 IHC I | |||||
| ≤10% negative & >10% weak | 82 | (73.9) | 90 | (79.6) | p=0.39 |
| >10% strong | 29 | (26.1) | 23 | (20.4) | |
| Not available | 7 | 0 | |||
| TP53 IHC according to grade/stage | |||||
| Inverted/Urothelial papilloma & PUNLMP & pTa low-grade | |||||
| ≤10% negative & >10% weak | 48 | (77.4) | 69 | (94.5) | p=0.005 |
| >10% strong | 14 | (22.6) | 4 | (5.5) | |
| Not available | 1 | 0 | |||
| pTa high-grade & pT1 | |||||
| ≤10% negative & >10% weak | 27 | (79.4) | 20 | (60.6) | p=0.16 |
| >10% strong | 7 | (20.6) | 13 | (39.4) | |
| Not available | 2 | 0 | |||
| ≥ pT2 | |||||
| ≤10% negative & >10% weak | 7 | (50.0) | 1 | (14.3) | p=0.17 |
| >10% strong | 7 | (50.0) | 6 | (85.7) | |
| Not available | 3 | 0 | |||
| TP53 IHC III | |||||
| ≤10% negative | 45 | (40.6) | 81 | (71.7) | p<0.0001 |
| >10% weak | 37 | (33.3) | 9 | (8.0) | |
| >10% strong | 29 | (26.1) | 23 | (20.3) | |
| Not available | 7 | 0 | |||
IHC, immunohistochemistry; PUNLMP, papillary urothelial neoplasm of low malignant potential.
IHC of TP53 was performed in 111 early-onset and all consecutive patients. The intensity was recorded as negative in 112, weak in 52 and strong in 60 cases. Similar percentages of positive cells were obtained in the different cohorts (data not shown). A >10% strong staining of TP53 was observed in 29 of the early-onset cases and was not significantly different to the 23 stained consecutive samples (p=0.39). When we distinguished the tumours according to grade/stage, we detected 14 (22.6%) out of 62 early-onset papillomas, PUNLMPs and pTa low-grade tumours with >10% strong staining. In contrast, only 4 (5.5%) out of 73 consecutive samples of the same grade/stage characteristics were stained (p=0.005). Remarkably, when we also considered a >10% weak staining, 66 (59.4%) early-onset cases showed a >10% weak or strong nuclear accumulation of TP53, whereas only 32 (28.3%) consecutive tumours presenting both intensity categories were included (p<0.0001).
LOH of chromosome 17 and mutations of the TP53 gene
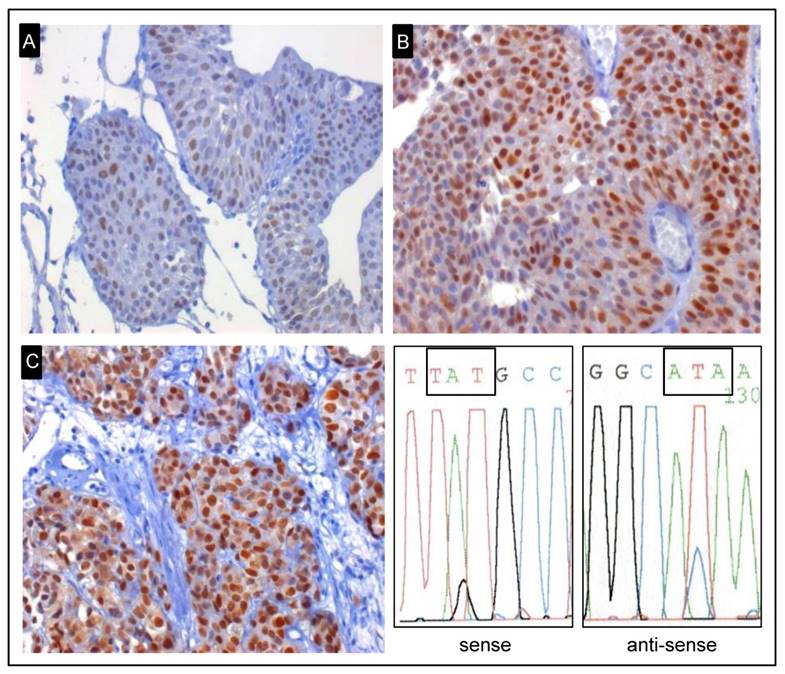
The analysis of LOH of chromosome 17 and the TP53 gene did not reveal any significant difference between the early-onset and unselected group (Table 3). All five TP53 mutations of the early-onset cases were transitions (G:C→A:T; A:T→G:C) and all but one (silent mutation) were miss-sense mutations in the exons 8, 6 and 5. Two mutation carriers were >10% strongly stained for TP53 (Figure 2). Furthermore, ten mutations were discovered in seven (6.2%) consecutive patients showing a >10% strong IHC. Nine transitions (G:C→A:T; C:T→T:A) and one transversion (G:A→C:T) were identified. Seven mutations in exon 8 led to a change in the corresponding amino-acid. One non-sense and two miss-sense mutations were detected in exons 6, 7 and 9.
Molecular changes of the studied cohorts.
| Early-onset group | Consecutive group | Group comparison | |||
|---|---|---|---|---|---|
| n | (%) | n | (%) | p-value | |
| LOH of chromosome 17 | |||||
| No | 63 | (92.6) | 62 | (91.2) | p=1.0 |
| Yes | 5 | (7.4) | 6 | (8.8) | |
| Not informative | 28 | 42 | |||
| Not available | 22 | 3 | |||
| TP53 gene | |||||
| Wild-type | 100 | (95.2) | 106 | (93.8) | p=0.87 |
| Mutated | 5 | (4.8) | 7 | (6.2) | |
| Not available | 13 | 0 | |||
| LOH of chromosome 9 | |||||
| No | 67 | (70.5) | 46 | (55.4) | p=0.053 |
| Yes | 28 | (29.5) | 37 | (44.6) | |
| 9p | 6 | (6.3) | 4 | (4.8) | |
| 9q | 13 | (13.7) | 24 | (28.9) | |
| 9p+q | 9 | (9.5) | 9 | (10.9) | |
| Not informative | 9 | 30 | |||
| Not available | 14 | 0 | |||
| FGFR3 gene | |||||
| Wild-type | 74 | (65.5) | 41 | (36.3) | p<0.0001 |
| Mutation | 39 | (34.5) | 72 | (63.7) | |
| p.S249C | 24 | (21.3) | 40 | (35.4) | |
| p.Y375C | 11 | (9.7) | 15 | (13.3) | |
| Other mutations | 4 | (3.5) | 17 | (15.0) | |
| Not available | 5 | 0 | |||
LOH; Loss of heterozygosity;
Representative examples of early-onset tumours for TP53 immunohistochemical staining. A, pTa low-grade tumour with a weak expression of TP53 and no TP53 mutation (x400). B, pTa low-grade tumour with strong expression of TP53 and not mutated (x400). C, Strong expression of TP53 and a mutation in exon 8 were identified in pT2 high-grade tumour (x400). Representative example of sequence analysis of the shown mutated early-onset tumour. The mutation p.C275Y (TGT → TAT) was detected by using Sanger sequencing.
LOH of chromosome 9 and FGFR3 mutation analysis
A borderline significantly lower percentage of deletions of chromosome 9 was found among the early-onset (29.5%) compared to the consecutive series (44.6%) (p=0.053, Table 3). Moreover, the distribution of chromosome 9 alterations (9p+q vs 9p vs 9q) was not different between the two cohorts (p=0.31). FGFR3 analysis revealed mutations in 39 (34.5%) of the 113 analysed early-onset tumours (Table 3). Among the unselected cohort, a significantly higher number of 72 (63.7%) patients were detected as mutated (p<0.0001). In both cohorts we identified p.S249C and p.Y375C mutations as the most frequent ones and no difference of the distribution was observed (p=0.24). The frequency of FGFR3 mutations in different age categories of early-onset patients are shown in Supplementary table 1. Due to low numbers of patients <30 years, we used the median age of 39 to stratify the early-onset cohort. A significantly lower frequency of 12 (23.5%) out of 51 patients aged <39 were mutated compared to 27 (43.5%) out of 62 early-onset patients ≥39 (p=0.04). In contrast, by using the median of 70 years for the consecutive cohort no difference of the distribution was observed (p=0.39, data not shown).
Summarized, any of the typical alterations of UBC were detected in 88 (77.9%) out of 113 analysed consecutive cases, whereas only half of the early-onset tumours were identified as mutated (p<0.0001, Supplementary figure 1). Mutations in FGFR3 and TP53 were mutually exclusive, only one consecutive patient presented with a simultaneous mutation.
Discussion
Only few studies have attempted to investigate the biological behaviour of early-onset tumours, with no clear definition regarding the maximum age of onset [9]. Some studies showed a better clinical outcome, especially for patients of the first two decades presenting low-grade and -stage tumours [9, 14, 23]. In contrast, other studies did not observe any difference at the clinicopathological level in the various groups of onset [24]. In other tumour entities the threshold between early-onset and consecutive cancer is mostly defined in the middle years. Early-onset colorectal cancer is mainly reported for patients younger than 50, with the average age of diagnosis of about 70 years [25, 26]. Compared to elderly colorectal cancer, some risk factors and molecular features differ in young patients [27]. As the peak of UBC presents in the seventh decade of life and due to low incidence rates below age 35, we included patients 45 years old or younger. In our study, the early-onset and the unselected group did not show any difference in the grade/stage distribution. The molecular features of young UBC are explored even less (summarized in Table 4).
Summary of the different molecular studies of early-onset patients.
| Author | Weyerer 2017 | Huang 2015 | Williamson 2014 | Wild 2007 | Migaldi 2004 | Linn 1998 |
|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Number of early-onset patients | 118 | 45 | 17 | 14 | 50 | 73 |
| Age, range (yr) | 17-45 | ≤30 | 6-26 | 4-19 | <45 | <30 |
| Abnormal expression of CK20a | 62/99 (62.6) | - | - | 2/13 (15.4) | - | - |
| High expression of Mib1/Ki-67 | 44/107 (41.1)b | - | - | 2/13 (15.4)b | 22/50 (44)c | - |
| TP53 positive staining | 29/111 (26.1)d | - | - | 1/13 (7.7)b | 28/50 (56)e | 49/73 (67)b |
| TP53 mutation | 5/105 (4.8)f | - | 0/17g | 2/12 (16.7)f | - | - |
| Deletion of chromosome 17 | 5/68 (7.4)h | - | 1/17 (5.9)i Gain | 0/4h | - | 15/72 (20.8)i |
| Deletion of chromosome 9 | 28/95 (29.5)j | - | 3/17 (17.7)k | 0/6j | - | 9/67 (13.4)k |
| FGFR3 mutation | 39/113 (34.5)l | 13/39 (33.3)m | 0/17n | 0/10l | - | - |
a In both studies 10% of stained cells were used as cut-off.
b Cut-off level was defined at 10%.
c Cut-off level was defined at 15%.
d Cut-off level was defined at 10% and the intensity was considered.
e Cut-off level was defined at 20%.
f Exons 5 to 9 were analysed.
g Exons 5, 7 and 8 were analysed.
h Marker at chromosome 17p.13.1 (p53Alu) was used.
i Chromosome enumeration probe/ centromeric probe for chromosome 17 was used.
j Two microsatellite markers at chromosomal region 9p21 (D9S304, D9S1751) and two markers at chromosome arm 9q (D9S303, D9S747) were used.
k Locus specific indicator probe for 9p21/ centromeric probe was used.
l Nine common mutations were tested by SNaPshot method.
m Immunohistochemistry was used for detection.
n Exons 7, 10 and 15 were analysed.
In elderly patients, mutations of TP53 occur commonly in muscle invasive tumours and display a key feature in the evolution of carcinoma in situ to an invasive disease [28]. In this study, we did not observe a difference of TP53 nuclear accumulation among the two cohorts using a >10% strong nuclear staining pattern as cut-off. Interestingly, when we compared these strong positive samples in correlation to grade/stage, we detected a distinct distribution among the two series. A significantly higher number of TP53 strongly positive papillomas, PUNLMPS and pTa low-grade tumours were found in the early-onset group. Our results confirmed a previous study by Linn et al. describing 73 patients aged 30 years old or younger, where 67% of early-onset tumours showed a high expression of TP53 by using immunohistochemistry [13]. In contrast to our study the intensity of the TP53 staining was not considered. Since almost all tumour samples were non-muscle invasive, the finding of a frequent TP53 overexpression was an unexpected result. When we also included a >10% weak together with a strong IHC staining, we detected as well a significantly higher frequency of positive cells among the early-onset cases.
By performing molecular analysis of TP53 we identified a similar percentage of mutations in both cohorts. As it has been shown that a strong staining correlates with TP53 mutations [29], we did not identify a >10% weak sample as mutated. In a study of atypical bronchial epithelium the lack of TP53 mutation accompanied with high expressed TP53 without considering the intensity was reported [30]. Due to the absence of mutations the authors suggested that the stained TP53 might be wild-type and might act as a protector against premalignant lesions. Considering both intensity categories and the very frequent staining in both early-onset studies, it appears that wild-type TP53 was identified and that young samples might also try to activate molecular protection mechanisms. Of course there might also be other mechanisms through which the TP53 staining may occur among young cases. For example, the accumulation might as well be due to an inactivation of degeneration pathways [31]. In summary, our results support the hypothesis that TP53 positivity and the distribution according to grade/stage subphenotypes may be dependent from age of disease onset.
Deletions of chromosome 9 are frequent numerical abnormalities in UBC, do not depend on grade/stage distribution and are recognized as one of the initial steps in UBC [6, 7]. LOH of chromosome 9 was only found in 29.5% of early-onset UBC, whereas tumours of the consecutive series showed numerical abnormalities with a frequency of 44.6%. This finding is in line with the two studies of Wild et al. and Williamson et al. in which the authors found infrequent alterations of chromosome 9 [10, 11]. These similar results suggest that early-onset UBC may be more genomically stable.
Activating mutations of FGFR3 are associated with low-grade non-muscle invasive UBC, with approximately 70% of these tumours carrying alterations [4]. In two recently published studies of urothelial neoplasms in patients <26 years, the total lack of FGFR3 mutations was reported [10, 11], although almost all tumours presented with a low-grade/stage. An interesting observation in our analysis is that within the early-onset series patients <39 were significantly and infrequently mutated. Therefore, it seems that the difference between early- and the regular-onset of the disease is most pronounced for very young patients. Huang et al. have used IHC of FGFR3 and have reported a frequency of 33.3% of positive stained cells among 45 patients ≤30. Due to clinical differences and since only 13.3% of patients aged 25 or younger were positively stained, they suggested that 25 may represent the cut-off level between early- and regular-onset UBC [12]. Compared to our study, a much lower sample size of early-onset cases was used and the FGFR3 status was not evaluated by molecular analysis. The authors did not compare the clinical behaviour and staining pattern with consecutive patients, thus they might miss part of the early-onset cases. Summarized, we do not think that 25 may indicate the cut-off for early-onset UBC. The low frequency of FGFR3 mutations in young cases adds to the evidence, that mutation profiles and the pathways of carcinogenesis of elderly patients may be less important and distinct molecular features may be involved in the development of early-onset UBC.
Taken together, in only half of the analysed early-onset tumours a typical molecular alteration of bladder cancer could be detected. In two recent studies of BC in young patients below the age of 20 a specific HRAS mutation associated with Costello syndrome was found as well as methylation of the PCDH7 gene in these and older BC patients, suggesting that this may represent a very early change [32-34]. In addition, it was recently shown that activating mutations in the core promoter of the TERT gene were detected more infrequently in early-onset compared to consecutive cases [35]. Thus, these results add to a different mechanism and more investigations in early-onset patients are needed to reveal the distinct molecular profile and to better characterize these specific patients.
In conclusion, we demonstrated in the largest cohort of early-onset cases studied, that the known molecular characteristics of UBC are differentially represented in this specific group. In comparison to patients younger or older than 45 years, we observed an infrequent number of FGFR3 mutations and chromosome 9 deletions and a distinct distribution of TP53 positivity according to grade/stage among young cases. Our data support the hypothesis that the typical profile of UBC is not independent of the age of disease onset and suggest that early-onset tumours represent a differentially biologically driven entity.
Supplementary Material
Supplementary figure 1 and table 1.
Acknowledgements
The present work was performed in fulfillment of the requirements for obtaining the degree “Dr. med.” (M.D.). Monika Kerscher, Andrea Lieschke, Nina Niessl, and Rudolf Jung are thanked for excellent technical assistance. We acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding programme Open Access Publishing.
VW was supported by a Mildred Scheel doctoral fellowship from the German Cancer Aid.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Malats N, Real FX. Epidemiology of bladder cancer. Hematology/oncology clinics of North America. 2015;29:177-89 vii
2. Babjuk M, Oosterlinck W, Sylvester R, Kaasinen E, Bohle A, Palou-Redorta J. et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder. European urology. 2008;54:303-14
3. Pasin E, Josephson DY, Mitra AP, Cote RJ, Stein JP. Superficial bladder cancer: an update on etiology, molecular development, classification, and natural history. Reviews in urology. 2008;10:31-43
4. Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nature reviews Cancer. 2005;5:713-25
5. Lindgren D, Frigyesi A, Gudjonsson S, Sjodahl G, Hallden C, Chebil G. et al. Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer research. 2010;70:3463-72
6. Sauter G, Moch H, Carroll P, Kerschmann R, Mihatsch MJ, Waldman FM. Chromosome-9 loss detected by fluorescence in situ hybridization in bladder cancer. International journal of cancer Journal international du cancer. 1995;64:99-103
7. Simon R, Burger H, Brinkschmidt C, Bocker W, Hertle L, Terpe HJ. Chromosomal aberrations associated with invasion in papillary superficial bladder cancer. The Journal of pathology. 1998;185:345-51
8. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C. et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer. 2013 Available from: http://globocan.iarc.fr, accessed on 20/06/2016
9. Williamson SR, Lopez-Beltran A, MacLennan GT, Montironi R, Cheng L. Unique clinicopathologic and molecular characteristics of urinary bladder tumors in children and young adults. Urologic oncology. 2013;31:414-26
10. Wild PJ, Giedl J, Stoehr R, Junker K, Boehm S, van Oers JM. et al. Genomic aberrations are rare in urothelial neoplasms of patients 19 years or younger. The Journal of pathology. 2007;211:18-25
11. Williamson SR, Wang M, Montironi R, Eble JN, Lopez-Beltran A, Zhang S. et al. Molecular characteristics of urothelial neoplasms in children and young adults: a subset of tumors from young patients harbors chromosomal abnormalities but not FGFR3 or TP53 gene mutations. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2014;27:1540-8
12. Huang H, Sun M, Li X, Jin J. Urothelial carcinoma of the bladder in patients aged 30 years or younger: clinicopathological analysis and expression of fibroblast growth factor receptor 3 (FGFR3) of 45 cases. Med Oncol. 2015;32:137
13. Linn JF, Sesterhenn I, Mostofi FK, Schoenberg M. The molecular characteristics of bladder cancer in young patients. The Journal of urology. 1998;159:1493-6
14. Migaldi M, Rossi G, Maiorana A, Sartori G, Ferrari P, De Gaetani C. et al. Superficial papillary urothelial carcinomas in young and elderly patients: a comparative study. BJU international. 2004;94:311-6
15. Humphrey PA, Moch H, Cubilla AL, Ulbright TM, Reuter VE. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part B: Prostate and Bladder Tumours. European urology. 2016;70:106-19
16. van Rhijn BW, Montironi R, Zwarthoff EC, Jobsis AC, van der Kwast TH. Frequent FGFR3 mutations in urothelial papilloma. The Journal of pathology. 2002;198:245-51
17. Harnden P, Eardley I, Joyce AD, Southgate J. Cytokeratin 20 as an objective marker of urothelial dysplasia. Br J Urol. 1996;78:870-5
18. Stoehr R, Zietz S, Burger M, Filbeck T, Denzinger S, Obermann EC. et al. Deletions of chromosomes 9 and 8p in histologically normal urothelium of patients with bladder cancer. European urology. 2005;47:58-63
19. Hartmann A, Rosner U, Schlake G, Dietmaier W, Zaak D, Hofstaedter F. et al. Clonality and genetic divergence in multifocal low-grade superficial urothelial carcinoma as determined by chromosome 9 and p53 deletion analysis. Lab Invest. 2000;80:709-18
20. Schlegel J, Bocker T, Zirngibl H, Hofstadter F, Ruschoff J. Detection of microsatellite instability in human colorectal carcinomas using a non-radioactive PCR-based screening technique. Virchows Archiv: an international journal of pathology. 1995;426:223-7
21. van Oers JM, Lurkin I, van Exsel AJ, Nijsen Y, van Rhijn BW, van der Aa MN. et al. A simple and fast method for the simultaneous detection of nine fibroblast growth factor receptor 3 mutations in bladder cancer and voided urine. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:7743-8
22. Hartmann A, Zanardo L, Bocker-Edmonston T, Blaszyk H, Dietmaier W, Stoehr R. et al. Frequent microsatellite instability in sporadic tumors of the upper urinary tract. Cancer research. 2002;62:6796-802
23. McCarthy JP, Gavrell GJ, LeBlanc GA. Transitional cell carcinoma of bladder in patients under thirty years of age. Urology. 1979;13:487-9
24. Yossepowitch O, Dalbagni G. Transitional cell carcinoma of the bladder in young adults: presentation, natural history and outcome. The Journal of urology. 2002;168:61-6
25. Siegel RL, Jemal A, Ward EM. Increase in incidence of colorectal cancer among young men and women in the United States. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009;18:1695-8
26. Liang J, Kalady MF, Church J. Young age of onset colorectal cancers. International journal of colorectal disease. 2015;30:1653-7
27. Silla IO, Rueda D, Rodriguez Y, Garcia JL, de la Cruz Vigo F, Perea J. Early-onset colorectal cancer: a separate subset of colorectal cancer. World journal of gastroenterology. 2014;20:17288-96
28. Cheng L, Zhang S, MacLennan GT, Williamson SR, Lopez-Beltran A, Montironi R. Bladder cancer: translating molecular genetic insights into clinical practice. Human pathology. 2011;42:455-81
29. Tashiro H, Isacson C, Levine R, Kurman RJ, Cho KR, Hedrick L. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. The American journal of pathology. 1997;150:177-85
30. Wakamatsu K, Nakanishi Y, Takayama K, Miyazaki H, Hayashi K, Hara N. Frequent expression of p53 protein without mutation in the atypical epithelium of human bronchus. American journal of respiratory cell and molecular biology. 1999;21:209-15
31. Ciechanover A, DiGiuseppe JA, Bercovich B, Orian A, Richter JD, Schwartz AL. et al. Degradation of nuclear oncoproteins by the ubiquitin system in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:139-43
32. Beukers W, Hercegovac A, Zwarthoff EC. HRAS mutations in bladder cancer at an early age and the possible association with the Costello Syndrome. European journal of human genetics: EJHG. 2014;22:837-9
33. Castillo-Martin M, Collazo Lorduy A, Gladoun N, Hyun G, Cordon-Cardo C. H-RAS mutation is a key molecular feature of pediatric urothelial bladder cancer. A detailed report of three cases. Journal of pediatric urology. 2016;12:91 e1-7
34. Beukers W, Hercegovac A, Vermeij M, Kandimalla R, Blok AC, van der Aa MM. et al. Hypermethylation of the polycomb group target gene PCDH7 in bladder tumors from patients of all ages. The Journal of urology. 2013;190:311-6
35. Giedl J, Rogler A, Wild A, Riener MO, Filbeck T, Burger M. et al. TERT Core Promotor Mutations in Early-Onset Bladder Cancer. Journal of Cancer. 2016;7:915-20
Author contact
![]() Corresponding author: Johannes Giedl, M.D., Institute of Pathology, University Hospital Erlangen, Friedrich-Alexander University Erlangen-Nürnberg, D-91054 Erlangen, Germany, E-mail: johannes.giedlde, Phone: +49 9131 8522524, Fax: +49 9131 8524745.
Corresponding author: Johannes Giedl, M.D., Institute of Pathology, University Hospital Erlangen, Friedrich-Alexander University Erlangen-Nürnberg, D-91054 Erlangen, Germany, E-mail: johannes.giedlde, Phone: +49 9131 8522524, Fax: +49 9131 8524745.