Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(13):3899-3906. doi:10.7150/jca.113576 This issue Cite
Review
The Role of P53 in Immune Evasion and Therapeutic Strategies in Hematologic Malignancies
Jing Wen1#, Linlin Fu2#, Hui Zhong2 ![]() , Hongxia Chen2
, Hongxia Chen2 ![]()
1. School of Clinical Medicine, Southwest Medical University, Luzhou, Sichuan, China.
2. Department of Hematology, Chongqing University Three Gorges Hospital, Chongqing, China.
# These authors contributed equally to the paper.
Received 2025-3-11; Accepted 2025-7-9; Published 2025-8-28
Abstract

P53 is a crucial tumor suppressor gene that plays an essential role in maintaining genomic stability, regulating cell cycle progression, and inducing apoptosis. In hematologic malignancies, P53 mutations are frequently associated with poor prognosis, treatment resistance, and immune evasion. Recent research has highlighted the impact of P53 dysfunction on tumor immune escape mechanisms, including impaired antigen presentation, altered cytokine signaling, and recruitment of immunosuppressive cells. This review integrates recent findings on P53 mutations in hematologic malignancies, focusing on their role in immune evasion and potential therapeutic strategies aimed at restoring P53 function or targeting associated pathways. Understanding these mechanisms may provide new insights into the development of effective immunotherapeutic approaches for hematologic cancers.
Keywords: P53, Immune Evasion, Hematologic Malignancies
Introduction
Hematologic malignancies are characterized by genomic instability, often driven by mutations in tumor suppressor genes such as TP53. The advent of Next-Generation Sequencing (NGS) has revolutionized patient stratification by identifying TP53 mutational status, which is critical for prognosis and therapeutic decision-making [1]. For instance, in acute myeloid leukemia (AML), TP53-mutated patients exhibit complex karyotypes and a dismal median survival of <6 months, necessitating first-line therapies distinct from conventional chemotherapy (e.g., hypomethylating agents or targeted drugs) [2]. Similarly, in myelodysplastic syndromes (MDS), TP53 mutations correlate with high-risk subtypes and rapid progression to AML, while in chronic lymphocytic leukemia (CLL), 17p deletions co-occurring with TP53 mutations predict resistance to BTK inhibitors [3]. This stratification underscores the need for mutation-specific therapeutic approaches to improve outcomes in these high-risk cohorts.
P53 Structure and Function
P53 is a transcription factor that regulates the expression of hundreds of genes involved in cell cycle arrest, apoptosis, DNA repair, and immune modulation [4]. Structurally, P53 consists of several functional domains: transactivation domain (TAD), proline-rich domain (PRD), DNA-binding domain (DBD), tetramerization domain (TD), and regulatory domain (RD) [5]. Wild-type P53 maintains cellular homeostasis by responding to various stress signals, including DNA damage and oncogene activation [6]. However, TP53 mutations often result in loss of function (LOF) or gain of function (GOF), contributing to tumorigenesis and immune escape. GOF mutations can lead to the activation of oncogenic pathways, further exacerbating tumor progression and immune suppression [7] (Figure 1, Table 1). In hematologic malignancies, TP53 mutations cluster in the DBD, with hotspot codons R175, R248, and R273 prevalent in AML and MDS. These mutations disrupt DNA binding, leading to loss of function (LOF) or gain of function (GOF). GOF mutants (e.g., R248Q) promote oncogenic signaling (e.g., NF-κB activation) and immune evasion. In AML, single-hit TP53 mutations (monoallelic) coexist with multi-hit alterations (bi-allelic mutations + 17p deletions), driving aggressive phenotypes and therapy resistance [8] (Table 2).
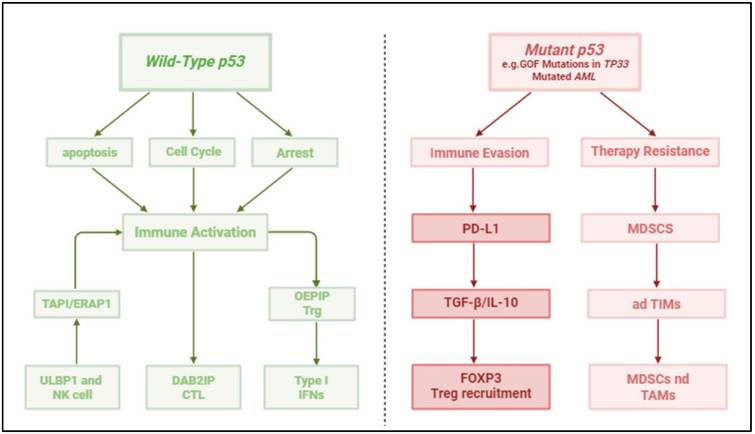
Contrasting Roles of Wild-Type and Mutant p53 in Hematologic Malignancies. This two-panel diagram illustrates the dual functions of p53 in the tumor microenvironment of hematologic cancers. The left panel depicts wild-type p53, which maintains genomic stability through induction of cell cycle arrest (via p21 and Gadd45), apoptosis (via Bax, PUMA, NOXA), and enhancement of anti-tumor immunity by promoting antigen presentation (TAP1, ERAP1) and stimulating immune effector cells (CTLs, NK cells). The right panel shows mutant p53, particularly gain-of-function (GOF) variants, contributing to immune evasion by downregulating antigen processing machinery, upregulating immune checkpoints (PD-L1), altering cytokine secretion (↑ TGF-β, IL-10, ↓ Type I IFNs), and promoting recruitment of immunosuppressive cells (Tregs, MDSCs, adTIMs: adaptive tumor-infiltrating myeloid cells). These opposing mechanisms highlight the importance of p53 status in shaping immune surveillance and therapy resistance in AML, CLL, and other hematologic malignancies.
Role of p53 in Normal Hematopoiesis and Immunity
| Function | Description | Key Molecular Targets | Immune Relevance |
|---|---|---|---|
| Cell Cycle Arrest | Halts the cell cycle at G1/S checkpoint upon DNA damage | p21 (CDKN1A), GADD45 | Prevents propagation of DNA-damaged HSCs |
| Apoptosis | Promotes mitochondrial-mediated apoptosis of damaged cells | BAX, PUMA, NOXA | Eliminates transformed hematopoietic cells |
| Senescence | Enforces terminal cell cycle arrest in pre-leukemic clones | p21, p16INK4A, pRB | Prevents clonal expansion of dysplastic cells |
| DNA Repair | Enhances nucleotide excision and base excision repair pathways | XPC, DDB2, p48 | Maintains genomic stability |
| Immune Surveillance | Promotes antigen presentation and modulates cytokines (e.g., IFN-γ, IL-12) | TAP1/2, MHC-I, IRF9 | Supports CD8+ T cell-mediated clearance |
Common TP53 Mutations in Hematologic Malignancies
| Mutation (Codon) | Amino Acid Change | Structural/Functional Class | Associated Disease(s) | Frequency (%) | Effect on p53 Function |
|---|---|---|---|---|---|
| R175H | Arginine → Histidine | Conformational/structural | AML, MDS | ~10-20 | Loss of DNA binding, gain-of-function (GOF) |
| R248Q | Arginine → Glutamine | DNA contact mutation | CLL, DLBCL | ~5-10 | Disrupts target gene activation |
| R273H | Arginine → Histidine | DNA contact mutation | MDS, AML | ~8-15 | Retains protein stability, impairs transcription |
| G245S | Glycine → Serine | Structural destabilization | AML | ~3-5 | Destabilizes p53 conformation |
| Y220C | Tyrosine → Cysteine | Surface mutation | MDS | ~2-4 | Creates a destabilizing surface crevice |
| R282W | Arginine → Tryptophan | DNA contact mutation | DLBCL, MDS | ~4-6 | Inhibits proper folding |
Mechanisms of P53-Mediated Immune Evasion
Impaired Antigen Presentation
P53 mutations disrupt the major histocompatibility complex (MHC) class I antigen presentation pathway, preventing immune cells from recognizing and eliminating tumor cells [9]. Mutant P53 downregulates key components of antigen processing, such as transporter associated with antigen processing (TAP1) and endoplasmic reticulum aminopeptidase 1 (ERAP1), leading to reduced surface expression of MHC-I molecules [10]. This impairment decreases CD8+ T cell recognition, allowing malignant cells to escape cytotoxic immune responses [11] (Table 3).
Immune Evasion Mechanisms Associated with p53 Dysfunction
| Mechanism | Effect of TP53 Loss or Mutation | Resulting Immune Impact |
|---|---|---|
| Downregulation of MHC-I | Reduces transcription of antigen-presentation machinery | Impaired CD8+ T cell recognition |
| Upregulation of PD-L1 | Disinhibition of PD-L1 via miR-34a loss | T cell exhaustion and anergy |
| Impaired NK cell ligands | Reduced expression of NKG2D ligands | Decreased NK cell-mediated cytotoxicity |
| Altered cytokine secretion | Aberrant IL-6/IL-10 expression | Promotes suppressive myeloid-derived suppressor cells (MDSCs) |
| Loss of miR-34 family | Disruption of immune checkpoint control | Overexpression of PD-L1, CD47 |
Suppression of Pro-Inflammatory Cytokines
Wild-type P53 enhances anti-tumor immunity by regulating cytokine production. However, P53 mutations are associated with increased secretion of immunosuppressive cytokines such as IL-10 and TGF-β, which inhibit T cell activation and promote an immunosuppressive TME [12]. Additionally, mutant P53 can upregulate PD-L1 expression, further dampening anti-tumor immune responses by suppressing T cell function [13] (Table 3).
Recruitment of Immunosuppressive Cells
P53 mutations contribute to immune evasion by promoting the accumulation of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) within the TME [14]. These cells inhibit cytotoxic T lymphocyte (CTL) function and facilitate tumor growth by secreting immunosuppressive factors and disrupting normal immune surveillance [15] (Table 3).
P53 in Specific Hematologic Malignancies
Acute Myeloid Leukemia (AML)
AML is a heterogeneous and aggressive malignancy characterized by clonal proliferation of myeloid precursors in the bone marrow [16]. P53 mutations occur in approximately 5-10% of AML cases but are disproportionately represented in patients with complex karyotypes and therapy-related AML [17]. These mutations are strongly associated with poor prognosis due to intrinsic resistance to conventional chemotherapy and a high risk of relapse [18]. Mechanistically, mutant P53 contributes to immune evasion by downregulating antigen presentation machinery, increasing PD-L1 expression, and modulating cytokine secretion to create an immunosuppressive microenvironment [19] (Table 4).
Hematologic Malignancies Frequently Involving TP53 Alterations
| Malignancy | TP53 Mutation Frequency | Chromosomal Abnormalities | Prognosis When Mutated | Standard Therapy Response | Notable Features |
|---|---|---|---|---|---|
| AML | ~10-20% | del(17p), complex karyotype | Very poor | Low CR rate (~20%) | Chemoresistance, early relapse |
| MDS | ~5-15% | -5q, -7, del(17p) | Poor | Resistance to HMAs | Progresses to AML frequently |
| CLL | ~8-15% | del(17p), del(11q) | Poor | Refractory to fludarabine | Prefer BTK/BCL2 inhibitors |
| DLBCL | ~15% | 17p deletions | Variable | Poor response to R-CHOP | Associated with double-hit lymphomas |
Myelodysplastic Syndromes (MDS)
MDS represents a group of hematologic disorders characterized by ineffective hematopoiesis and a high propensity for transformation into AML [20]. TP53 mutations are found in approximately 7-11% of MDS cases and are associated with increased genomic instability and poor overall survival [21]. The immunological consequences of P53 mutations in MDS include reduced MHC class I expression, leading to impaired antigen presentation, and increased secretion of immunosuppressive cytokines such as IL-6 and TGF-β. These factors contribute to a permissive bone marrow microenvironment that fosters malignant progression [22] (Table 4).
Multiple Myeloma (MM)
Multiple myeloma (MM) is a malignancy of plasma cells characterized by clonal proliferation within the bone marrow, resulting in immune dysfunction and bone destruction [23]. P53 mutations are present in 5-10% of newly diagnosed MM cases but become more frequent in relapsed and refractory disease, particularly in patients with chromosome 17p deletion (del(17p)) [24]. In MM, IL-6 is excessively produced in the bone marrow, promoting the proliferation and survival of myeloma cells [25]. Elevated IL-6 levels are associated with increased tumor burden, resistance to apoptosis, and poorer prognosis [26]. Mutant P53 in MM may facilitate immune evasion by enhancing IL-6-driven tumor growth and suppressing T-cell responses. Furthermore, MM cells with impaired P53 function demonstrate heightened resistance to immune surveillance, underscoring the need for further research into potential counteracting strategies (Table 4).
Lymphoma
P53 mutations are frequently observed in aggressive lymphomas, such as diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and Burkitt lymphoma, where they correlate with high tumor proliferation rates and immune escape [27]. In DLBCL, TP53 mutations are associated with reduced immune surveillance due to impaired antigen presentation and increased expression of immune checkpoint molecules like PD-L1[28]. In MCL, TP53 mutations are a defining feature of blastoid and pleomorphic variants, which are highly aggressive and show increased immune evasion capabilities [29]. Given the role of P53 in regulating immune responses, its dysfunction in lymphoma contributes significantly to tumor progression and resistance to immune-mediated tumor suppression.
Chronic Lymphocytic Leukemia (CLL)
CLL is a malignancy of mature B lymphocytes and is one of the most common leukemias in adults [30]. TP53 mutations occur in approximately 5-15% of newly diagnosed CLL cases but are significantly more prevalent in relapsed or refractory disease [31]. These mutations often co-occur with 17p deletions, further impairing P53 function and leading to aggressive disease behavior and poor survival outcomes [31]. P53 mutations in CLL contribute to immune evasion by altering antigen presentation, downregulating MHC class I expression, and increasing PD-L1 levels on leukemic cells [32]. This results in impaired cytotoxic T cell activity and enhanced immune suppression within the tumor microenvironment [33]. Additionally, mutant P53 influences cytokine secretion, promoting an immunosuppressive milieu through increased levels of TGF-β and IL-10, which inhibit T cell proliferation and function [34]. Future research should focus on identifying biomarkers that predict disease progression and immune evasion mechanisms in CLL patients with TP53 mutations (Table 4).
Therapeutic Strategies Targeting P53 and Immune Evasion
The tumor suppressor gene TP53, commonly referred to as the "guardian of the genome," plays a central role in maintaining cellular integrity by regulating the cell cycle, apoptosis, and genomic stability. Mutations or functional impairments in TP53 are present in more than 50% of human cancers and are often associated with poor prognosis, resistance to therapy, and increased immune evasion [35]. Recent advancements have revealed novel strategies to reactivate or compensate for defective p53 signaling, with the dual goal of suppressing tumor proliferation and restoring anti-tumor immune responses. This section explores emerging therapeutic strategies focused on both wild-type and mutant forms of p53, with an emphasis on how these interventions modulate immune evasion in hematological malignancies such as acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), chronic lymphocytic leukemia (CLL), and diffuse large B-cell lymphoma (DLBCL) (Table 5).
Small-Molecule Therapies Targeting p53 Pathway
| Drug Name | Mechanism of Action | Cancer Type | Trial Phase | Clinical Trial ID | Response Rate | Additional Notes |
|---|---|---|---|---|---|---|
| Idasanutlin | MDM2 inhibitor (p53 stabilization) | AML (WT p53) | Phase II | NCT02545283 | ORR: 40% | Synergistic with cytarabine |
| APR-246 | Reactivates mutant p53 (R175H etc.) | MDS, AML | Phase III | NCT03745716 | CR: 30% | Reduces PD-L1; combines with azacitidine |
| Arsenic Trioxide | Promotes degradation of mutant p53 | AML | Phase I/II | NCT03855371 | Ongoing | Synergy with proteasome inhibitors |
| Eprenetapopt | Re-folds mutant p53 to active form | MDS, AML | Phase II/III | Multiple | Varies | Induces ferroptosis |
Small-Molecule Activators
MDM2 Inhibitors (e.g., Idasanutlin), is one of the most promising classes of small-molecule activators targets MDM2, a negative regulator of p53. In normal cells, MDM2 binds to p53, promoting its ubiquitination and proteasomal degradation [36]. In TP53 wild-type cancers, MDM2 is often overexpressed, leading to functional inactivation of p53 despite its intact genetic sequence [37]. Idasanutlin, a potent and selective MDM2 antagonist, has shown efficacy in TP53 wild-type AML. By preventing MDM2-p53 interaction, Idasanutlin stabilizes p53, enhancing its transcriptional activity and promoting tumor cell apoptosis [38]. Notably, Idasanutlin also restores MHC class I (MHC-I) expression on leukemia cells, which is essential for recognition by cytotoxic CD8+ T cells [39]. This immune-modulating effect opens the door for synergistic combinations with immune checkpoint inhibitors or adoptive cell therapies. In the clinical trial NCT02545283, Idasanutlin achieved an overall response rate (ORR) of 40%, highlighting its potential in reactivating p53-dependent tumor suppressive and immune pathways [40].
APR-246 (Eprenetapopt), also known as Eprenetapopt, targets mutant p53 by restoring its proper folding and function [41]. It is especially effective against common structural mutations such as R175H, which result in loss of DNA-binding capability. APR-246 promotes the refolding of mutant p53 into a functional conformation, allowing it to resume transcription of downstream targets involved in cell cycle arrest and apoptosis [42]. Beyond its pro-apoptotic effects, APR-246 also modulates the immune landscape of tumors. It has been shown to downregulate PD-L1, a key immune checkpoint molecule that suppresses T cell activity [43]. In a phase II clinical trial involving patients with MDS and TP53 mutations (Clinical trial NCT03745716), APR-246 demonstrated a complete remission (CR) rate of 30%, signifying its dual role in correcting p53 defects and mitigating immune escape mechanisms [44].
Gene Therapy
CRISPR/Cas9-Mediated Gene Editing technologies such as CRISPR/Cas9 have opened new avenues for directly correcting TP53 mutations [45]. In CLL models, CRISPR-based correction of TP53 mutations has led to restoration of wild-type p53 activity [46]. This reactivation reinstates expression of miR-34a, a well-known p53-regulated microRNA that functions as a tumor suppressor and immune modulator. miR-34a targets several components of the PD-1/PD-L1 axis, and its expression correlates with reduced levels of PD-L1, thereby enhancing T cell-mediated anti-tumor responses [47]. Though still preclinical, these findings suggest that gene therapy approaches could offer a curative potential by repairing the genetic defect at its source while simultaneously reawakening immune surveillance (Table 6).
Gene Therapy Approaches Targeting TP53
| Strategy | Methodology | Disease Target | Outcome in Preclinical/Clinical Studies | Limitations |
|---|---|---|---|---|
| CRISPR/Cas9 | Homology-directed repair of TP53 | CLL, AML models | Restored p53 function, ↑miR-34a, ↓PD-L1 | Delivery, off-target editing |
| AAV-p53 Delivery | Wild-type p53 via viral vector | MDS, AML | Induced apoptosis, tumor suppression | Immune clearance of vector |
| siRNA Knockdown | Silencing MDM2 or mutant TP53 | CLL (in vitro) | Resensitization to chemotherapy | Transient effects |
Adoptive Cell Transfer and Mutant Degraders
CAR-T Cell Therapy
Chimeric Antigen Receptor (CAR)-T cells represent a breakthrough in immunotherapy. However, patients with TP53 mutations often show resistance to conventional CAR-T approaches due to enhanced immune evasion and defective apoptosis [48]. In DLBCL, tumors harboring TP53 mutations have been particularly refractory to CD19-targeted CAR-T cells [49]. To overcome this, dual-targeting strategies such as CD19/CD22 CAR-T have been developed. These modified CAR-T cells recognize two different antigens simultaneously, reducing the risk of antigen escape [50]. In the clinical trial NCT04007029, this dual-targeting approach resulted in a complete response rate of 58%, even in the presence of p53 dysfunction, suggesting that broadening antigen coverage can compensate for underlying genomic instability [51] (Table 7).
Immunotherapy Strategies in p53-Mutant Hematologic Malignancies
| Strategy | Target(s) | Disease | Clinical Trial/Study | Outcome | Considerations |
|---|---|---|---|---|---|
| CAR-T (CD19/CD22) | Dual-antigen CARs | DLBCL (TP53-mut) | NCT04007029 | CR: 58% | Overcomes antigen escape |
| Checkpoint Blockade | PD-1/PD-L1, CTLA-4 | AML, MDS | Multiple | Limited in TP53-mut | Often requires combination therapy |
| Liposomal miR-34a | miRNA replacement | CLL | NCT05084365 | Phase I underway | Reduces PD-L1, targets oncogenes |
| BiTEs | CD3/CD19 | B-ALL, CLL | Preclinical | Enhanced cytotoxicity | TP53 status may affect durability |
Arsenic Trioxide
Originally used in acute promyelocytic leukemia, arsenic trioxide has recently been investigated for its ability to degrade mutant p53 proteins in AML via the proteasomal pathway [52]. This degradation eliminates the dominant-negative or gain-of-function effects of mutant p53, restoring the balance of cell cycle regulation and apoptosis [53]. In trial NCT03855371, early data suggest that arsenic trioxide could selectively target mutant p53, potentially reversing immune evasion in AML by removing dysfunctional p53 isoforms that suppress immune gene expression [54].
GSK2830371, a selective inhibitor of PPM1D, has shown promise in preclinical AML models by restoring p53 function and increasing the transcription of pro-apoptotic genes [57-58]. Interestingly, PPM1D inhibition also appears to reduce the expression of immune-inhibitory molecules, thereby sensitizing tumors to immune-mediated clearance [59]. This strategy holds potential for AML patients with wild-type TP53 but dysregulated p53 signaling due to upstream suppressors like PPM1D (Table 8).
Emerging Small Molecules and Biological Agents in TP53 Pathway
| Agent | Target | Disease Context | Mechanism | Development Status |
|---|---|---|---|---|
| GSK2830371 | PPM1D (negative p53 regulator) | AML, MDS (WT p53 + PPM1D overexpression) | Restores p53-mediated apoptosis | Preclinical |
| COTI-2 | Mutant p53, PI3K/mTOR | MDS, AML | Refolds mutant p53, inhibits mTOR | Phase I/II |
| Dual MDM2/MDMX Inhibitors | Nutlin-based derivatives | AML, CLL | Prevent full p53 degradation | Preclinical/early trials |
sEVs Delivering miR-34a
Small extracellular vesicles (sEVs) are emerging as efficient carriers for therapeutic nucleic acids [60]. A novel strategy under investigation involves delivering miR-34a via liposomal formulations of sEVs. This approach takes advantage of miR-34a's ability to regulate both oncogenic pathways and immune checkpoint expression [61]. In a Phase I trial (NCT05084365) involving patients with TP53-mutated CLL, systemically delivered miR-34a-containing sEVs have shown potential to reduce PD-L1 expression, suppress oncogenes, and promote immune activation [62]. As this approach bypasses the need to correct the p53 gene directly, it represents an elegant method to re-establish downstream p53-like effects in tumors with irreversible TP53 mutations (Table 7).
Targeting TP53 and immune evasion mechanisms is a promising and rapidly evolving field, particularly for hematologic malignancies that have historically been difficult to treat due to p53 dysfunction. The strategies discussed—ranging from small-molecule reactivators and gene editing tools to advanced cell therapies and RNA-based interventions—highlight the diverse and complementary ways in which the p53 pathway can be modulated. A deeper understanding of p53's role in immune regulation is critical for designing effective combination therapies that harness both direct tumor suppression and immune activation, offering hope for improved outcomes in patients with p53-related malignancies.
Discussion
The tumor suppressor p53 plays a pivotal role in maintaining genomic integrity, and its dysregulation is a hallmark of various hematologic malignancies, including AML, MDS, CLL, and DLBCL. While the clinical relevance of TP53 mutations has been extensively documented, our current study contributes to the field by highlighting emerging therapeutic strategies specifically designed to target p53 dysfunction and its downstream consequences on immune evasion.
One of the novel insights presented in this work is the therapeutic potential of MDM2 inhibitors, particularly Idasanutlin, in restoring MHC-I expression and enhancing immune surveillance in TP53 wild-type AML. This finding not only reinforces the immunomodulatory role of p53 but also supports the rationale for combining MDM2 inhibitors with immune checkpoint blockade therapies to augment antitumor immunity [36-40]. Additionally, our discussion of APR-246 (Eprenetapopt) in correcting structural p53 mutations—especially in the context of PD-L1 downregulation in MDS—extends previous work by integrating its immunologic consequences, which had not been comprehensively addressed in earlier literature. By linking p53 restoration with checkpoint molecule suppression, we emphasize the dual impact of APR-246 on both tumor cell apoptosis and immune activation [41-44]. Our study also underscores the therapeutic innovation of CRISPR/Cas9-mediated p53 repair, with a particular focus on its ability to re-induce miR-34a and suppress PD-L1 expression in CLL models. This mechanistic link between gene editing and immune checkpoint regulation provides a promising avenue for precision oncology in p53-mutant hematologic cancers [45-47]. Moreover, we highlight dual-antigen targeting CAR-T strategies (CD19/CD22) as an effective approach to overcome immune resistance in TP53-mutated DLBCL. This represents a significant evolution beyond traditional CAR-T therapies, particularly for patients who fail standard treatments due to p53-related resistance pathways [48-51]. Another novel aspect of our analysis includes the repurposing of arsenic trioxide for proteasomal degradation of mutant p53, expanding its utility beyond acute promyelocytic leukemia. This underexplored mechanism offers a valuable alternative for patients harboring dominant-negative or gain-of-function TP53 mutations [52-54]. We also discuss the implications of targeting PPM1D overexpression, a common event in TP53 wild-type AML, with agents like GSK2830371. This sheds light on a non-mutational form of p53 suppression and illustrates how pharmacologic inhibition of p53 repressors may restore tumor suppressive function [55-59]. Finally, we bring attention to a novel phase I trial of small extracellular vesicles (sEVs) delivering miR-34a, which represents a first-in-human effort to re-establish p53-related immune control via RNA-based nanomedicine in TP53-mutant CLL [60-62].
Conclusion and Future Directions
P53 plays a pivotal role in hematologic malignancies by regulating immune surveillance and tumor suppression. Its mutations enable immune evasion, contributing to disease progression and therapy resistance. Understanding the mechanisms by which P53 mutations alter the immune landscape provides opportunities for developing targeted therapies. Future research should focus on refining combination strategies that integrate P53-targeted treatments with immunotherapy to improve patient outcomes in hematologic cancers.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (82300190), New Chongqing Youth Innovative Talent Project, Chongqing Medical High-End Talent Project and Chongqing University Medical and Industrial Integration Project, Project number 2022CDJYGRH-012.
Author contributions
HZ and HC designed the study. JW and HC wrote and edited the manuscript. JW drew the figures. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ghoreyshi N, Heidari R, Farhadi A, Chamanara M, Farahani N, Vahidi M. et al. Next-generation sequencing in cancer diagnosis and treatment: clinical applications and future directions. Discov Oncol. 2025;16(1):578
2. Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH. et al. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022;12(11):2516-2529
3. Shahzad M, Amin MK, Daver NG, Shah MV, Hiwase D, Arber DA. et al. What have we learned about TP53-mutated acute myeloid leukemia? Blood Cancer J. 2024;14(1):202
4. Zhang H, Xu J, Long Y, Maimaitijiang A, Su Z, Li W, Li J. Unraveling the Guardian: p53's Multifaceted Role in the DNA Damage Response and Tumor Treatment Strategies. International Journal of Molecular Sciences. 2024;25(23):12928
5. Joerger AC, Fersht AR. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb Perspect Biol. 2010;2(6):a000919
6. Yasser A, Wu H, Chai C, Yousef H, Persad S, Sergi C. et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp Mol Med. 2022 54, 1658-1669
7. Alvarado-Ortiz E, de la Cruz-López KG, Becerril-Rico J, Sarabia-Sánchez MA, Ortiz-Sánchez E, García-Carrancá A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front Cell Dev Biol. 2021;8:607670
8. Ahmadi SE, Rahimian E, Rahimi S, Zarandi B, Bahraini M, Soleymani M. et al. From regulation to deregulation of p53 in hematologic malignancies: implications for diagnosis, prognosis and therapy. Biomark Res. 2024 12, 137
9. Wu HH, Leng S, Eisenstat DD, Sergi C, Leng R. Targeting p53 for immune modulation: Exploring its functions in tumor immunity and inflammation. Cancer Lett. 2025;617:217614
10. Braun MW, Iwakuma T. Regulation of cytotoxic T-cell responses by p53 in cancer. Transl Cancer Res. 2016;5(6):692-697
11. Philip M, Schietinger A. CD8+ T cell differentiation and dysfunction in cancer. Nat Rev Immunol. 2022;22(4):209-223
12. Liu S, Liu T, Jiang J, Guo H, Yang R. p53 mutation and deletion contribute to tumor immune evasion. Front Genet. 2023;14:1088455
13. Liu N, Jiang X, Guo L, Zhang C, Jiang M, Sun Z. et al. Mutant p53 achieved Gain-of-Function by promoting tumor growth and immune escape through PHLPP2/AKT/PD-L1 pathway. Int J Biol Sci. 2022;18(6):2419-2438
14. Zhu KL, Su F, Yang JR, Xiao RW, Wu RY, Cao MY, et. al. TP53 to mediate immune escape in tumor microenvironment: an overview of the research progress. Mol Biol Rep. 2024;51(1):205
15. Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S. et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020;30(2):481-496.e6
16. Vakiti A, Reynolds SB, Mewawalla P. Acute Myeloid Leukemia. 2024 Apr 27. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2025 1
17. Weinberg OK, Siddon A, Madanat YF, Gagan J, Arber DA, Dal Cin P. et al. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022;6(9):2847-2853
18. Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute Myeloid Leukemia: The Good, the Bad, and the Ugly. Am Soc Clin Oncol Educ Book. 2018;38:555-573
19. Vadakekolathu J, Rutella S. Escape from T-cell-targeting immunotherapies in acute myeloid leukemia. Blood. 2024;143(26):2689-2700
20. Zhou J, Orazi A, Czader MB. Myelodysplastic syndromes. Semin Diagn Pathol. 2011;28(4):258-72
21. Shah MV, Tran ENH, Shah S, Chhetri R, Baranwal A, Ladon D. et al. TP53 mutation variant allele frequency of ≥10% is associated with poor prognosis in therapy-related myeloid neoplasms. Blood Cancer J. 2023;13(1):51
22. Barakos GP, Hatzimichael E. Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Diseases. 2022;10(2):33
23. Abduh MS. An overview of multiple myeloma: A monoclonal plasma cell malignancy's diagnosis, management, and treatment modalities. Saudi J Biol Sci. 2024;31(2):103920
24. Lodé L, Eveillard M, Trichet V, Soussi T, Wuillème S, Richebourg S. et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica. 2010;95(11):1973-6
25. Harmer D, Falank C, Reagan MR. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front Endocrinol (Lausanne). 2019;9:788
26. Sang YB, Yang H, Lee WS, Lee SJ, Kim SG, Cheon J. et al. High Serum Levels of IL-6 Predict Poor Responses in Patients Treated with Pembrolizumab plus Axitinib for Advanced Renal Cell Carcinoma. Cancers (Basel). 2022;14(23):5985
27. Yu L, Yu TT, Young KH. Cross-talk between Myc and p53 in B-cell lymphomas. Chronic Dis Transl Med. 2019;5(3):139-154
28. Pascual M, Mena-Varas M, Robles EF, Garcia-Barchino MJ, Panizo C, Hervas-Stubbs S. et al. PD-1/PD-L1 immune checkpoint and p53 loss facilitate tumor progression in activated B-cell diffuse large B-cell lymphomas. Blood. 2019;133(22):2401-2412
29. Balsas P, Veloza L, Clot G, Sureda-Gómez M, Rodríguez ML, Masaoutis C. et al. SOX11, CD70, and Treg cells configure the tumor-immune microenvironment of aggressive mantle cell lymphoma. Blood. 2021;138(22):2202-2215
30. Darwiche W, Gubler B, Marolleau JP, Ghamlouch H. Chronic Lymphocytic Leukemia B-Cell Normal Cellular Counterpart: Clues from a Functional Perspective. Front Immunol. 2018;9:683
31. Chauffaille MLLF, Zalcberg I, Barreto WG, Bendit I. Detection of somatic TP53 mutations and 17p deletions in patients with chronic lymphocytic leukemia: a review of the current methods. Hematol Transfus Cell Ther. 2020;42(3):261-268
32. Vom Stein AF, Hallek M, Nguyen PH. Role of the tumor microenvironment in CLL pathogenesis. Semin Hematol. 2024;61(3):142-154
33. Kwok M, Agathanggelou A, Davies N, Stankovic T. Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives. Cancers (Basel). 2021;13(18):4681
34. Carlsen L, Zhang S, Tian X, De La Cruz A, George A, Arnoff TE. et al. The role of p53 in anti-tumor immunity and response to immunotherapy. Front Mol Biosci. 2023;10:1148389
35. Hernández B, El-Deiry WS. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer. 2021;1876(1):188556
36. Huang Y, Li W, Zhou Y, Bai J, Li N, Su Z. et al. Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction. Cells. 2025;14(8):583
37. Wang HL, Guo M, Wei HD, Chen YH. Targeting p53 pathways: mechanisms, structures and advances in therapy. Sig Transduct Target Ther. 2023 8, 92
38. Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther. 2019;12:2903-2910
39. Ingelshed K, Melssen MM, Kannan P, Chandramohan A, Partridge AW, Jiang L. et al. MDM2/MDMX inhibition by Sulanemadlin synergizes with anti-Programmed Death 1 immunotherapy in wild-type p53 tumors. iScience. 2024;27(6):109862
40. Montesinos P, Beckermann BM, Catalani O, Esteve J, Gamel K, Konopleva MY. et al. MIRROS: a randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020;16(13):807-815
41. Wang Z, Hu H, Heitink L, Rogers K, You Y, Tan T. et al. The anti-cancer agent APR-246 can activate several programmed cell death processes to kill malignant cells. Cell Death Differ. 2023;30(4):1033-1046. Erratum in: Cell Death Differ. 2023;30(4):1096
42. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9(5):439
43. Ghosh A, Michels J, Mezzadra R, Venkatesh D, Dong L, Gomez R. et al. Increased p53 expression induced by APR-246 reprograms tumor-associated macrophages to augment immune checkpoint blockade. J Clin Invest. 2022;132(18):e148141
44. Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA. et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J Clin Oncol. 2021;39(14):1584-1594
45. Kolanu ND. CRISPR-Cas9 Gene Editing: Curing Genetic Diseases by Inherited Epigenetic Modifications. Glob Med Genet. 2024;11(1):113-122
46. Quijada M, Pérez C, Hernández M, Rodríguez AE, Herrero AB, Hernández JM. et al. Dissecting the role of TP53 alterations in del(11q) chronic lymphocytic leukemia. Clin Transl Med. 2021Feb;11(2):e304
47. Pan W, Chai B, Li L, Lu Z, Ma Z. p53/MicroRNA-34 axis in cancer and beyond. Heliyon. 2023;9(4):e15155
48. Mueller J, Schimmer RR, Koch C, Schneiter F, Fullin J, Lysenko V. et al. Targeting the mevalonate or Wnt pathways to overcome CAR T-cell resistance in TP53-mutant AML cells. EMBO Mol Med. 2024;16(3):445-474
49. Cai Z, Zou D, Ma Q, Sun W, Guo Y. Combined autologous hematopoietic stem cell transplantation and CD19 CAR T-cell therapy for relapsed/refractory diffuse large B-cell lymphoma with TP53 mutation: A case report. SAGE Open Med Case Rep. 2025;13:2050313X241306236
50. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S. et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797-1805
51. Larson SM, Walthers CM, Ji B, Ghafouri SN, Naparstek J, Trent J. et al. CD19/CD20 Bispecific Chimeric Antigen Receptor (CAR) in Naive/Memory T Cells for the Treatment of Relapsed or Refractory Non-Hodgkin Lymphoma. Cancer Discov. 2023;13(3):580-597
52. Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9(8):e103497
53. Zhu G, Cai J, Zhong H. TP53 signal pathway confers potential therapy target in acute myeloid leukemia. Eur J Haematol. 2023;110(5):480-489
54. Zawacka JE. p53 biology and reactivation for improved therapy in MDS and AML. Biomark Res. 2024;12(1):34
55. Zhang L, Hsu JI, Goodell MA. PPM1D in Solid and Hematologic Malignancies: Friend and Foe? Mol Cancer Res. 2022;20(9):1365-1378
56. Stoyanov M, Martinikova AS, Matejkova K, Horackova K, Zemankova P, Burdova K. et al. PPM1D activity promotes cellular transformation by preventing senescence and cell death. Oncogene. 2024;43(42):3081-3093
57. Chamberlain V, Drew Y, Lunec J. Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma. Cancers (Basel). 2021;14(1):14
58. Esfandiari A, Hawthorne TA, Nakjang S, Lunec J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol Cancer Ther. 2016;15(3):379-91
59. Kojima K, Maeda A, Yoshimura M, Nishida Y, Kimura S. The pathophysiological significance of PPM1D and therapeutic targeting of PPM1D-mediated signaling by GSK2830371 in mantle cell lymphoma. Oncotarget. 2016;7(43):69625-69637
60. Joo HS, Suh JH, So CM, Jeon HJ, Yoon SH, Lee JM. Emerging Roles of Using Small Extracellular Vesicles as an Anti-Cancer Drug. Int J Mol Sci. 2023;24(18):14063
61. Sharma P, Dando I, Strippoli R, Kumar S, Somoza A, Cordani M. et al. Nanomaterials for Autophagy-Related miRNA-34a Delivery in Cancer Treatment. Front Pharmacol. 2020;11:1141
62. Patel B, Greenland JC, Williams-Gray CH. Clinical Trial Highlights: Anti-Inflammatory and Immunomodulatory Agents. J Parkinsons Dis. 2024;14(7):1283-1300
Author contact
![]() Corresponding authors: Hui Zhong, email: 13896976866com and Hongxia Chen, email: hongxiachenedu.cn.
Corresponding authors: Hui Zhong, email: 13896976866com and Hongxia Chen, email: hongxiachenedu.cn.