Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(12):3673-3683. doi:10.7150/jca.115715 This issue Cite
Review
SETD2 and EZH2: Two epigenetic drivers of prostate cancer
Jiamin Wang1, Longquan Xiang2, Haiyan Zhang3 ![]() , Xiangyu Zhang2
, Xiangyu Zhang2 ![]()
1. School of Clinical Medicine, Shandong Second Medical University, Weifang, Shandong 261053, P.R. China.
2. Department of Pathology, Jining No.1 People's Hospital, Jining, Shandong 272000, P.R. China.
3. Zoucheng People's Hospital, Zoucheng, Shandong 273500, P.R. China.
Received 2025-4-14; Accepted 2025-6-29; Published 2025-7-28
Abstract

Prostate cancer is a prevalent malignancy among men, characterized by complex mechanisms underlying metastasis and treatment resistance. Epigenetic modifications play a crucial role in regulating prostate cancer progression, particularly involving histone methyltransferases such as SET-domain containing 2 (SETD2) and Enhancer of Zeste homolog 2 (EZH2). SETD2 contributes to chromatin stability by catalyzing the trimethylation of histone H3 lysine 36 (H3K36me3), and its downregulation is strongly correlated with increased invasiveness and epithelial-mesenchymal transition in prostate cancer. Conversely, EZH2, the catalytic subunit of Polycomb Repressive Complex 2, mediates gene silencing through H3K27me3 modification and is frequently overexpressed in advanced disease, promoting tumor metastasis and resistance to therapy. Notably, SETD2 regulates EZH2 stability through direct protein interactions, highlighting a coordinated epigenetic regulatory axis. Multi-omics studies have revealed that SETD2 loss induces aberrant DNA methylation and activates oncogenic signaling pathways, whereas EZH2 overexpression cooperates with PI3K-AKT pathway dysregulation to drive castration-resistant prostate cancer. Although inhibitors targeting SETD2 (e.g., EZM0414) and EZH2 (e.g., tazemetostat) demonstrate antitumor activity in preclinical models, their clinical efficacy remains constrained by drug resistance and tumor microenvironment heterogeneity. Emerging evidence suggests that combining epigenetic therapies with immunotherapy may enhance therapeutic outcomes. This review comprehensively systematically examines the molecular mechanisms underlying the SETD2/EZH2 axis in prostate cancer, providing a theoretical foundation for developing precision therapies based on SETD2- or EZH2-mediated epigenetic modifications.
Keywords: SETD2, EZH2, Prostate cancer, Inhibitor, Epigenetic modifications.
1. Introduction
Prostate cancer remains one of the most commonly diagnosed malignancies in men and ranks among the leading causes of cancer-related mortality worldwide [1]. Among the numerous mechanisms involved in its development and progression, epigenetic regulation has emerged as a significant contributor. The concept of epigenetics was proposed by Conrad Waddington in 1939. Epigenetic modifications refer to heritable changes in gene expression that do not involve alterations in the DNA sequence itself. These changes—such as DNA methylation, histone modification (methylation and acetylation), and chromatin remodeling—play essential roles in development and disease, often preceding genetic mutations in cancer initiation. Epigenetic changes manifest earlier than genetic changes. Studies exploring the mechanisms underlying prostate cancer metastasis have focused on the role of epigenetic regulation and its modifications. Histone methyltransferases (HMTs) are comprised of histone lysine methyltransferases (KMTs) and protein arginine methyltransferases (PRMTs). Both SETD2 and EZH2 are KMTs, and involved in prostate cancer progression.
One of the key players in epigenetic regulation is the histone methyltransferase SET-domain containing 2 (SETD2), the sole enzyme known to catalyze histone H3 trimethylation at lysine 36 (H3K36me3). The SETD2 gene is located on chromosome 3, p21.31 [2], and is widely recognized for its tumor-suppressive functions. Aberrant expression of SETD2 has been linked to poor prognosis, chromatin instability, increased tumor aggressiveness, increased metastatic potential, and resistance to therapy across various cancers, including prostate cancer [3-5]. In particular, reduced or absent SETD2 expression has been associated with enhanced invasiveness and epithelial-mesenchymal transition (EMT), underscoring its critical role in disease progression [6].
By contrast, Enhancer of Zeste homolog 2 (EZH2), the catalytic subunit of Polycomb Repressive Complex 2 (PRC2), is responsible for catalyzing mono-, di-, and tri-methylation of histone H3 lysine 27 (H3K27me1-3) [7-8]. EZH2 is a well-established oncogene whose overexpression leads to transcriptional silencing of tumor suppressor genes, thereby promoting proliferation, metastasis, and resistance to hormonal therapies in prostate cancer [9-11]. Studies have uncovered that SETD2 binds to EZH2 and promotes its degradation through lysine 735 (K735) methylation, thereby preventing a shift toward a hyper-repressive H3K27me3 chromatin state [12].
This review aims to elucidate the epigenetic regulation of SETD2 and EZH2 in prostate cancer, focusing on their molecular mechanisms of action, involvement in metastasis, effects on histone modification, and impact on gene expression regulation to promote or inhibit prostate cancer metastasis, to offer a basis for prostate cancer diagnosis and treatment, as well as for the research and development of novel drugs.
2. SETD2 and Prostate Cancer
2.1 Structure and function of SETD2
SETD2 is located on the human chromosome 3p21.31 region [2]. SETD2 contains multiple exons and introns, a structural feature that enables the generation of multiple transcripts during post-transcriptional processing of genes through variable splicing and other means. These different transcripts may perform diverse functions across cellular environments or under different physiopathological conditions. Human SETD2 protein possesses multiple functional domains that are more conserved, mainly including the AWS (SET-related)-SET-PS (post-SET) structural domain, the WW structural domain, SRI (Set2-Rpb1 interacting structural domain), the SETD2-hnrnp interacting (SHI) structural domain and a large unstructured n-terminal structural domain [13]. Different structural domains have their functions and missions. Among them, the n-terminal region of SETD2 plays a decisive role in maintaining the stability of SETD2. The removal and loss of the n-terminal fragment results in compromised stability of SETD2, which significantly decreases the expression of H3K36me3 [14]. SRI domain of SETD2 is indispensable for SETD2 functions, SRI domain's direct association with phosphorylated Pol II (RNAPII-pCTD) can lead to targeted gene transcription elongation [15]. SRI domain contain 15 amino acids, can recognize the linker DNA of chromatin. SRI can control substrate specificity. SRI domain can affect the half-life of SETD2 protein through proteasome-dependent pathway, and influence the catalytic activity of SET domain. And SRI domain affect SETD2's ability to methylate non-histone substrates. A pathogenic mutation (R2510H) in the SRI domain impairs SETD2 ability to methylate α-tubulin at lysine 40 during mitosis [16]. Cryptic transcription is the process of transcription occurring at unexpected or non-canonical sites within the genome. SETD2 can regulate cryptic transcription and pre-mRNA splicing to alter target gene functions [17]. SETD2-mediated H3K36me3 serve as a binding site for various chromatin regulators that can inhibit cryptic transcription. One study shows that cryptic 5' splice sites are activated when they bind U1 snRNP much stronger than authentic 5' splice sites [18]. SETD2 prevents cryptic transcription using a different H3K36me3-mediated mechanism, and is independent of histone deacetylation. This process is closely related to interaction of SETD2 with DNA methyltransferase 3B (DNMT3B). But one study showed that SETD2-mediated H3K36me3 could induce histone deacetylation in higher eukaryote to inhibit cryptic transcription [19]. SETD2 mutation often leads to defects in transcript processing, including cryptic transcription. SETD2 interacts with hnRNP L to regulate alternative splicing of pre-mRNA. One study shows that SETD2 knockdown can induce alternative splicing in PKM2, TPM1 and TPM2 genes [20]. SETD2 plays an invaluable role in mammalian epigenetic regulation. SETD2 catalyzes histone methylation and interacts with RNA polymerase II to mediate transcriptional elongation. We noted that the development of several tumors is associated with mutations in SETD2; for instance, gastric, kidney, and lung cancers [6]. SETD2 is the only histone methyltransferase (HMTase) that catalyzes the trimethylation of lysine 36 on histone H3 (H3K36me3), which results in the epigenetic marking of H3K36me3 [21]. Among the epigenetic marks, H3K36me3 plays a role in transcription elongation, selective splicing, and DNA repair (Figure 1) [21-22]. Sometimes, low expression of SETD2 can confer advantages of proliferation, colony formation, migration and invasion for cancer cells.
Molecular structure diagram and functional schematic of SET-domain containing 2 (SETD2). Activation is indicated by arrows and suppression by blocking lines.
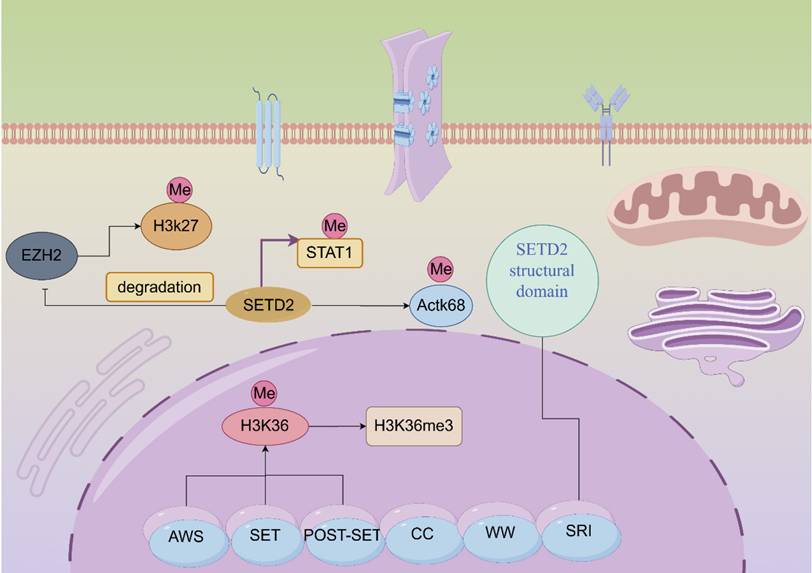
2.2 Epigenetic regulation of SETD2 in prostate cancer and metastatic mechanisms
The majority of SETD2 mutations were heterogenous, and almost half of these mutations were nonsense, frameshift, splice site mutations or deletions, which were loss-of-function alternations. SETD2 mutant patients usually harbored high mutation burden and microsatellite instability. SETD2 mutation occurred in a dispersed manner across the whole sequence. SETD2 mutation of loss-of-function was related to prostate cancer progression [23]. Several studies have demonstrated significantly lower levels of SETD2 expression in prostate tissues when compared to that in normal prostate tissues. The downregulation of SETD2 is closely associated with prostate cancer development, which reinforces the idea of SETD2 exerting a biological function as a tumor suppressor. Moreover, decreased SETD2 expression decreases the H3K36me3. In contrast, the SETD2-H3K36me3-signaling axis is closely related to epigenetic regulation, which, in turn, affects chromatin status. In addition, SETD2 loss may be associated with pancreatic cancer because its loss leads to the ectopic expression of H3K27me3 and H3K27ac, which would contribute to the occurrence of immune escape in pancreatic ductal carcinoma [24-25].
DNA methyltransferase 3β (DNMT3B) is a methyltransferase, and SETD2 promotes DNMT3B recruitment [24]. SETD2, H3K36me3, DNMT3B, and DNA methylation interact in epigenetic regulation, enabling the fidelity of gene transcription [26]. SETD2 deficiency disrupts this balance, resulting in hypermethylation or hypomethylation of histone proteins in vivo, which can induce malignant changes as cancer cells gain the ability to invade and migrate.
Moreover, in the absence of the key oncogenes, SETD2-H3K36me3 is similarly absent, which then constructs a possible association between SETD2-H3K36me3 and carcinogenesis [24]. There are several important structural domains in the SETD2 protein that act synergistically to enable SETD2 to recognize and bind to the histone H3 tail, which, in turn, catalyzes H3K36me3 [27]. When SETD2 is deficient, it inhibits the expression of the epithelial marker E-cadherin, while upregulating the expression of mesenchymal markers N-cadherin and Vimentin, which promotes the EMT of prostate cancer cells, so that the cells acquire a stronger migratory and invasive ability and thus become prone to metastasis. SETD2 can maintain a normal epithelial cell phenotype by regulating the histone methylation status of the promoter regions of the related genes. When the SETD2 expression is reduced, it fails to effectively inhibit the expression of mesenchymal-related genes, leading to EMT in prostate cancer cells and thereby promoting tumor metastasis. SETD2 mutation often leads to resistance to DNA-damaging agents, such as doxorubicin, cytarabine, and etoposide. SETD2 loss decreases the activation of the DNA damage response after exposure to cytotoxic agents. One study showed that SETD2 missense mutation (p.T1171K) affect CREB1 phosphorylation and mediate the cisplatin resistance [28]. There are also a study showing that SETD2 regulate MSH6 gene expression to mediate temozolomide resistance [29]. SETD2 inactive mutation is involved in sunitinib resistance in renal cell carcinoma, may be attributed to reduced signal transduction of MCL-1. SETD2 knockout can inhibit ERK activation induced by cisplatin and upregulate Bcl-xL to induce cisplatin resistance. One study indicates that SETD2 loss in cancer cell can shape cancer-associated fibroblasts heterogeneity to support cancer progression [30]. SETD2 inactivation can affect tumor immune microenvironment to enhance neutrophil recruitment. SETD2 may influence the functions of CD8+ T cell, CD4+ T cell and macrophages. SETD2 can suppress the Th17 cell development and promote iTreg cell polarization through phospholipid remodeling. One study shows that SETD2 mutation was associated with the efficacy of immunotherapy, this is due to higher tumor mutation burden [31]. It may be used as potential biomarker for cancer immunotherapy in future. SETD2 inactivation in cancer cells can sensitizes cancer cells to immune checkpoint inhibitors. SETD2 was included into a SIGP model to predict immunotherapy outcomes [32].
3. EZH2 and Prostate Cancer
3.1 Structure and function of EZH2
EZH2 is localized on chromosome 7q35 and contains 20 exons encoding 746 amino acids [33]. EZH2 is an important polycomb group (PcG) protein, which is closely associated with epigenetic regulation and epigenetically silences histones by modifying them during transcription. Initially, we discovered PcG proteins in Drosophila, where they play a negative regulatory role in gene expression during growth and development [34]. In mammals, PcG proteins form two major multiprotein complexes called polycomb repressive complex 1 (PRC1) and PRC2, EZH2 is a key enzyme that catalyzes the trimethylation (H3K27me3) of H3 lysine 27 (H3K27), while EZH2 is the catalytic subunit of PRC2, which mediates histone trimethylation [35-37]. H3K27me3 modification can lead to downstream tumor suppressor gene inhibition. The specific trimethylation of H3K27 by EZH2 may be attributed to the fact that EZH2 contains a c-terminal SET structural domain, which ensures that HMTase functions, leading to denser chromatin and epigenetic silencing of the target genes [38-41]. In addition to the methyltransferase activity, EZH2 can independently act as a transcription factor involved in epigenetic regulation [42-43]. As a major regulator in cell-cycle progression, autophagy, and apoptosis, EZH2 promotes DNA damage repair and inhibits cellular senescence [44-46]. Moreover, in gastric cancer cells, EZH2 can bind to the promoter region of tumor suppressor gene P21 and mediate H3K27me3 modification, resulting in the transcriptional silencing of P21, which, in turn, promotes the proliferation of gastric cancer cells. As P21 is the key tumor suppressor gene, its function inhibition prevents it from exerting the normal inhibitory effect on tumor cells, which ultimately leads to the abnormal proliferation of gastric cancer cells [47]. In addition to histones, EZH2 methylates non-histone proteins in a PRC2-dependent manner (Figure 2) [48].
Structure and function of Polycomb Repressive Complex 2 (PRC2). Schematic illustration of the function of enhancer of zeste homolog 2 (EZH2). Activation is indicated by arrows and suppression by blocking lines.
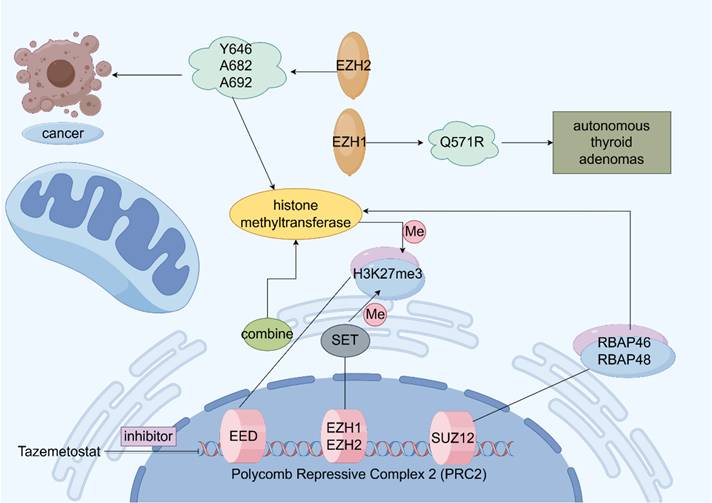
3.2 EZH2 and prostate cancer metastasis
Gain-of-function mutations or loss-of-function mutations may occur in EZH2, and both mutation type was involved in the development of prostate cancer. EZH2 functions as an epigenetic writer and AR coregulator, these activities were related to oncogenic activity of EZH2 [49]. EZH2 usually overexpressed in prostate cancer compared with normal prostate tissue. EZH2 mediate IFN-γ-JAK-STAT1 signaling pathway inactivation to promote prostate cancer progression [50]. EZH2 also involves in neuroendocrine prostate cancer progression, this is due to cooperation of EZH2 and N-Myc [51]. High expression of EZH2 was also correlated with high risk of recurrence of prostate cancer after radical prostatectomy. EZH2 can activate Ras and NF-κB pathway by epigenetically silencing DAB2IP [52]. EZH2 can regulate CDH1 gene expression to affect EMT process of prostate cancer cell. Enzalutamide can induce prostate cancer cell neuroendocrine differentiation via EZH2-STAT3 signaling pathway. One study showed that androgen deprivation can induce neuroendocrine differentiation using CREB-EZH2-TSP1 pathway in prostate cancer [53]. Another study demonstrated that T350 phosphorylated EZH2 act with AR to promote lineage plasticity and neuroendocrine differentiation [54]. MALAT1, a long-stranded non-coding RNA (lncRNA), is overexpressed in both prostate cancer (PCa) and castration-resistant prostate cancer (CRPC). Studies have demonstrated that MALAT1 binds to EZH2 [55] and plays a crucial role in recruiting EZH2 to tumor suppressor gene loci, thereby facilitating PCa cell migration and invasion [55]. One of the most prevalent genetic alterations in prostate cancer is the TMPRESS2-ERG fusion, caused by chromosomal translocation on chromosome 21, which has been shown to activate EZH2 expression [56-57]. EZH2 is frequently overexpressed in prostate cancer cells and interacts with several DNA methyltransferases (DNMT1, DNMT3A, DNMT3B), reinforcing its role in epigenetic silencing [58]. A pivotal cDNA microarray study in 2002 first established the association between EZH2 and prostate cancer, identifying it as the most upregulated gene in metastatic cases [59]. Notably, EZH2 overexpression is also detected in localized tumors with a high risk of recurrence after radical prostatectomy [59]. EZH2 can act as a coactivator for AR, and promote CRPC progression. Furthermore, EZH2 and BRCA1 cooperate to regulate prostate cancer stem cell phenotype and properties [60].
As a key epigenetic regulator, EZH2 is tightly controlled at transcriptional, translational, and post-translational levels. MicroRNAs (miRNAs) also regulate EZH2 expression post-transcriptionally. For instance, miR-101, miR-26a, and miR-26b —known to suppress EZH2—are significantly downregulated in prostate cancer [61-63]. EZH2 can block pro-apoptotic pathway and enhance the anti-apoptotic pathway to induce drug resistance. Loss-of-function mutations in EZH2 promoted resistance to the chemotherapeutic agent Ara C in acute myeloid leukemia. And EZH2 can induce c-Myc overexpression to induce cisplatin resistance in ovarian cancer. And EZH2 is involved in tamoxifen resistance by silencing the expression of GREB1. Inhibition of EZH2 functions can reverse drug resistance to some extent. EZH2 overexpression was one important cause of docetaxel and enzalutamide resistance in prostate cancer [64]. Inhibition of EZH2 can enhance the anti-tumor effect of metformin in prostate cancer. EZH2 can regulate the innate and adaptive immune systems of the tumor microenvironment, EZH2 was a driver of resistance to immunotherapies [65]. EZH2 overexpressed in tumor cells can inhibit T cell activation by upregulating PD-L1 expression. Otherwise, EZH2 suppression leads to increased activated CD8+ T cells, increased M1 tumor-associated macrophages and enhanced response to PD-1 therapy in prostate cancer. EZH2 also can regulate cytokines and chemokine in the tumor microenvironment [66]. EZH2 in cancer cells can regulate NK cell activation in the tumor microenvironment. EZH2 can change the tumor bone microenvironment by regulating the osteoblast and osteoclast when prostate cancer bone metastasis occurs [67]. Inhibition of EZH2 can positively activate the immune system toward tumor suppression.
4. Inhibitors
4.1 Inhibitors of SETD2
SETD2, a lysine N-methyltransferase and the sole enzyme responsible for H3K36 trimethylation, has emerged as a promising therapeutic target due to its role in epigenetic regulation and tumor suppression. It is implicated in various cancers, including multiple myeloma characterized by t (4,14) translocations [68]. A selective, potent, and orally bioavailable small-molecule SETD2 inhibitor, EZM0414, has recently shown promise in preclinical studies [68]. SETD2 loss is associated with aggressive forms of renal, gastric, colon, and pancreatic cancers [69], and it has been implicated in the development of aggressive gastrointestinal mesenchymal tumors [70]. In physiological processes, SETD2 inhibits intestinal epithelial damage by regulating oxidative stress-related factors and plays an inhibitory role in epithelial repair processes [71-72]. WEE1 is a class of inhibitory tyrosine kinases, and SETD2 deficiency reduces H3K36me3 levels during cellular physiology, leading to downregulation of the regulatory subunit of ribonucleotide reductase subunit M2 (RRM2) and a subsequent decrease in dNTP availability [73-74]. In preclinical experiments, adding a WEE1 kinase inhibitor inhibited the RRM2 expression level, leading to further depletion of the dNTP pool in SETD2-deficient cells (which serves as the single DNA unit), resulting in impaired DNA replication and cell death [75]. Recent studies have unveiled that renal cancer cells harboring SETD2 mutations or downregulated SETD2 expression are highly sensitive to histone demethylase inhibitors (HDMIs) in both in vitro and in vivo models [76-77]. Of note, a combination of HDMIs) and poly(adenosine diphosphate-ribose) polymerase inhibitors (PARPi, i.e., HMA + PARPi) exerted synergistic antitumor effects on cell growth by enhancing DNA damage and immune signaling, especially in DNA repair-deficient cancers, such as BRCA-mutated breast cancer [78-79]. PARP1 plays a crucial role in homologous recombination (HR)-mediated DNA double-strand break (DSB) repair, which contributes to cancer cell survival in a sustained DNA damage environment. PARPis block PARP1 functions, leading to the accumulation of cytotoxic DSBs and tumor cell death [80-81]. Given SETD2's key role in HR and DSB repair, its deficiency may sensitize cancer cells to PARP inhibition, positioning SETD2 as a compelling therapeutic target for such strategies [82-83].
4.2 Inhibitors of EZH2
EZH2 overexpression is a hallmark of various cancers and is associated with disrupted methylation patterns that promote tumorigenesis. By silencing tumor suppressor genes, EZH2 enhances cell proliferation, invasion, and progression of cancer cells [84-90]. In prostate and breast cancers, EZH2 is phosphorylated by AKT at Ser21, which suppresses its methyltransferase activity by reducing its affinity for histone H3 and leading to diminished H3K27me3 levels. This phosphorylation pEZH2-S21 also results in increased expression of EZH2-repressed genes [91]. EZH2 regulates numerous genes that contribute to prostate cancer development by transcriptionally inhibiting tumor metastasis. In prostate cancer, numerous genes have been identified as direct EZH2 targets and are associated with cancer progression and metastasis in the silenced state, which further confirms that EZH2 is a true oncogene. EZH2 can epigenetically silence the dysfunctional homology interaction protein (DAB2IP), a tumor suppressor involved in regulating Ras and NF-κB pathways, thereby driving tumorigenesis and metastasis in prostate cancer [92].
For more than two decades, the compound 3-deazepane-A (DZNep) has been widely recognized as a potent inhibitor of S-adenosine-L-homocysteine (SAH) hydrolase, a cofactor required for EZH2-dependent methylation. Subsequent studies have revealed that DZNep reduces intracellular EZH2 levels and inhibits H3K27me3 [93]. These findings established DZNep as the first compound to target EZH2. In vivo experiments demonstrated that DZNep selectively induces apoptosis in cancer cells without affecting normal cells [93-94]. Beyond its anti-proliferative activity, EZH2 inhibition has been shown to suppress migration and invasion of prostate cancer cells [94].
Tazemetostat (Tazverik), is an oral EZH2 inhibitor approved by the U.S. Food and Drug Administration (FDA), for the treatment of follicular lymphoma (FL) and epithelioid sarcoma (ES). In FL, EZH2 activity is enhanced through interactions between EZH2 mutants and wild-type EZH2 (WT EZH2) with oncogenes. Currently, clinical trials are investigating the combination of EZH2 inhibitors with other therapeutic modalities, including immunotherapy, conventional chemotherapy, and targeted therapies [95]. Notably, EZH2 inhibitors may exhibit synergistic effects when combined with immunotherapeutic or chemotherapeutic agents.
5. Signaling Pathways in Prostate Cancer
EZH2, as the catalytic core subunit of PRC2, is markedly overexpressed in prostate cancer. Moreover, the androgen receptor (AR) is a central therapeutic target in prostate cancer and remains highly expressed even in advanced disease stages [96]. AR expression is widespread in both primary and metastatic prostate cancer tissues. Importantly, EZH2 regulates AR protein levels and modulates the transcriptional activity of AR downstream target genes by directly interacting with AR [97]. Prostate cancer progression is regulated by the androgens testosterone and 5α-dihydrotestosterone, which exert their effects through binding to AR [98]. Ligand binding to AR induces a conformational change in the receptor, releasing auxiliary proteins and promoting AR dimerization. These dimers subsequently translocate to the nucleus and bind to androgen-responsive elements in the promoters of specific genes involved in cell proliferation and apoptosis suppression—key drivers of prostate cancer progression [99-100]. The interaction between AR signaling and the tumor microenvironment is highly complex, exhibiting both oncogenic and tumor-suppressive influences [101]. Stromal cells play a critical role in prostate development and cancer initiation, mediated in part by AR signaling. Remarkably, AR is undetectable in prostate epithelial tissues during early development, whereas its expression in stromal cells is significantly upregulated [102-103]. Consequently, androgen deprivation therapy (ADT) remains a frontline treatment for prostate cancer. Although ADT is initially effective against prostate cancer, resistance inevitably develops, leading to CRPC, which is associated with poor prognosis [104-106].
Recent studies have shown that the PI3K-AKT signaling pathway is significantly aberrantly activated in prostate cancer, particularly in CRPC, and its overactivation is strongly associated with disease progression [107-108]. Phosphatidylinositol 3-kinase (PI3K) enzymes, part of the lipid kinase superfamily, are categorized into three classes (I, II, and III) based on their substrate specificity and subunit composition. Their structural differences are predominantly reflected in the combination of catalytic and regulatory subunits [109-110]. Class IA PI3K catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), a key second messenger that activates downstream signaling pathways governing key biological processes such as cell proliferation, autophagy, and apoptosis [111]. PIP3 accumulation recruits and activates phosphatidylinositol-dependent kinase 1 (PDK1), which in turn phosphorylates AKT serine/threonine kinase at the Thr308 site. Activated AKT further phosphorylates several downstream effector molecules, such as FOXO transcription factors, glycogen synthase kinase 3β (GSK3β), nuclear factor κB (NF-κB), and tuberous sclerosis complex 2 (TSC2) [112-114]. For example, AKT phosphorylates TSC2, inhibiting its GTPase activity and thereby activating Ras homologous enriched enkephalin (RHEB), which in turn prevents the inhibitory effect of RHEB on mTORC1. AKT also suppresses cell autophagy by phosphorylating Unc-51-like autophagy-activating kinase 1 and ribosomal S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) to synergistically regulate cell growth and ribosome biosynthesis. S6K and 4EBP1 synergistically regulate cell growth, protein translation, and ribosome biosynthesis. Multi-omics analyses have uncovered extensive molecular abnormalities in the PI3K pathway in prostate cancer, including copy number variations, mutations, and transcriptional dysregulation. These abnormalities are present in up to 42% of primary tumor samples and are even more prevalent in metastatic lesions [115-117]. The PI3K-AKT-mTOR signaling axis—one of the most frequently deregulated pathways in prostate cancer—contributes to tumor growth and therapy resistance by regulating cell proliferation, survival, and metabolic reprogramming. Despite the development of various targeted agents, including pan-PI3K inhibitors, PI3K subtype-specific inhibitors, AKT inhibitors, mTOR inhibitors, and dual-target inhibitors, their clinical efficacy has been limited. Preclinical studies and early-phase clinical trials have highlighted challenges such as compensatory pathway activation, off-target toxicity, and the complex interplay between tumor cells and the microenvironment [118-120].
6. Conclusion
This paper focuses on prostate cancer, examining the epigenetic regulation of SETD2 and EZH2, their roles in metastasis, and the progress of related inhibitors. The discussion provides a multi-dimensional theoretical foundation and practical guidance for the diagnosis, treatment, and development of novel therapeutic strategies for prostate cancer.
SETD2, a critical HMTase, plays an essential and irreplaceable role in epigenetic regulation. In prostate cancer, the SETD2 expression level is significantly reduced and is closely associated with prostate cancer development and metastasis. Its downregulation leads to reduced H3K36me3 levels, disrupting the epigenetic balance and triggering abnormal protein methylation in vivo. This dysregulation promotes EMT, enhancing the migratory and invasive capacity of prostate cancer cells and ultimately facilitating metastasis. Additionally, SETD2 loss compromises the fidelity of gene transcription, which, combined with the dysregulation of key oncogenes, further contributes to tumorigenesis. With growing insight into SETD2's mechanisms, it has emerged as a promising therapeutic target. The development of small-molecule SETD2 inhibitors such as EZM0414, along with promising results from combination therapies, offers new avenues for prostate cancer treatment. Combination regimens involving SETD2 inhibitors with WEE1 inhibitors, HDMIs, or PARPis have demonstrated encouraging therapeutic efficacy in preclinical cancer models and are expected to form the basis for new treatment strategies for prostate cancer.
EZH2, the catalytic subunit of PRC2, functions as an oncogene in prostate cancer. It catalyzes H3K27 trimethylation, condensing chromatin and silencing gene expression. It also serves as a transcription regulator involved in diverse biological processes such as cell cycle control, autophagy, and apoptosis. EZH2 is commonly overexpressed in prostate cancer and interacts with various molecular partners. For instance, it binds to the lncRNA MALAT1, promoting PCa cell migration and invasion, and associates with DNA methyltransferases, modulating gene expression. EZH2 expression is tightly regulated at transcription, translation, and post-translational modification levels, with miRNAs playing a notable role in post-transcriptional control. EZH2 upregulation disrupts normal methylation patterns, silences tumor suppressor genes, promotes cell proliferation and invasion, and accelerates cancer progression. The compound DZNep was the first EZH2-targeting agent, known to reduce PRC2 complex protein levels, inhibit H3K27me3, and selectively induce apoptosis in cancer cells without affecting normal cells. Tazemetostat (Tazverik), a U.S. FDA-approved oral EZH2 inhibitor, is already in clinical use for the treatment of specific cancers. Ongoing trials are investigating EZH2 inhibitors in combination with immunotherapy, chemotherapy, and targeted agents. These combination therapies hold promise due to their potential synergistic effects and could offer new hope for patients with advanced disease.
EZH2 also contributes to prostate cancer development by directly interacting with AR, enhancing AR activity, and modulating the transcription of its downstream target genes. While the AR signaling pathway remains a central therapeutic target in prostate cancer, long-term reliance on ADT often leads to CRPC, which is associated with poor clinical outcomes. Concurrently, the PI3K-AKT-mTOR pathway is aberrantly activated in prostate cancer, driving tumor growth and treatment resistance by regulating proliferation, survival, and metabolic reprogramming. Despite the development of numerous inhibitors—targeting PI3K, AKT, mTOR, or combinations thereof—clinical success has been limited. Challenges include compensatory pathway activation, off-target effects, and the complexity of the tumor microenvironment. Consequently, while EZH2, AR, and PI3K-AKT-mTOR represent vital therapeutic targets, their intricate regulatory networks remain significant obstacles to effective treatment.
In summary, SETD2 and EZH2 play distinct but critical roles in prostate cancer development, progression, and metastasis. SETD2 functions as a tumor suppressor, and its loss promotes cancer progression and metastasis. By contrast, EZH2 acts as an oncogene, with its overexpression driving tumor aggressiveness. Deepening our understanding of their molecular mechanisms and advancing targeted therapies against these regulators are crucial for uncovering the pathogenesis of prostate cancer and enabling precision medicine. Future research should focus on exploring the complex regulatory networks of SETD2 and EZH2 in prostate cancer, refining current inhibitor-based treatment regimens, and enhancing combination treatment strategies to improve therapeutic outcomes and patient quality of life.
7. Future Directions
The study of epigenetics for carcinogenesis is becoming more and more promising. SETD2 and EZH2 are two important members of HMTs, and plays important roles in prostate cancer progression and metastasis. Mechanistically, SETD2 or EZH2 regulate prostate cancer metastasis and drug resistance warrant further study. And the effect of SETD2 or EZH2 on tumor microenvironment, especially tumor immune microenvironment needs to de deeply studied. Furthermore, tumor cell metabolism is a hot topic in recent years, and whether SETD2 or EZH2 influence tumor cell metabolism and its corresponding mechanism needs to be studied.
Acknowledgements
Funding
This work was financially supported through grants from the Natural Science Foundation of Shandong Province (Nos. ZR2023MH260 and ZR2017QH005), the National Natural Science Foundation of China (Grant No. 81803097), Doctoral Fund of Jining No.1 People's Hospital (2022-BS-002) and Key Research and Development Plan of Jining (2022YXNS113).
Consent to publication
All authors have consented to publication of this manuscript. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shin HJ, Hua JT, Li H. Recent advances in understanding DNA methylation of prostate cancer. Front Oncol. 2023;13:1182727
2. Gamallat Y, Felipe Lima J, Seyedi S, Li Q, Rokne JG, Alhajj R. et al. Exploring The Prognostic Significance of SET-Domain Containing 2 (SETD2) Expression in Advanced and Castrate-Resistant Prostate Cancer. Cancers (Basel). 2024;16(7):1436
3. Kumari S, Singh M, Kumar S, Muthuswamy S. SETD2 controls m6A modification of transcriptome and regulates the molecular oncogenesis of glioma. Med. Oncol. 2023;40(9):249
4. Battaglin F, Krause H, Elliot A, Abraham J, Soni S, Algaze S. et al. SETD2 gene expression and the molecular landscape of colorectal cancer (CRC). J. Clin. Oncol. 2023;41:4
5. Chen Z, Raghoonundun C, Chen W, Zhang Y, Tang W, Fan X. et al. SETD2 indicates favourable prognosis in gastric cancer and suppresses cancer cell proliferation, migration, and invasion. Biochem Biophys Res Commun. 2018;498(3):579-585
6. He J, Xu T, Zhao F, Guo J, Hu Q. SETD2-H3K36ME3: an important bridge between the environment and tumors. Front Genet. 2023;14:1204463
7. Yi Y, Li Y, Chen K, Cao Q. Unveiling the non-canonical functions of EZH2 in prostate cancer. Oncotarget. 2023;14:127-128
8. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343-349
9. Yi Y, Li Y, Meng Q, Li Q, Li F, Lu B. et al. A PRC2-independent function for EZH2 in regulating rRNA 2′-O methylation and IRES-dependent translation. Nat Cell Biol. 2021;23(4):341-354
10. Yi Y, Li Y, Li C, Wu L, Zhao D, Li F. et al. Methylation-dependent and -independent roles of EZH2 synergize in CDCA8 activation in prostate cancer. Oncogene. 2022;41(11):1610-1621
11. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y. et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(4):444-456
12. Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y. et al. SETD2 Restricts Prostate Cancer Metastasis by Integrating EZH2 and AMPK Signaling Pathways. Cancer Cell. 2020;38(3):350-365.e7
13. Yu M, Qian K, Wang G, Xiao Y, Zhu Y, Ju L. Histone methyltransferase SETD2: An epigenetic driver in clear cell renal cell carcinoma. Front Oncol. 2023;13:1114461
14. Bhattacharya S, Workman JL. Regulation of SETD2 stability is important for the fidelity of H3K36me3 deposition. Epigenetics Chromatin. 2020;13(1):40
15. Boulanger C, Haidara N, Yague-Sanz C, Larochelle M, Jacques PÉ, Hermand D. et al. Repression of pervasive antisense transcription is the primary role of fission yeast RNA polymerase II CTD serine 2 phosphorylation. Nucleic Acids Res. 2024;52(13):7572-7589
16. Kearns S, Mason FM, Rathmell WK, Park IY, Walker C, Verhey KJ. et al. Molecular determinants for α-tubulin methylation by SETD2. J Biol Chem. 2021;297(1):100898
17. Ryu HY. Histone Modification Pathways Suppressing Cryptic Transcription. Epigenomes. 2024;8(4):42
18. Nelson KK, Green MR. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc Natl Acad Sci U S A. 1990;87(16):6253-7
19. Molenaar TM, van Leeuwen F. SETD2: from chromatin modifier to multipronged regulator of the genome and beyond. Cell Mol Life Sci. 2022;79(6):346
20. Zhu K, Lei PJ, Ju LG, Wang X, Huang K, Yang B. et al. SPOP-containing complex regulates SETD2 stability and H3K36me3-coupled alternative splicing. Nucleic Acids Res. 2017;45(1):92-105
21. Bajusz D, Bognár Z, Ebner J, Grebien F, Keserű GM. Discovery of a Non-Nucleoside SETD2 Methyltransferase Inhibitor against Acute Myeloid Leukemia. Int J Mol Sci. 2021;22(18):10055
22. Skucha A., Ebner J., Grebien F. Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond. Int. J. Mol. Sci. 2019;20:1029
23. Zheng X, Lin J, Xiong J, Guan Y, Lan B, Li Y. et al. SETD2 variation correlates with tumor mutational burden and MSI along with improved response to immunotherapy. BMC Cancer. 2023;23(1):686
24. Weng Y, Xue J, Niu N. SETD2 in cancer: functions, molecular mechanisms, and therapeutic regimens. Cancer Biol Med. 2024;21(9):725-30
25. Niu N, Shen X, Zhang L, Chen Y, Lu P, Yang W. et al. Tumor Cell-Intrinsic SETD2 Deficiency Reprograms Neutrophils to Foster Immune Escape in Pancreatic Tumorigenesis. Adv Sci (Weinh). 2023;10(2):e2202937
26. Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G. et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543(7643):72-77
27. Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27(2):406-20
28. Kim IK, McCutcheon JN, Rao G, Liu SV, Pommier Y, Skrzypski M. et al. Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene. 2019;38(2):180-193
29. Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL. et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15(14):4622-9
30. Niu N, Shen X, Wang Z, Chen Y, Weng Y, Yu F. et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell. 2024;42(5):869-884.e9
31. Lu M, Zhao B, Liu M, Wu L, Li Y, Zhai Y. et al. Pan-cancer analysis of SETD2 mutation and its association with the efficacy of immunotherapy. NPJ Precis Oncol. 2021;5(1):51
32. Yu L, Gong C. Pancancer analysis of a potential gene mutation model in the prediction of immunotherapy outcomes. Front Genet. 2022;13:917118
33. Cardoso C, Mignon C, Hetet G, Grandchamps B, Fontes M, Colleaux L. The human EZH2 gene: genomic organisation and revised mapping in 7q35 within the critical region for malignant myeloid disorders. Eur J Hum Genet. 2000;8(3):174-80
34. Park SH, Fong KW, Mong E, Martin MC, Schiltz GE, Yu J. Going beyond Polycomb: EZH2 functions in prostate cancer. Oncogene. 2021;40(39):5788-5798
35. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343-9
36. Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011;2(9):e204
37. Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv. 2015;1(11):e1500737
38. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039-43
39. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893-905
40. Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111(2):197-208
41. Nouruzi S, Tabrizian N, Zoubeidi A. Beyond Expression: Role of Phosphorylated Residues of EZH2 in Lineage Plasticity in Prostate Cancer. Endocrinology. 2023;164(4):bqad023
42. Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H. et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27(14):5105-19
43. Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H. et al. Paf and EZH2 induce Wnt/betacatenin signaling hyperactivation. Mol Cell. 2013;52(2):193-205
44. Nutt SL, Keenan C, Chopin M, Allan RS. EZH2 function in immune cell development. Biol Chem. 2020;401(8):933-943
45. Yao Y, Hu H, Yang Y, Zhou G, Shang Z, Yang X. et al. Downregulation of Enhancer of Zeste Homolog 2 (EZH2) is essential for the Induction of Autophagy and Apoptosis in Colorectal Cancer Cells. Genes (Basel). 2016;7(10):83
46. Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018;22(13):3480-3492
47. Xu J, Wang Z, Lu W, Jiang H, Lu J, Qiu J. et al. EZH2 promotes gastric cancer cells proliferation by repressing p21 expression. Pathol Res Pract. 2019;215(6):152374
48. Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13(1):104
49. Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q. et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018;25(10):2808-2820.e4
50. Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014;8(1):204-16
51. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L. et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell. 2016;30(4):563-577
52. Bellazzo A, Di Minin G, Collavin L. Block one, unleash a hundred. Mechanisms of DAB2IP inactivation in cancer. Cell Death Differ. 2017;24(1):15-25
53. Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N. et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun. 2018;9(1):4080
54. Xin L. EZH2 accompanies prostate cancer progression. Nat Cell Biol. 2021;23(9):934-936
55. Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ. et al. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget. 2015;6(38):41045-55
56. Schulz WA, Hoffmann MJ. Epigenetic mechanisms in the biology of prostate cancer. Semin Cancer Biol. 2009;19(3):172-80
57. Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell. 2013;4(5):331-41
58. Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7(3):299-313
59. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624-9
60. Gorodetska I, Lukiyanchuk V, Peitzsch C, Kozeretska I, Dubrovska A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int J Cancer. 2019;145(11):2974-2985
61. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D. et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834-8
62. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257-61
63. Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27(12):1788-93
64. Bai Y, Zhang Z, Cheng L, Wang R, Chen X, Kong Y. et al. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J Biol Chem. 2019;294(25):9911-9923
65. Mortezaee K. EZH2 regulatory roles in cancer immunity and immunotherapy. Pathol Res Pract. 2025;270:155992
66. Sun S, Yu F, Xu D, Zheng H, Li M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim Biophys Acta Rev Cancer. 2022;1877(2):188700
67. Zhang L, Qu J, Qi Y, Duan Y, Huang YW, Zhou Z. et al. EZH2 engages TGFβ signaling to promote breast cancer bone metastasis via integrin β1-FAK activation. Nat Commun. 2022;13(1):2543
68. Alford JS, Lampe JW, Brach D, Chesworth R, Cosmopoulos K, Duncan KW. et al. Conformational-Design-Driven Discovery of EZM0414: A Selective, Potent SETD2 Inhibitor for Clinical Studies. ACS Med Chem Lett. 2022;13(7):1137-1143
69. Chen R, Zhao WQ, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer. 2020;11(11):3349-3356
70. Huang KK, McPherson JR, Tay ST, Das K, Tan IB, Ng CC. et al. SETD2 histone modifier loss in aggressive GI stromal tumours. Gut. 2016;65(12):1960-1972
71. Liu M, Rao H, Liu J, Li X, Feng W, Gui L. et al. The histone methyltransferase SETD2 modulates oxidative stress to attenuate experimental colitis. Redox Biol. 2021;43:102004
72. Li X, Liu C, Zhu Y, Rao H, Liu M, Gui L. et al. SETD2 epidermal deficiency promotes cutaneous wound healing via activation of AKT/mTOR Signalling. Cell Prolif. 2021;54(6):e13045
73. Elbæk CR, Petrosius V, Sørensen CS. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat Res. 2020;819-820:111694
74. Maldonado E, Rathmell WK, Shapiro GI, Takebe N, Rodon J, Mahalingam D. et al. A Phase II Trial of the WEE1 Inhibitor Adavosertib in SETD2-Altered Advanced Solid Tumor Malignancies (NCI 10170). Cancer Res Commun. 2024;4(7):1793-1801
75. Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G. et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell. 2015;28(5):557-568
76. Li HT, Jang HJ, Rohena-Rivera K, Liu M, Gujar H, Kulchycki J. et al. RNA mis-splicing drives viral mimicry response after DNMTi therapy in SETD2-mutant kidney cancer. Cell Rep. 2023;42(1):112016
77. Zhou X, Sekino Y, Li HT, Fu G, Yang Z, Zhao S. et al. SETD2 Deficiency Confers Sensitivity to Dual Inhibition of DNA Methylation and PARP in Kidney Cancer. Cancer Res. 2023;83(22):3813-3826
78. Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y. et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer Cell. 2016;30(4):637-650
79. Baer MR, Kogan AA, Bentzen SM, Mi T, Lapidus RG, Duong VH. et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin Cancer Res. 2022;28(7):1313-1322
80. Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI. et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518(7538):258-62
81. Li H, Tu J, Zhao Z, Chen L, Qu Y, Li H. et al. Molecular signatures of BRCAness analysis identifies PARP inhibitor Niraparib as a novel targeted therapeutic strategy for soft tissue Sarcomas. Theranostics. 2020;10(21):9477-9494
82. Li F, Mao G, Tong D, Huang J, Gu L, Yang W. et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153(3):590-600
83. Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ. et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7(6):2006-18
84. Tonini T, D'Andrilli G, Fucito A, Gaspa L, Bagella L. Importance of Ezh2 polycomb protein in tumorigenesis process interfering with the pathway of growth suppressive key elements. J Cell Physiol. 2008;214(2):295-300
85. Chen Y, Xie D, Yin Li W, Man Cheung C, Yao H, Chan CY. et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett. 2010;297(1):109-16
86. Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;21(2):209-20
87. Zoroddu S, Marchesi I, Bagella L. PRC2: an epigenetic multiprotein complex with a key role in the development of rhabdomyosarcoma carcinogenesis. Clin Epigenetics. 2021;13(1):156
88. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323-35
89. Liu L, Xu Z, Zhong L, Wang H, Jiang S, Long Q. et al. Enhancer of zeste homolog 2 (EZH2) promotes tumour cell migration and invasion via epigenetic repression of E-cadherin in renal cell carcinoma. BJU Int. 2016;117(2):351-62
90. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338(6113):1465-9
91. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT. et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310(5746):306-10
92. Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280(23):22437-44
93. Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL. et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050-63
94. Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE. et al. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer. 2011;10:40
95. Eich ML, Athar M, Ferguson JE 3rd, Varambally S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020;80(24):5449-5458
96. Liu Q, Wang G, Li Q, Jiang W, Kim JS, Wang R. et al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int J Cancer. 2019;145(2):415-426
97. Guo L, Luo X, Yang P, Zhang Y, Huang J, Wang H. et al. Ilicicolin A Exerts Antitumor Effect in Castration-Resistant Prostate Cancer Via Suppressing EZH2 Signaling Pathway. Front Pharmacol. 2021;12:723729
98. Aurilio G, Cimadamore A, Mazzucchelli R, Lopez-Beltran A, Verri E, Scarpelli M. et al. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells. 2020;9(12):2653
99. Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem. 2006;99(2):333-44
100. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20(13):3001-15
101. McAllister MJ, Underwood MA, Leung HY, Edwards J. A review on the interactions between the tumor microenvironment and androgen receptor signaling in prostate cancer. Transl Res. 2019;206:91-106
102. Hayward SW, Haughney PC, Lopes ES, Danielpour D, Cunha GR. The rat prostatic epithelial cell line NRP-152 can differentiate in vivo in response to its stromal environment. Prostate. 1999;39(3):205-12
103. Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD. et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61(22):8135-42
104. Crawford ED, Heidenreich A, Lawrentschuk N, Tombal B, Pompeo ACL, Mendoza-Valdes A. et al. Androgen-targeted therapy in men with prostate cancer: evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019;22(1):24-38
105. Culig Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr Mol Biol Rep. 2017;3(4):230-235
106. Rice MA, Malhotra SV, Stoyanova T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front Oncol. 2019;9:801
107. Crumbaker M, Khoja L, Joshua AM. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers (Basel). 2017;9(4):34
108. Pearson HB, Li J, Meniel VS, Fennell CM, Waring P, Montgomery KG. et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018;8(6):764-779
109. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075-83
110. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329-41
111. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627-44
112. Gagliardi PA, Puliafito A, Primo L. PDK1: At the crossroad of cancer signaling pathways. Semin Cancer Biol. 2018;48:27-35
113. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169(3):381-405
114. Berenjeno IM, Piñeiro R, Castillo SD, Pearce W, McGranahan N, Dewhurst SM. et al. Oncogenic PIK3CA induces centrosome amplification and tolerance to genome doubling. Nat Commun. 2017;8(1):1773
115. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239-43
116. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM. et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-1228
117. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I. et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428-11436
118. de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A. et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res. 2019;25(3):928-936
119. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15(5):273-291
120. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int J Mol Sci. 2020;21(12):4507
Author contact
![]() Corresponding author: Haiyan Zhang, Zoucheng People's Hospital, Zoucheng, Shandong 272000, P.R. China; Tel./Fax: 0086 537 5250811; Email: baby530319196com. Xiangyu Zhang, Department of Pathology, Jining No.1 People's Hospital, Jining, Shandong 272000, P.R. China; Tel./Fax: 0086 537 6626309; Email: zhangxiangyu666com.
Corresponding author: Haiyan Zhang, Zoucheng People's Hospital, Zoucheng, Shandong 272000, P.R. China; Tel./Fax: 0086 537 5250811; Email: baby530319196com. Xiangyu Zhang, Department of Pathology, Jining No.1 People's Hospital, Jining, Shandong 272000, P.R. China; Tel./Fax: 0086 537 6626309; Email: zhangxiangyu666com.