Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(11):3425-3449. doi:10.7150/jca.108163 This issue Cite
Review
Strategies to Overcome PD-1/PD-L1 Blockade Resistance: Focusing on Combination with Immune Checkpoint Blockades
Dinglin Liu1,2#, Haoyue Xiao2#, Ying Xiang1,2 ![]() , Dian Zhong2, Yuchen Liu2, Yunfei Wang2, Weijia Zhang1
, Dian Zhong2, Yuchen Liu2, Yunfei Wang2, Weijia Zhang1 ![]()
1. Department of Oncology, First Affiliated Hospital of Yangtze University, Jingzhou, Hubei 434023, China.
2. Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, 1 Nanhuan Road, Jingzhou, Hubei 434023, China.
# The authors contributed to this work equally.
Received 2024-12-4; Accepted 2025-6-27; Published 2025-7-24
Abstract

In recent years, immune checkpoint blockades (ICBs) have made rapid progress in the field of cancer treatment, providing significant therapeutic effects and survival benefits, especially in patients with advanced refractory tumors. PD-1/PD-L1 blockade is one of the most widely used ICBs. However, its application is limited by low response rate and drug resistance. It is of great significance to investigate the complex mechanisms of PD-1/PD-L1 blockade resistance. In this review, we outline some crucial aspects, including lack of effector T cells, lack of target PD-1/PD-L1, poor immunogenicity of tumors, immunosuppressive TME, and other mechanisms (such as metabolism, epigenetic alterations, and gut microbiota). Combination therapy has become a promising strategy to overcome drug resistance. Based on the upregulation of other immune checkpoints after PD-1/PD-L1 blockade treatment, we focus on the combination with other ICBs, including CTLA-4, TIM-3, LAG-3, TIGIT, VISTA, and some emerging immune checkpoints, so as to provide evidence for improving the benefit of ICBs in cancers.
Keywords: PD-1, PD-L1, immune checkpoint blockade, resistance, combination therapy
Introduction
For a long time, various strategies have been employed to boost the immune response to fight against cancer. However, the frequent adverse effects associated with these treatments highlight their limitations. Notably, over the past decade, the blockade of programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) has transcended the limitations of previous cancer immunotherapy [1]. Specifically, this innovative treatment is capable of defending against tumors by restoring the innate anti-tumor immune response with a minor or no increase in adverse effects, a concept described as “Normalized Cancer Immunotherapy” [2]. Mechanistically, malignant tumors may have the ability to stimulate the expression of various immune checkpoints, such as PD-1 and PD-L1, thereby inhibiting the normal activation of T cells within the tumor microenvironment (TME), which ultimately allows the tumor to escape immune attack. Through reactivating immune cells, immune checkpoint therapy can achieve immune normalization without excessively amplifying the immune response, thereby reducing the occurrence of severe toxic effects [3].
The first antibody against PD-1 (nivolumab) was approved by the FDA in 2014. Since then, blocking antibodies against PD-1 or PD-L1 (anti-PD-(L)1) have been approved for application in multiple tumors. Additionally, clinical trials have demonstrated that PD-1/PD-L1 blockade can provide a durable clinical response in specific tumor types and patient populations and may lead to long-term tumor non-progression after treatment discontinuation in some patients, thereby potentially enhancing overall survival [4, 5]. However, the efficacy is confined to a small portion of individuals. Many patients quickly acquire resistance and experience different immune-related adverse events to some extent [6, 7]. Furthermore, the lack of effective biomarkers makes it challenging to predict which patients will benefit from anti-PD-1/PD-L1 treatment [8]. Additionally, the human immune system is a dynamic environment, contributing to the considerable challenges of immunotherapy [9]. Even if there are many challenges, on the whole, for patients with late-stage malignant tumors that have undergone extensive metastasis or are unresponsive to traditional anti-cancer therapies (radiation, chemotherapy, surgery, targeted therapies), immunotherapy provides a promising and innovative option.
Considering the low response rate to monotherapy and the issue of resistance, some experts propose that the combinations of multiple immune checkpoint blockades (ICBs) might expand the group of patients benefiting from treatment, lengthen the objective response rate, and reduce the occurrence of resistance. This has been supported by the findings from multiple preclinical and clinical trials [10]. The current review delves into the molecular mechanisms of PD-1/PD-L1 blockade resistance, emphasizing the synergistic anti-tumor effects of anti-PD-1/PD-L1 combined with other ICBs, so as to provide evidence for improving the benefits of ICBs in cancers.
PD-1 Pathway and Clinical Applications in Cancer Therapy
Structure and function of the PD-1 pathway
PD-1 (CD279) was discovered by Tasuku Honjo and his team in 1992, who found that PD-1 levels increased during a classic type of programmed cell death process in mouse T cell hybridomas [11]. Later on, PD-1 was recognized as an immune checkpoint that not only negatively regulated the peripheral immune response but also participated in maintaining immune tolerance [12]. Structurally, PD-1 belongs to the CD28 family and is a transmembrane glycoprotein with the extracellular domain, the transmembrane segment, and the cytoplasmic tail domain. The cytoplasmic tail domain contains two motifs: an immune receptor tyrosine-based inhibitory motif (ITIM) and an immune receptor inhibitory tyrosine-based switch motif (ITSM). The ITSM motif is the key structure that mediates the suppression of immune response [13].
PD-1 pathway not only includes PD-1 but also its ligands, PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273). Among these, PD-L1, as the primary ligand for PD-1, is commonly upregulated in tumor cells and also expressed in B cells, T cells, dendritic cells, macrophages, bone marrow-derived mast cells, and some non-immune cells [14]. In contrast, PD-L2, as the secondary ligand for PD-1, is more confined in antigen-presenting cells (APCs, for example, macrophages and dendritic cells) stimulated by cytokines, and can also be induced in other immune cells, non-immune cells, and tumor cells [15]. Mechanistically, the interaction between PD-1 and its ligands is mainly mediated by the tyrosine phosphatase SHP-2, which dephosphorylates signaling molecules downstream of the T-cell receptor (TCR), thereby blocking the activation of T-cell activation [1]. Furthermore, PI3K/Akt/mTOR signaling pathway and RAS/MEK/ERK signaling pathway are the main downstream of PD-1 and its ligands, both of which are associated with the decreased T cell function and immunosuppression [16].
Cancer immune evasion via PD-1/PD-L1
PD-1/PD-L1 pathway, involved in protecting cells against T cell attack, is considered to be one of the major mechanisms of tumor immune escape. The PD-L1 expression levels in various types of cancers have been confirmed to be correlated with negative outcomes [17, 18]. Mechanistically, tumor cells upregulate the expression of PD-L1, and subsequently, the overexpressed PD-L1 binds to and transmits inhibitory signals to PD-1-expressing T cells, especially CD8+ T cells, thus evading the attack of the immune system [19].
The expression of PD-L1 in tumors is mainly regulated by inflammatory mediators, and interferon (IFN)-γ is a notable one. Paradoxically, to fight tumor cells, anti-tumor immune cells secrete IFN-γ, but IFN-γ, in turn, induces the expression of genes (such as PD-L1) involved in tumor immune evasion [20]. The natural expression of the PD-L1 protein is limited to specific cancer tissues, which is induced by IFN-γ in the TME [21]. However, when tumor cells that do not naturally express PD-L1 protein are treated with IFN-γ, most of them are induced to express PD-L1 protein [22]. PD-L1 expression is negative in the majority of cancer cell lines cultured in vitro. When melanoma cells were implanted into IFN-γ-deficient mice, the unsuccessful upregulation of PD-L1 demonstrated that IFN-γ was required for PD-L1-induced expression in tumor cells [23]. On the one hand, IFN-γ can upregulate the expression of major histocompatibility complex (MHC)-I and promote T cell differentiation, thereby enhancing anti-tumor immune response. On the other hand, induced PD-L1 expression by IFN-γ also helps tumors achieve immune evasion by binding with the PD-1 on T cells [21].
Cancer immunotherapy with PD-1/PD-L1 blockade
To date, many anti-PD-1 antibodies (Abs) and anti-PD-L1 Abs have been developed to block PD-1/PD-L1 signaling. Anti-PD-1 Abs (nivolumab, pembrolizumab, and cemiplimab) and anti-PD-L1 antibodies (atezolizumab, avelumab, and durvalumab) have been approved by FDA for some solid tumor and hematologic cancers [24]. Some clinical trials have confirmed the anti-tumor efficacy of PD-1/PD-L1 blocking therapies and have consistently demonstrated clinical therapeutic benefits across a wide range of cancer types [25-27]. Mechanistically, blocking PD-1/PD-L1 led to increased proliferation of CD8+ T cells [28]. Another research showed that the reinvigoration of CD8+ T cells after anti-PD-1 therapy was associated with clinical outcomes [29]. Patients with tumor-infiltrating CD8+ T cells exhibited a greater response to anti-PD-1 treatment [30]. PD-1 blocking therapy could increase CD8+ T cells in the peripheral blood of patients with non-small cell lung cancer (NSCLC) [31]. PD-1 blocking could not only strengthen the activity of T cells that target cancer cells but also boost the activity of other immune cells in the TME, such as NK cells and B cells [32].
However, researchers and clinicians also pay attention to the immune-related adverse events associated with checkpoint blockade treatment and strive to balance the risks and benefits [33].
Immune-related adverse events associated with PD-1/PD-L1 blockade
Although PD-1/PD-L1 blockades exhibit anti-tumor effects by activating the immune system, they also lead to an attack of the immune system on normal tissues, and these types of drug-related adverse reactions, mediated by immune mechanisms and which can involve different systems, are referred to as “immune-related adverse events (irAEs)”. The most common irAEs for PD-1/PD-L1 blockade are endocrine (thyroid disorders such as hypothyroidism and hyperthyroidism), gastrointestinal (diarrhea, colitis, nausea), lung (pneumonitis), skin (rash, pruritus, and vitiligo) and musculoskeletal (arthralgia, arthritis, and myalgia), and constitutional symptoms (fatigue, pyrexia, and anorexia) [34], as shown in Figure 1. Approximately 20% of patients with PD-1/PD-L1 blockade therapy develop some mild version of gastrointestinal inflammation, with 2-5% developing more severe inflammation [35]. Endocrine toxicities are also common with PD-1/PD-L1 blockade therapy. Clinically significant thyroiditis (hypothyroidism) occurs in 8% of patients on PD-1/PD-L1 blockade. Other endocrine toxicities, including autoimmune diabetes and adrenal insufficiency, are rare but are extremely important to recognize because they can be deadly [36]. A recent study found that α-myosin was a direct target of cytotoxic CD8+ T cells, and α-myosin reactive cells could be expanded from the peripheral blood of patients with immune checkpoint blockade-induced myocarditis [37].
The most clinically important immune-related adverse events (irAEs) for PD-1/PD-L1 blockade therapy. IrAEs affect multiple systems and organs, leading to hypothyroidism, hyperthyroidism, pneumonitis, autoimmune diabetes, rash, vitiligo, myocarditis, diarrhoea, colitis, arthralgia, arthritis, myalgia, etc. (By Figdraw.)
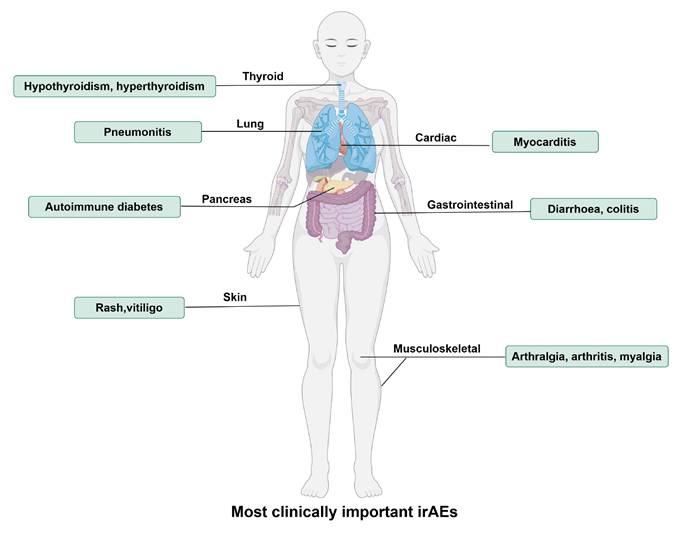
In a meta-analysis regarding PD-1/PD-L1 blockade therapies, among 6,507 patients, 1,111 (17.1%) experienced irAEs of any grade. Among 4,921 patients, 196 (4.0%) experienced irAEs of Grade 3 or higher. Moreover, compared with the use of PD-L1 antibodies, the risk of irAEs occurrence might be higher with the use of PD-1 antibodies [38]. Compared to anti-PD-L1, anti-PD-1 therapies are more frequently associated with pneumonitis (2.4% vs 0%), rash (12.2% vs 5.5%), vitiligo (4.0% vs 0%), colitis (0.7% vs 0%), hepatitis (0.4% vs 0%), hypothyroidism (5.1% vs 2.2%), hyperthyroidism (1.6% vs 0%), and anaemia (4.8% vs 0.7%) [34]. Current conventional therapy for irAEs includes discontinuation of immune checkpoint blockades and administration of glucocorticoids or infliximab [39].
Mechanisms of PD-1/PD-L1 Blockade Resistance
Although the PD-1 blockade therapy has shown enormous potential in cancers, in fact, only a small number of patients benefit from its application. For example, when applied to treat advanced recurrent ovarian cancers, PD-1 blockade showed low objective response rates [40]. The unsatisfactory response rate limits the application in clinical settings.
The key to the success of PD-1 blocking therapy lies in whether there is resistance to it. Therefore, it is essential to investigate the mechanisms of resistance and low response rates so as to discuss relevant strategies effectively. Based on the clinical response of patients and in order to provide better clinical guidance, the Society for Immunotherapy of Cancer has defined three types of resistance to anti-PD-(L)1 treatment: (1) Primary resistance. It describes disease progression in patients who have been exposed to PD-(L)1 checkpoint inhibitor for at least 6 weeks and have a stable disease period (SD) < 6 months. (2) Secondary resistance. It occurs in patients who have received anti-tumor treatment and have documented, confirmed objective response or extended SD (> 6 months) and then later progress despite continued treatment. (3) The resistance that develops after discontinuation of therapy. It is mainly used to consider other adjuvant therapies after stopping anti-PD-1 treatment and to weigh risk and maximum benefit when deciding whether to cease treatment [41].
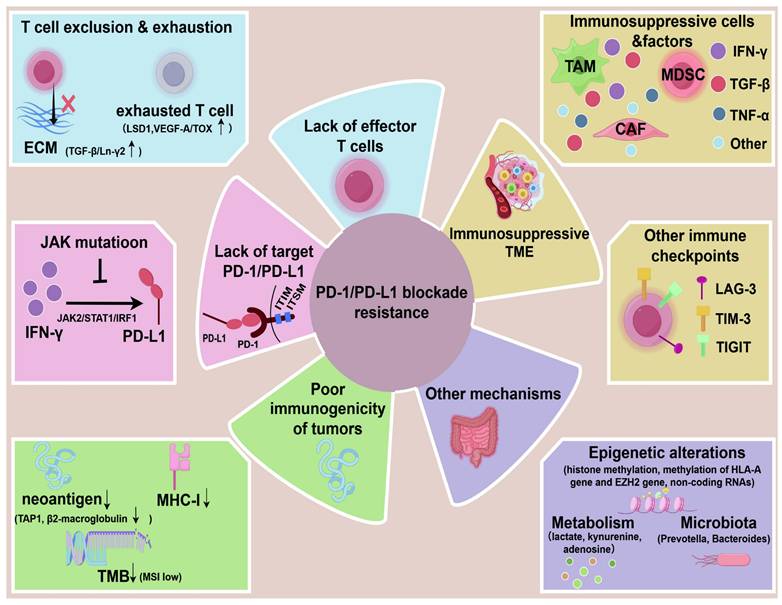
Some scientists have classified the immune resistance mechanisms in the TME into direct, indirect, or other mechanisms [42]. Direct resistance is defined as the lack of one or two necessary targets for anti-PD1 therapy: PD-L1 expression and tumor-infiltrating lymphocytes (TILs). For example, compared to type Ⅱ (PD-L1+TIL+), the types Ⅰ, Ⅲ, and Ⅴ of melanoma may not benefit much from anti-PD1 therapy. It is also referred to as “target-missing” resistance [10]. Indirect resistance is non-specific and not unique to anti-PD1 treatments but may be part of the resistance mechanisms in all tumors, such as antigen loss and lack of effective antigen presentation [42]. Additionally, there are some other novel mechanisms, such as the gut microbiota, epigenetics, and metabolism. Here, we propose some crucial aspects as below, and also illustrate in Figure 2.
The mechanisms of PD-1/PD-L1 blockade resistance. The main mechanisms include lack of target PD-1/PD-L1(e.g. JAK mutation in tumor inhibit PD-L1 induction by IFN-γ), lack of effector T cells (T cell exclusion and exhaustion), poor immunogenicity of tumors (low neoantigen, low MHC-I, low tumor mutation burden), immunosuppressive TME (immunosuppressive cells, factors, other immune checkpoints), other mechanisms (metabolism, epigenetic alterations, microbitota). (By Figdraw.)
Lack of effector T cells
T cell exclusion
Clinically, some “cold” tumors, such as ovarian, prostatic, and pancreatic cancers, show low response to anti-PD-(L)1 therapy. These cold tumors can be immune-desert or immune-excluded. The former lacks T cell infiltration into the tumor and its surroundings, while the latter is defined by T cells being trapped around the tumor periphery or within the stroma, preventing them from sufficient infiltration, which results in a low level of T cells within the tumor [43]. Critically, to achieve an effective anti-tumor immune response, there must be sufficient T cell infiltration, making direct physical contact with the tumor [44]. Generally, T cell infiltration into the TME correlates with a better prognosis for some cancers. It was found that the 5-year overall survival rate of ovarian cancer patients with T cell infiltration was much higher (38.0%) than those lacking T cell infiltration (4.5%) [45].
Several mechanisms are involved in T cell exclusion, including physical barriers within the tumor stroma, overexpression of TGF-β, and the accumulation of harmful metabolic products within the TME [43]. Notably, the stromal cells within the TME contribute to the T cell exclusion, which include cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and cancer-associated mesenchymal stem cells (CA-MSCs) [44, 46]. TGF-β can drive the T cell exclusion within tumors. On the one side, exposure to TGF-β reduces MHC-I expression on cancer cells, while suppression of TGF-β can recover MHC-I expression. On the other side, TGF-β stimulation can activate CAFs, thereby improving the extracellular matrix (ECM), a physical barrier restricting T cell infiltration [47]. Moreover, the TGF-β1 signaling derived from CAFs leads to T cell exclusion by upregulating the expression of Ln-γ2, a subunit of laminin, which is a key component of the ECM, thereby constructing a protective barrier for the tumor. This barrier blocks the immune cells' infiltration into the tumor and then reduces the efficacy of anti-PD-1 therapy [48]. CA-MSCs can also promote the T cell exclusion. It was confirmed that there was a reverse relationship between CA-MSCs and the CD8+ T cell infiltration. CA-MSCs promoted CD8+ T cell exclusion within the TME by secreting various chemokines (e.g., CCL2, CX3CL1, and TGF-β1) and restricted T cells within the stroma to reduce the efficacy of PD1 blockade. Under certain situations, CA-MSCs can also differentiate into CAFs [46]. Additionally, it was found that the activation of the β-catenin pathway was associated with T cell exclusion in mice with melanoma [49], which was thought to increase PD-1/PD-L1 blockade resistance.
T cell exhaustion
T cell exhaustion is a form of dysfunction that T cells gradually acquire when continuously exposed to antigen stimulation. It is a special type of T cells with low reactivity characterized by the loss of effector function (reduced production of IL-2, TNF-α, and IFN-γ, etc.) and the overexpression of various inhibitory receptors on the cell surface (PD-1, CTLA-4, TIM-3, etc.). Blocking these inhibitory receptors can partially reverse T cell exhaustion [50]. However, PD-1 blocking therapy can also induce T cell exhaustion, which may be one of the reasons for resistance. It was found that anti-PD-1 therapy promoted the progenitor exhausted CD8+ T cells' proliferation and differentiation in vivo, ultimately leading to an increase in terminally exhausted cells but short lifespans [51]. Additionally, it was revealed that increased collagen levels in lung tumors were involved in PD-(L)1 blockade resistance. This process was associated with the leukocyte-associated immunoglobulin (Ig)-like receptor (LAIR1), inhibiting lymphocytes through SHP-1 signaling. The reduction of tumor collagen deposition increased T cell infiltration, diminished T cell exhaustion, and eliminated resistance to anti-PD-L1 therapy [52]. Another study discovered immunosuppressive CD10+ALPL+ neutrophils mediated resistance to anti-PD-1 immunotherapy by inducing an irreversible T cell exhaustion [53]. The lysine-specific demethylase 1 (LSD1) could inhibit the progenitor pool and promote T cell exhaustion within the TME by antagonizing TCF1-mediated transcription. Inhibition of LSD1 could enhance the progenitor phenotype, thereby promoting a sustained response to anti-PD-1 and avoiding resistance [54]. In addition, researchers found that in microsatellite stable colorectal cancers that were lowly responsive to PD-1 blockade, VEGF-A induced the expression of transcription factor TOX-mediated T cell exhaustion. T cell exhaustion could be recovered after knocking down TOX, and the combination with anti-VEGF-A treatment could improve the response to PD-1 blockade therapy [55].
Lack of target PD-1/PD-L1
During tumor progression, once tumor cells express tumor antigens that can be recognized by immune cells, especially T cells, they will face the immune attack. To escape this immune attack, PD-1 and PD-L1 are upregulated under TME stimulation, which contribute to the resistance to immune response and immune surveillance evasion. Currently, the prevailing view is that PD-1 protein is rapidly expressed on activated effector T cells by TCR stimulation and is also regulated by various factors and pathways like TGF-β, IL-12, IL-6, IFN-α, TNF-α, etc[56]. PD-L1 protein is seldom constitutively expressed in normal tissues and cultured tumor cell lines, but it can be found in the majority of cancer specimens, suggesting a latent role of the TME in regulating PD-L1 expression [22]. Specifically, PD-L1 is selectively upregulated within the TME by IFN-γ released from T cells, which may be due to various mechanisms, such as IFN-γ leads to the activation of JAK2/STAT1/IRF1, which subsequently upregulates PD-L1 expression [21]. However, the signaling pathways through which IFN-γ induces PD-L1 differ among various tumor types [56].
Mechanistically, PD-1 and PD-L1 blockades exert anti-tumor effects by inhibiting the negative regulatory effects on T cells via binding with PD-1 and PD-L1, respectively. Therefore, the expression of PD-1 or PD-L1 is indispensable for this therapy. A study indicated that melanoma patients with high tumor burden and JAK1/2 mutations are insensitive to PD-1/PD-L1 blockade, partly because the JAK1/2 mutation hindered the adaptive upregulation of PD-L1 when exposed to IFN-γ[57]. Consistently, patients with melanoma who are both PD-L1 positive and TILs positive are the most effective to PD-1 blockade therapy [58]. Currently, it is imperative to identify specific patient types before initiating anti-PD-(L)1 therapy to avoid ineffective treatment. For instance, patients with PD-L1 negative may need to consider other immunotherapies in addition to PD-1/PD-L1 blockade [10]. Although PD-L1 positive is suggestive for therapeutic efficacy, it is still worth noticing that the expression levels of PD-L1 are incorrectly measured sometimes due to tumor heterogeneity, dynamic changes of PD-L1, lack of validated biomarkers, and inaccurate biopsy sampling [8, 59].
Although PD-L1 positive expression is considered as a marker of better response to anti-PD-(L)1 therapy, PD-L1 in exosomes may contribute to resistance to anti-PD-(L)1 therapy. Higher levels of circulating exosomal PD-L1 before treatment are negatively correlated with the response, indicating T cell exhaustion at a stage that is irreversible by anti-PD-(L)1 therapy. Thus, the levels of exosomal PD-L1 can distinguish patients who will respond to anti-PD-(L)1 treatment [60]. It was found that either the exosome inhibitor GW4869 or the suppression of Rab27a to reduce exosome release improved the efficacy of anti-PD-L1 treatment [61]. This might be due to the fact that exosomal PD-L1 depleted antibodies, prevented the availability of surplus antibodies to inhibit PD-L1, which eventually affected the efficacy of anti-PD-L1 therapy [62].
Poor immunogenicity of tumors
It is well known that antigens that are normally presented do not lead to any immune attack. However, when anomalies such as DNA mutations happen and these antigens are presented on the cell surface, immune cells could identify the tumor cells, eliciting the anti-tumor response. The tumor immunogenicity influences the identification of T cells. Therefore, the tumor immunogenicity is crucial to the efficacy of PD-1/PD-L1 blockade.
Loss of neoantigens and antigen presentation
“Tumor immunoediting” includes three stages: immune elimination, immune equilibrium, and immune escape. At the immune elimination stage, T cells eliminate tumor cells that strongly express antigens. In contrast, at the immune evasion stage, in the face of intense selective pressure by the immune system, tumors may evade attack from the immune system by losing the expression of antigens [63]. To trigger an effective anti-tumor immune response, two crucial steps are necessary. Firstly, tumor cells process and present tumor antigens via MHC-I. Secondly, APCs take up the antigen and mediate cross-presentation to activate CD8+ T cells [64].
Neoantigens, as a type of tumor-specific antigen, can enhance the sensitivity to immune checkpoint blockade therapy, while the deficiency often leads to immune escape. Cancers with abundant mutated neoantigens show increased sensitivity to PD-1 inhibitors [65]. Many mechanisms can influence antigen presentation, such as regulating the function of dendritic cells, HLA expression levels, and other genes' expressions that are involved in antigen presentation. It was found that downregulated MHC-I may be a marker of resistance to PD-1 blockade [66]. Furthermore, there is an opinion that a dysfunction within the components of the Antigen Processing Machinery (APM), such as low expression or silencing of TAP1, β2-microglobulin, or HLA-A, HLA-B, and HLA-C, may hinder the presentation of neoantigens on the cell surface and also affect the infiltration of CD8+ cells [67]. A study found that Nintedanib enhanced the efficacy of PD-L1 blockade therapy by increasing the levels of PD-L1 and MHC-1 expressed on tumor cells [68]. Another research discovered that the EZH2 inhibition enhanced the presentation of antigens by upregulating MHC-1 expression so as to increase the sensitivity to PD-1 blockade therapy [69].
Low tumor mutation burden
There is an apparent correlation between the tumor mutation burden (TMB) and the objective response rate of PD-1/PD-L1 blockade. TMB may be one of the reasons for the different outcomes in different cancers [70]. Generally, errors in DNA replication can be corrected in normal cells through a mechanism called mismatch repair (MMR). However, the absence of MMR can lead to easier accumulation of mutations, thereby increasing tumor immunogenicity. Microsatellite instability (MSI) is one of the important indicators of MMR deficiency [71]. Clinically, in patients with metastatic MSI-High-dMMR colorectal cancer, pembrolizumab (anti-PD-1) treatment extended the duration of progression-free survival [6]. A multi-center study on advanced NSCLC patients revealed that elevated TMB increased the response to PD-1/PD-L1 blockade and improved overall survival in those with high PD-L1 expression [72]. Similarly, in patients with advanced solid tumors, it revealed that objective response to pembrolizumab was observed in 29% (30 out of 102) with high TMB and in 6% (43 out of 688) without high TMB, suggesting that high TMB levels may be applied to distinguish patients who would benefit significantly from anti-PD-1 treatment [73]. In addition, TMB was found to be significantly higher in melanoma patients who responded to combination blockades with anti-CTLA-4 and anti-PD-1 than in those who did not respond [74]. All of the above suggest that the elevated TMB is positively correlated with the efficacy of anti-PD-1, and patients with high TMB show high sensitivity, while those with a low TMB often show low sensitivity.
Immunosuppressive TME
To resist the anti-tumor immune response, the tumor educates an immunosuppressive TME, whose components can affect anti-tumor immunotherapy, such as immunosuppressive cells, cytokines, and some co-inhibitory receptors. The immunosuppressive TME partly accounts for the resistance to PD-1/PD-L1 blockade therapy [42].
Immunosuppressive cells
Currently, it is known that there are some immunosuppressive cells in the TME, such as TAMs, CAFs, and MDSCs. TAMs exist as M1 and M2 types, usually the M2 type, which is often associated with tumor progression and poor outcomes [75]. TAMs can influence immunotherapy and contribute to immune resistance through various mechanisms [76]. (1) Upregulation of immune checkpoints, primarily PD-L1. One study found that M2-type TAMs promoted PD-L1 overexpression in gastric cancer cells via M2-Exos exosomes, which eventually promoted the growth and invasion of cancer cells [77]. Another study found that TAMs were the primary source of PD-L1 in the murine cholangiocarcinoma model, with approximately 60% of TAMs expressing PD-L1[78]. High expression of PD-L1 in TAMs indicated an activated immune microenvironment, often accompanied by abundant CD8+ T cell infiltration as well as high expression of immune-related genes [79]. In addition, TAMs also expressed other immune checkpoints that collaborate with PD-L1 to promote immune evasion, such as VISTA [75]. (2) Crosstalk between regulatory T cells (Tregs) and TAMs: On the one hand, TAMs recruit Tregs into the TME through some chemokines (such as CCL20), cytokines, and exosomes. On the other hand, Tregs can promote the immunosuppressive function of TAMs [76]. (3) Hijacking anti-PD-1 antibody: TAM can capture anti-PD-1 antibody through binding to the Fc region of the antibody with the FcγR expressed on the macrophages [80]. (4) Influencing T cell activation and function. TAMs express the transcription factor IRF8, which is necessary for antigen presentation by TAMs, and can lead to the depletion of cytotoxic T lymphocytes (CTLs) within the TME. The specific absence of IRF8 in TAMs can prevent the exhaustion of cancer-cell-reactive CTLs and inhibit tumor growth [81]. (5) Secreting regulatory cytokines. TAMs can promote immune suppression and affect the PD-(L)1 blockade efficacy by secreting regulatory cytokines (such as TGF-β, PGE2, IL-6) [75].
CAFs can affect PD-1/PD-L1 immunotherapy by secreting various cytokines (such as WNT2, TGF-β1, CXCL5) and extracellular vesicles. They also regulate other immune cells within the TME and participate in the ECM remodeling, eventually leading to the failure of anti-PD-(L)1 therapy [82]. MDSCs, as a group of immature myeloid cells, are considered to be negatively correlated with the efficacy of immune checkpoint blockade due to their ability to suppress T cell activity and express PD-L1. Targeting MDSCs could boost the efficacy of anti-PD-1 therapy [78].
Immunosuppressive factors
Within the TME, there are cytokines that can negatively regulate the efficacy of anti-PD-(L)1 therapy. For instance, cytokines such as IFN-γ, TGF-β, TNF-α, ECF and GM-CSF can promote tumor immune evasion by regulating the expression of PD-L1[56].
IFN-γ is essential in regulating PD-L1 expression, commonly secreted by CD4+ T cells differentiated into Th1. It plays a vital role in innate and adaptive immune responses against pathogens and tumors [83]. To exhibit its function, IFN-γ must bind to its receptor IFNGR (includes IFNGR-1 and IFNGR-2), and the activation of IFN-γ signaling is mainly mediated through the JAK-STAT pathway [83]. IFN-γ is essential for “tumor immunoediting” [83, 84]. At the stage of immune elimination, IFN-γ acts as an immune-stimulating molecule, together with other components in the TME, promoting the immune system to recognize and eliminate the tumor cells that strongly express antigens. For instance, IFN can induce the conversion of TAMs from pro-tumor M2 type to the anti-tumor M1 type [85]. At the stage of immune equilibrium, IFN-γ is essential for maintaining the dormancy and balance of tumors. At the stage of immune evasion, IFN-γ acts as an immunosuppressive molecule, promoting tumor cells to evade the immune system surveillance. Besides PD-L1, IFN-γ also promotes the expression of other immune checkpoints on T cells, such as LAG-3 and CTLA-4[83]. The modulation of tumor immunity by the interferon signaling pathway is complex. It was revealed that when exposed to IFN-γ or IFN-β, B16 tumor cells with an IFN-γ deficiency showed a more significant growth tendency compared to wild-type tumor cells. Ptpn2 negatively regulated IFN-γ signaling by dephosphorylating JAK1 and STAT1. That loss of Ptpn2 enhanced IFN-γ signaling and antigen presentation to T cells, leading to increased sensitivity to immunotherapy [86]. Another study found that the deletion of JAK2 led to a decrease of tumor suppressor gene CDKN2A expression, which increased the susceptibility of tumors to develop resistance to IFN-γ and immunotherapy [87].
Similarly, TGF-β can shape an immunosuppressive TME resistant to anti-PD-(L)1 therapy by regulating the activity of various immune cells, promoting epithelial-mesenchymal transition, and promoting T cell exclusion [88]. That dual inhibition of TGF-β and PD-1 could increase the anti-tumor efficacy. Moreover, some chemokine receptors, such as CCR1, CCR2, CCR4, and CCR5, have been proven to be associated with tumor immunosuppression. Combined inhibition of these chemokines with anti-PD-(L)1 treatment can improve the therapeutic efficacy [89].
Other immune checkpoints
Within the TME, other immune checkpoints, such as TIM-3, LAG-3, and TIGIT, are also considered to be related to anti-PD-(L)1 resistance. Specifically, one study indicated that PD-1 blockade upregulated other immune checkpoints, such as LAG-3 and CTLA-4, which may affect the efficacy of monotherapy [90]. However, this compensatory upregulation is likely to be overcome by combination blocking strategies, and even to some extent, the upregulated immune checkpoints may provide more targets for antibody blockade. Therefore, combination therapy may exhibit significant synergistic or combined effects. In mouse model of NSCLC, the upregulation of TIM-3 was found to be related to anti-PD-1 resistance, and blocking TIM-3 could boost the efficacy of anti-PD-1 therapy [91]. The following section will discuss the application of other ICBs in combination with PD-1/PD-L1 blockade in cancer therapy.
Other mechanisms
Metabolism
The rapid generation of ATP meets the high metabolic growth demands of the tumor, while the substantial accumulation of lactic acid helps to construct an immunosuppressive TME [92]. For instance, studies showed that lactate induced the conversion of macrophages into M2 type and promoted lung cancer progression, and the accumulation of lactic acid prevented T cell infiltration into the TME and reduced IFN-γ expression [93]. It is also worth noting that the high consumption of glucose in tumor cells may restrict the glycolytic activity in T cells, leading to T cell dysfunction. In addition, tumor cells also promote immune suppression by secreting some inhibitory metabolic products, such as kynurenic acid, adenosine, and PGE2[92]. Collectively, all of these factors are conducive to tumor immune evasion and are related to anti-PD-(L)1 resistance.
Epigenetic alterations
Epigenetic changes refer to the regulation of gene expression without altering the DNA sequence, mainly involving DNA methylation, histone modification, RNA modification, and chromatin remodeling. These epigenetic changes can be involved in anti-tumor immunity [94]. For example, it was found that the downregulation of MHC-I in ovarian cancer cells may be related to epigenetic mechanisms. Specifically, there was a strong inverse correlation between the expression of the HLA-A gene and the level of promoter methylation [47]. Another study found that histone methylation and DNA methylation of EZH2 suppressed the production of Th1 chemokines CXCL9 and CXCL10, and reduced effector T cell infiltration into the TME in mice with ovarian cancer. Consequently, inhibition of this epigenetic process was believed to boost anti-PD-L1 efficacy [95]. In recent years, some non-coding RNAs exhibited potential effects on the efficacy of PD-1/PD-L1 blockade therapy [96]. For instance, circHMGB2 could remodel the TME by upregulating the expression of CARM1 by sponging miR-181a-5p and limiting anti-PD-1 efficacy in NSCLC. Inhibition of CARM1 improved the sensitivities of NSCLC cells with high circHMGB2 expression to anti-PD-1 treatment [97].
Microbiota
Gut microbiome is thought to influence the efficacy of PD-1/PD-L1 blockade therapy [98]. For example, an analysis of the gut microbiome in patients with gastrointestinal tumors treated with anti-PD-1/PD-L1 showed that responders had an increased ratio of the relative abundance of Prevotella and Bacteroides, and the relative abundant types of bacteria varied with different tumors [99]. PD-L1 blockade was significantly effective in melanoma mice with microbiome transplantation from responders to anti-PD-1 therapy, but was completely ineffective in mice with microbiome transplantation from non-responders [98]. Additionally, it was revealed that antibiotics suppressed the efficacy of anti-PD-1 therapy in patients with advanced cancer. Notably, fecal microbiota transplantation (FMT) from patients who responded to anti-PD-1 therapy into antibiotic-treated mice improved the efficacy of anti-PD-1, while FMT from non-responders did not improve the efficacy [100].
PD-1/PD-L1 Blockade in Combination with Other ICBs
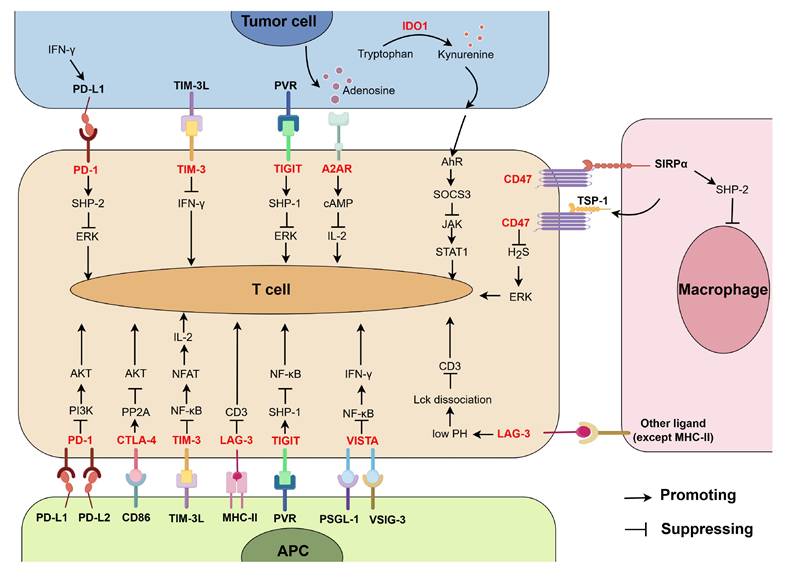
Combined application of PD-1/PD-L1 blockade with other treatments offers potential promising prospects for improving the low response rate of PD-1/PD-L1 blocking therapy and overcoming the drug resistance. Below, we will focus on the synergistic anti-tumor response of the combined blockades of PD-1/PD-L1 and other immune checkpoints, and the distribution of these immune checkpoints and their function are summarized in Figure 3.
The distribution T cell-related immune checkpoints (highlighted in red) and their function. Receptors PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, VISTA, A2AR, and CD47 interact with their ligands, then lead to suppression of T cell activation through different pathways. IDO1 promotes the production of kynurenine, which in turn binds with receptor AhR and suppresses T cell activation via SOCS3/JAK/STAT1 axis. (By Figdraw.)
CTLA-4
CTLA-4 (cytotoxic T-lymphocyte associated antigen-4, CD152), a receptor that is upregulated on activated T cells. CTLA-4 has a high resemblance to CD28 and binds to B7-1 (CD80) and B7-2 (CD86) on APCs with a higher affinity than CD28. As a co-inhibitory receptor, it competes with CD28, negatively regulating T cell activation [101]. Furthermore, CTLA-4 is mainly constitutively expressed on Tregs or induced after T cell activation in response to CD28 and TCR signaling [102]. Additionally, CTLA-4 induces the expression of indoleamine 2,3-dioxygenase 1 (IDO1) by activating the noncanonical NF-κB pathway, then the expression of IDO further promotes the differentiation of Tregs, which could inhibit the activation of other T cells, thereby forming an immunoregulatory network [103].
The blockade of CTLA-4's suppressive function allows and enhances the effective immune response against tumor cells [104]. In 2011, the first monoclonal antibody against CTLA-4 ipilimumab received FDA approval. Anti-CTLA-4 may exert its anti-tumor effects by increasing the activity of effector CD4+ T cells and inhibiting the Tregs-dependent immune suppression [32]. It was found that anti-CTLA-4 treatment mainly affected CD4+ T cells [32], and induced cytotoxic CD4+ T cells in melanoma patients [105]. Moreover, Tregs, a subset of CD4+ T cells, are the primary targets of anti-CTLA-4 for anti-tumor efficacy. High frequencies of Tregs before anti-CTLA-4 treatment are correlated with better response to anti-CTLA-4 therapy [106]. Additionally, the reduction in FoxP3/Tregs levels during treatment with ipilimumab treatment was linked to a better clinical outcome and survival rate [107].
The advantage of the combination of anti-CTLA-4 and anti-PD(L)1 therapy is that they affect T cell activation through different mechanisms, which may complement each other in mechanism and efficacy. (1) Affecting different subsets of TILs. Anti-PD-1 predominantly leads to the proliferation of exhausted-like CD8+ T cells within the tumor, whereas CTLA-4 inhibitor mainly results in the amplification of CD4+ effector cells and exhausted-like CD8+ T cells [19]. It was found that the combination of anti-PD-L1 and anti-CTLA-4 treatment had a stronger inhibitory effect in colon cancer than monotherapy. Both the anti-CTLA-4 monotherapy and the combined treatment increased CD4+ and CD8+ T cells significantly in the tumors, and decreased intra-tumoral Tregs [108]. (2) Affecting the activation of T cells at different stages. CTLA-4 is rarely expressed on naïve T cells, but is upregulated on activated T cells and reaches a peak within 48-72 hours. Thus, CTLA-4 may play a role after the initial T cell activation [109]. In addition, in another study, the expression levels of CTLA-4 in active T cells peaked on the third day and returned to baseline levels by the seventh day, while the PD-1 expression gradually increased over time, reaching the maximum level on the tenth day [110]. (3) Affecting T cell activation in different regions. PD-1 is highly expressed on activated T cells and requires binding to its ligand PD-L1 to exert its effects, which mainly restricts T cell activity at specific tissue sites, such as the TME. In contrast, CTLA-4's binding is not dependent on ligands, primarily competing with CD28 for the binding of B7-1 and B7-2. A study found that CTLA-4 on Tregs interacted with CD80 expressed on dendritic cells within the lymph nodes surrounding the tumor to regulate CD4+ T cell infiltration into tumors. Treatment with anti-CTLA-4 stimulated CD4+ T infiltration into the tumor, suggesting the inhibitory effect of CTLA-4 on T cells in lymph nodes [111]. Similarly, another study demonstrated that the expansion of TCR in melanoma patients from CTLA-4 blockade (tremelimumab) is related to the priming encounter between T cells and APCs in the lymph nodes, whereas the inhibitory signal of PD-1/PD-L1 with PD-1 blockade (pembrolizumab) occurs in peripheral tissues [112]. (4) Inhibiting the PI3K/Akt pathway by different ways. PD-1 blocks the induction of PI3K activity, which is essential for the activation of Akt, and this inhibition by PD-1 is dependent on its ITSM motif. In contrast, CTLA-4 directly inhibits Akt through the activation of phosphatase PP2A, while retaining the activity of PI3K [113]. These findings indicate that the two blockades may have synergistic or additive effects in inhibiting the PI3K/Akt pathway. (5) Overcoming the upregulation of immune checkpoints induced by monotherapy. For instance, a study showed that the CTLA-4 expression in CD4+ Tregs increased after the administration of the blocking antibody, and PD-1 expression in CD8+ T cells also increased, especially in the group treated with one blockade [114]. (6) Remodeling a more advantageous tumor immune microenvironment. It was revealed that the dual blockades of PD-1 and CTLA-4, in contrast to individual inhibition, significantly enhanced the CD8+ T cells/Tregs and CD8+ T cells/MDSCs ratios within the tumor in mice with melanoma. The increased effector T-cell (Teff) within the tumor seemed to be associated with high levels of inflammatory cytokine levels such as IFN-γ and TNF-α in the relevant tissues induced by the dual blockade [114]. Furthermore, in another study, it was discovered that dual blockades of PD-1 and CTLA-4 increased IFN-γ production, reduced the secretion of immunomodulatory cytokines such as TGF-β and IL-10, promoted TIL proliferation and its cytolytic function through upregulation of ribosomal S6 kinase (S6K), and two T-box transcription factors T-bet, and Eomes which regulated Th1 and cytolytic function of CD8+ cells [115].
To sum up, CTLA-4 expression in T cells is increased after anti-PD-(L)1 treatment. The combination of anti-CTLA-4 and anti-PD-(L)1 therapy is because that they affect T cell activation through different mechanisms, affecting different subsets of TILs at different stages and in different regions, so that they play more effective anti-tumor effects.
TIM-3
T cell immunoglobulin-3 (TIM-3, CD366, HAVCR2), serving as a co-inhibitory receptor, is involved in the immune regulation of autoimmune diseases, transplantation tolerance, tumors, and infectious diseases [116]. Initially, it was identified to negatively regulate the generation of Th1 and Tc1 cells that secrete IFN-γ, reducing their apoptosis. Subsequently, the research revealed its expression on innate immune cells, including dendritic cells, macrophages, and NK cells [117]. It has been confirmed that the ligands for TIM-3 include Galectin-9, PtdSer, CEACAM1, and HMGB1. Unlike classic immune checkpoints such as PD-1, TIM-3 does not have the inhibitory signaling motifs in its cytoplasmic tail, such as ITIMs or ITSMs, but it contains five conserved tyrosine residues [118]. TIM-3 could inhibit TCR-mediated signaling by suppressing the NF-κB/NFAT signaling pathway in Jurkat T cells and primary human CD8+ T cells, thereby inhibiting IL-2 secretion and T cell activation [119]. The high expression level of TIM-3 was found to be associated with T cell exhaustion and dysfunction within tumors [120].
Interestingly, TIM-3 was found to be co-expressed with PD-1, and the combined blocking of PD-1 and TIM-3 had a collaborative effect on enhancing effector T cells' function and their ability to destroy tumor cells [118]. The combined blocking increased the frequency of IFN-γ, TNF, IL-2-producing NY-ESO-1-specific CD8+ T cells and the frequency of proliferating and total NY-ESO-1-specific CD8+ T cells, suggesting a collaborative effect between TIM-3 and PD-1 blockade [120]. The co-expression of TIM-3 and PD-1 was found to be associated with poor prognosis of colorectal cancer [121] and gallbladder cancer [122]. Additionally, in mouse model of NSCLC, it was revealed that the upregulation of TIM-3 might be one of the mechanisms of anti-PD-1 resistance. Treatment with TIM-3 blockade after the failure of PD-1 blockade therapy can significantly enhance the anti-tumor efficacy and survival benefit. Furthermore, Galectin 9 (one of the ligands of TIM-3) was found to be significantly elevated in PD-1-resistant tumor samples at both RNA and protein levels. In addition, the combination therapy increased IFN-γ production and proliferation of TIM-3+CD8+ T cells from PD-1-resistant mice, as well as decreased the expression of some tumor-promoting cytokines, such as IL-6 and progranulin [91]. Studies showed that dual blocking of PD-1 and TIM-3 simultaneously exerted better anti-tumor effects compared to blocking either one alone. In a study about mice with acute myeloid leukemia, it was discovered that the combination of TIM-3-Fc fusion protein and anti-PD-L1 significantly reduced tumor burden at all time points and extended the survival of wild-type mice with high tumor load, compared to single blockade treatment [123].
In addition, it was found that the expression of PD-1 and TIM-3 was significantly upregulated in CD4+ and CD8+ T cells isolated from tumor tissues and ascites of patients with hepatocellular carcinoma. The combined blocking exhibited a more significant anti-tumor effect than a single antibody blockade in mice with hepatocellular carcinoma through increased production of T cell effector cytokines (IFN-γ and TNF-α) and TILs, decreased levels of immunosuppressive cytokines (IL-10 and IL-6), and the amounts of PD-1+TIM-3+CD8+ T cells within the TME, which are related to tumor immune evasion [124]. Thus, dual blocking of PD-1 and TIM-3 exerts better anti-tumor effects compared to blocking one alone, and the co-expressed TIM-3 with PD-1 is the possible mechanism.
LAG-3
The lymphocyte activation gene 3 (LAG-3), also known as CD223, is frequently expressed on various activated immune cells, including CD4+ and CD8+ T cells, Tregs, NK cells, B cells, and dendritic cells. It binds with several classical ligands, such as MHC-II, Galectin-3, LSECtin, alpha-synuclein, and FGL1. The engagement with these ligands can induce a state of exhaustion in the respective immune cells [125]. Specifically, LAG3 binds to MHC-II and inhibits T cell proliferation and cytokine production through the association with CD3 in the TCR-CD3 complex [126]. Moreover, LAG3 may serve as a signal disruptor in the absence of its canonical ligand MHC-II. Mechanistically, a tandem glutamic acid-proline repeat in the LAG3 cytoplasmic tail lowered the pH at the immune synapse and caused dissociation of the tyrosine kinase Lck from the CD4 or CD8 co-receptor, which resulted in a loss of co-receptor-TCR signaling and limited T cell activation [127]. Therefore, the blockade of LAG-3 has shown promising therapeutic effects in various types of malignant tumors, and anti-LAG-3 immunotherapeutic agents have been utilized to restore T cell function.
Notably, the combination of LAG-3 with other ICBs may yield better outcomes. In cancers, T cells are persistently stimulated by antigens, leading to the continuous high expression of LAG3 along with other co-inhibitory receptors such as PD-1, CTLA-4, and TIM-3. This results in T cell exhaustion, which is characterized by decreased cytokine production, reduced proliferative capacity, and diminished ability to kill tumor cells [128]. In 2022, the FDA approved Opdualag (a fixed-dose combination of LAG-3 blocking antibody relatlimab and PD-1 blocking antibody nivolumab) for the treatment of unresectable or metastatic melanoma [129]. One study showed that LAG3 and PD-1 were upregulated and co-expressed in TILs from mice bearing ovarian tumors. Dual blockade or gene knockout of LAG-3 and PD-1 produced high levels of IL-2, IFN-γ, TNF-α, and granzyme B, increased the percentage of CD8+ and CD4+ TILs, enhanced the effector function of CD8+ T cells, reduced the frequency of suppressive Tregs within the TME so that it delayed tumor growth and extended the life span of mice significantly [130]. Similarly, another study found that the expression of multiple immune checkpoints was upregulated in tumor-associated lymphocytes (TALs) isolated from patients with ovarian cancer, and the co-expression of PD-1 and LAG-3 was the most significant in CD8+ TALs, similar to the findings observed in mouse model [90]. Furthermore, in patients with advanced NSCLC, overexpression of LAG-3 was found to be negatively correlated with the survival benefit from PD-1/PD-L1 blockade [131]. To sum up, the combination of LAG-3 blockade with PD-1/PD-L1 blockade exhibits better anti-tumor effects than PD-1/PD-L1 blockade alone in clinic and preclinical studies, and this might be due to the co-expression of LAG3 and PD-1 at high levels in TILs within the TME.
TIGIT
T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), also known as Vstm3 or VSIG9, is an immune checkpoint receptor expressed on T cells and NK cells. Its structure includes an immunoglobulin variable domain, a transmembrane domain, and an ITIM [132]. The ligands identified for TIGIT include poliovirus receptor (PVR, CD155), PVRL2 (CD112), and PVRL3 (CD113), and PVR shows the highest affinity to TIGIT. Studies have demonstrated that TIGIT effectively competes with both CD226 (an activating receptor) and CD96 (an inhibitory receptor) for binding to their shared ligand PVR.[132]. TIGIT has a variety of actions: (1) After binding to PVR, TIGIT recruits SHIP-1 to inhibit the activation of the NF-κB and ERK signaling pathways, thereby reducing cytokine production and leading to the exhaustion of CD8+ T cells [133]. (2) The interaction between TIGIT and PVR induces IL-10 production in dendritic cells, thereby suppressing T cell activation [132]. (3) TIGIT competes with CD226 to bind PVR, thus inhibiting CD226-mediated T cell activation. (4) The expression of TIGIT in Tregs enhances their immunosuppressive function and stability [134]. (5) Tumors exploit the Fap2 protein of Fusobacterium nucleatum to interact with TIGIT, inhibiting the activation of T cells and NK cells [135].
Recent studies found that the combined targeting TIGIT and PD-1 outperformed single blocking in terms of tumor suppression. For example, one study found that TIGIT and PD-1 co-expressed on most NY-ESO-1-specific CD8+ T cells isolated from melanoma patients, and dual blockades of TIGIT and PD-1 further increased NY-ESO-1-specific CD8+ T cell counts. Additionally, it found that PD-1 blockade increased TIGIT expression, but TIGIT blockade could not increase PD-1 expression [136]. Moreover, another study found that in the surgical resection samples of glioblastoma, the TILs exhibited similar expression levels of PD-1 and TIGIT. Additionally, in mice with implanted tumors, it was observed that the long-term survival rate for the group treated with single PD-1 blockade was 16.7%, while the control group and the single TIGIT blockade group had a long-term survival rate of 0%. Surprisingly, the dual blockade of both targets increased the long-term survival rate to 48.0%, providing a greater survival benefit. The combined blockade and PD-1 single blockade could establish anti-tumor immune memory, and the combination blockade therapy provided more significant efficacy, which may be related to increased infiltration of CD8+ and CD4+ T cells, increased production of cytokines IFN-γ and TNF-α, and decreased tumor-infiltrating dendritic cells [137]. In a word, the dual blockade of TIGIT and PD-1 provides a greater survival benefit than PD-1/PD-L1 blockade alone. This might be due to the fact that PD-1 blockade increases TIGIT expression.
VISTA
V-domain Ig suppressor of T cell activation (VISTA), a transmembrane protein, is also known as PD-1 homologue (PD1H) due to the high homology of its IgV domain with the CD28 and B7 families [125]. There are several identified ligands for VISTA, such as PSGL-1, VSIG-3, Galectin-9, LRIG1, and Syndecan-2[138]. VISTA has been discovered with high expression levels in tumor-infiltrating myeloid cells, including myeloid dendritic cells and MDSCs. Moreover, blockade of VISTA was also found to impair the inhibitory effect of Tregs, but it was not clear whether this was related to the expression of VISTA on Tregs. Additionally, VISTA was constitutively expressed on naïve T cells, maintaining their resting state and inhibiting T cell activation [139]. After binding to PSGL-1, VISTA was able to inhibit the phosphorylation of NF-κB in T cells, thereby reducing the production of cytokines (such as IFN-γ) and suppressing the proliferation and activation of T cells [140]. VSIG-3 also acted as a ligand for VISTA. The interaction between VSIG-3 and VISTA significantly reduced the production of cytokines and chemokines by T cells, including IFN-γ, IL-2, IL-17, CCL5/Rantes, CCL3/MIP-1α, and CXCL11/I-TAC, and also inhibited the proliferation of T cells activated by anti-CD3 antibodies [141].
Interestingly, VISTA blockade was found to exhibit a cooperative effect with anti-PD-(L)1 therapy in multiple cancer types. For example, the combined administration of anti-VISTA and anti-PD-L1 after inoculation of colon cancer cells into mice resulted in significant anti-tumor effects, whereas the effects of monotherapy were not pronounced. This was due to the fact that combined blockade resulted in higher levels of cytokines (IFN-γ, TNF-α, and granzyme B) production by tumor-specific CD8+ T cells from tumor-draining lymph nodes compared to single blockade or control group [142]. In addition, it was reported that anti-VISTA reduced the resistance to anti-PD-(L)1 therapy since anti-VISTA increased antigen presentation and the expression of IFN-regulated genes, reduced myeloid cell-mediated suppression, and simultaneously improved T cell infiltration [143]. All in all, VISTA blockade exhibits synergistic anti-tumor effects with anti-PD-(L)1 treatment, and this might be due to that VISTA blockade improves T cell infiltration and reduces the resistance to anti-PD-(L)1 treatment via increasing the production of IFN-γ.
IDO
Indoleamine 2,3-dioxygenase 1 (IDO1), as the most widely studied indoleamine 2,3-dioxygenase, is the primary rate-limiting enzyme of tryptophan metabolism through the kynurenine pathway, which may be dysregulated in various disease states [144]. Through tryptophan depletion and kynurenine production, IDO plays a role in suppressing T cell response and maintaining immune tolerance in various pathological processes [145]. IDO1 metabolizes tryptophan to kynurenine, which subsequently activates AhR, resulting in the upregulation of SOCS3. This, in turn, inhibited the activation of the JAK-STAT1 signaling pathway and reduced the secretion of CXCL9 and CXCL10, thereby decreasing T-cell infiltration [146]. Moreover, the metabolism of tryptophan into kynurenine also promotes the expansion of Tregs, which inhibits inflammatory response by secreting anti-inflammatory cytokines, and suppresses the production of pro-inflammatory cytokines and the infiltration of neutrophils and Th1/Th17 cells [147].
In the field of cancer therapy, IDO1 has attracted more attention than IDO2. IDO1 is often silent in normal tissues but is expressed in restricted tissues [148]. However, overexpression of IDO is observed in various tumors and is associated with poor prognosis [149, 150]. Additionally, IFN-γ is believed to induce the expression of IDO [149].
It was found that the IDO1 inhibitor PF-06840003, when used in combination with anti-PD-L1, could induce a higher proportion of T cells secreting IFN-γ and the expression of the cytolytic enzyme granzyme A, thereby enhancing anti-tumor effects. Additionally, it was found that treatment with anti-CTLA-4 and anti-PD-L1 induced IDO1 expression, and this might be due to the secretion of IFN-γ by activated T cells after treatment, as IFN-γ was identified as an inducer of IDO1[151]. Similarly, another study found that treatment with PD-L1 blockade led to upregulation of IDO expression in vivo. The combination therapy of PD-L1 blockade and IDO inhibitor can improve anti-tumor effects by increasing the frequency of IL-2-producing and proliferating polyfunctional T cells within the tumor, and prolonging the duration and frequency of peripheral tumor-reactive lymphocytes at a later stage. However, the combination therapy did not increase the early anti-tumor CD8+ T cell counts in the tumor-draining lymph nodes [152]. When combined with PD-L1 blockade, IDO nano-inhibitor enhanced the anti-tumor efficacy of anti-PD-L1 via decreasing the proportion of immune suppressive cells (Tregs) and increasing the proportion of immune effector cells (IFN-γ secreting tumor-infiltrating T cells) [153]. Moreover, the co-expression of IDO-1 and PD-L1 might be related to the enhanced anti-tumor efficacy in NSCLC clinical trials with dual blocking of PD-1/PD-L1 and IDO-1[154]. To sum up, the combination of IDO1 inhibitor and anti-PD-L1 shows enhanced anti-tumor effects. This might be due to the fact that anti-PD-L1 induces IDO1 expression, then IDO1 inhibitor decreases proportion of Tregs and increases the proportion of immune effector cells.
CD47
CD47, also known as integrin-associated protein (IAP), is often utilized by cancer cells to evade immune system surveillance and attack. CD47 is generally expressed in human tissues, but the mRNA levels vary across different tissues. Form 2 is commonly expressed in bone marrow-derived cells, endothelial cells, and fibroblasts, and form 4 is most commonly expressed in neural tissues [155]. The CD47-SIRPα axis plays a role in tumor evasion in the process of phagocytosis mediated by macrophages, dendritic cells, and other phagocytic cells [156]. It was found that SHP2 deneddylation, through the CD47/SIRPα axis, mediated tumor immune suppression in colon cancer, and the administration of allosteric SHP2 inhibitors sensitized immunotherapy-resistant colorectal cancer to immunotherapy [157]. Moreover, CD47 could induce compartmental remodeling of tumor-infiltrating immune cells within the pancreatic cancer microenvironment [158]. Blocking the CD47-SIRPα axis was considered to be a promising option for cancer treatment [156]. In addition, CD47 could also bind to thrombospondin-1 (TSP-1), thereby limiting the production of H2S and inhibiting H2S-induced ERK1/2 phosphorylation, thus suppressing the activation of T cells [159].
Interestingly, PD-L1 and CD47 are co-expressed in various cancers [160]. The dual blockade of CD47 and PD-1/PD-L1 shows synergistic anti-tumor effects. It was found that CD47 absence markedly improved anti-tumor efficacy mediated by anti-PD-1 therapy [86]. Compared to PD-L1 blockade, CD47/PD-L1 bispecific antibody has superior anti-tumor efficacy, through increasing the levels of pro-inflammatory cytokines (IL-1β, IL-12, and IL-18), effector cytokines (IFN-γ) and T cell-recruiting chemokines (CXCL9, CXCL10, and CCL5), amplifying the systemic CD8+ T cell response, expanding the pool of intra-tumoral CD8+ T cells, reprogramming myeloid population, driving innate activation within the TME, increasing the frequency of stem-like progenitor and effector CD8+ T cells in the tumor, and promoting the differentiation of progenitor CD8+ T cells into an effector-like state[161]. Furthermore, CD47 and PD-L1 bispecific antibody (6MW3211) was found to exhibit lower toxicity reactions and synergistic anti-tumor effects via increasing IFN-γ levels and promoting phagocytosis of macrophages [160]. In a study on B-cell lymphoma, dual blockade with anti-CD47 and anti-PD-L1 therapy activated CD8+ T cells, increased the secretion of perforin, granzyme B, and IFN-γ, and enhanced macrophage infiltration [162]. To sum up, dual blockade of CD47 and PD-1/PD-L1 shows synergistic anti-tumor effects, and this might be due to the fact that CD47 is co-expressed with PD-L1 in various cancers, and CD47 blockade promotes phagocytosis of macrophages and activates CD8+ T cells.
Adenosine A2AR
Adenosine A2A receptor (A2AR) is expressed in immune tissues and various immune cells. A2AR was initially identified as a critical and non-redundant negative regulatory factor that protected normal tissues from inflammatory damage. Later, it was found to play a role in shielding tumors from attack by anti-tumor T cells [163]. The current perspective is that A2AR agonists regulate immune cells. For T cells, adenosine/A2AR can inhibit CD4+ T cells, increase the generation of Foxp3+ Tregs, and block the cytotoxic function of CD8+ T cells. For NK cells, the adenosine/A2AR signal limits the NK cell maturation and proliferation. For macrophages, adenosine/A2AR can induce the conversion of macrophages into the M2 phenotype, which promotes tumor growth [163, 164]. Consequently, the blockade of A2AR or the absence of A2AR can lead to anti-tumor effects and the improvement of tumor-induced immunosuppression. Jenabian and colleagues found that A2AR increased intracellular cAMP levels in Jurkat cell line and CD4+ T cells, thereby inhibiting the demethylation of the IL-2 gene promoter region, and consequently suppressing the proliferation of CD4+ T cells and the production of IL-2 in CD4+ T cells [165]. Additionally, it was also observed that A2AR antagonist could rescue tumor-reactive T cells (mainly CD8+ T cells) by reducing cAMP levels, freeing anti-tumor T cells from adenosine-mediated suppression, and enhancing the production of pro-inflammatory cytokines [166]. In a clinical study on renal cell carcinoma, A2AR antagonists led to significant tumor regression, and longer disease control time was associated with CD8+ T cell infiltration into the TME [167].
The combination of A2AR blockade with anti-PD-1/PD-L1 can enhance the therapeutic effect. PD-1 blockade upregulated A2AR expression on CD8+ TILs, which might account for the dual blockade combination therapy being more effective. Additionally, the combination blockade of PD-1 and A2AR increased the production of IFN-γ by tumor-infiltrating CD8+ T cells both in vitro and in vivo [168]. Furthermore, the primary tumors with higher PD-L1 expression and lower A2AR expression showed better outcomes and longer overall survival in patients treated with anti-PD-1 antibody alone or in combination with anti-PD-1 and anti-CTLA4; whereas increased expression of A2AR was associated with poor outcomes and short survival [169]. CPI-444 (an A2AR antagonist) moderately suppressed tumor growth, while combined with anti-PD-1 showed significant anti-tumor efficacy in vivo (causing tumor regression and improving survival), which might be related to the inhibition of PD-1 and LAG-3 expression by CPI-444 on CD8+ T cells and Tregs [170].
In summary, the preclinical studies and clinical trials on PD-1/PD-L1 blockade combination with other ICBs in cancer therapy have been summarized in Table 1A, B, C, D and Table 2A, B, C, D, showing synergistic anti-tumor efficacy to some extent. However, the combinations often resulted in increased toxicity. For instance, in a clinical trial on metastatic melanoma, the rate of confirmed objective response was 61% (44 of 72 patients) in the group that received both ipilimumab and nivolumab (combination group) versus 11% (4 of 37 patients) in the group that received ipilimumab and placebo (ipilimumab-monotherapy group), with complete responses reported in 16 patients (22%) in the combination group and no patients in the ipilimumab-monotherapy group. Notably, the incidence of grade 3-4 drug-related adverse events was found to be 54.3% for combination therapy (nivolumab and ipilimumab) compared to ipilimumab monoblockade of 23.9% [171]. Similarly, in another clinical trial (phase Ib) on resectable esophageal/gastroesophageal junction cancer, the incidence of grade 3 or higher treatment-related adverse events was 43.8% in the neoadjuvant nivolumab-relatlimab group compared to 18.8% in the nivolumab group [172]. It has been suggested that immune checkpoint blockade-induced irAEs may correlate with their anti-tumor efficacy and may even serve as a marker of response to immune checkpoint blockade therapy [7, 33]. Thus, it can be seen that the therapeutic potential of PD-1/PD-L1 blockade in combination with other ICBs, but the combinations often lead to an increase in the incidence of severe irAEs. More effective management of irAEs will be a prerequisite for the combined application of ICBs in clinical practice.
The preclinical studies of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Tumor types | Model | Findings (mechanisms) | References | |
|---|---|---|---|---|---|---|---|
| PD-L1×CTLA-4 | Anti-PD-L1 antibody | Anti-CTLA-4 antibody | Colon Cancer | BALB/c mice with CT26 orthotopic Colon Cancer | Dual CTLA-4 and PD-L1 blockade increased CD4+ and CD8+ T cells significantly in the tumors, and decreased intra-tumoral Tregs. | [108] | |
| PD-1/PD-L1×CTLA-4 | Anti-PD-1 (RMP1-14); anti-PD-L1 (9G2) | Anti-CTLA-4 (9D9) | Melanoma | C57BL/6 mice with B16 Melanoma | Dual CTLA-4 and PD-1 blockade enhanced the CD8+ T cells/Tregs and CD8+ T cells/MDSCs ratios in melanoma mice. The elevated Teff appeared to correlate with high levels of inflammatory cytokines like IFN-γ and TNF-α in the tumor tissues. | [114] | |
| PD-1/PD-L1×CTLA-4 | Anti-PD-1/αPD-L1 | Anti-CTLA-4 | Colon carcinoma; ovary cancer | BALB/c or C57BL/6 mice bearing with colon carcinoma cell line CT26 or ovarian carcinoma cell line ID8-VEGF. | Dual blockades of PD-1 and CTLA-4 increased IFN-γ production, reduced the secretion of TGF-β and IL-10, and promoted TIL proliferation and its cytolytic function through upregulation of ribosomal S6 kinase, T-bet, and Eomes. | [115] |
The preclinical studies of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| PD-1/PD-L1 blockade | Other ICB | Tumor types | Model | Findings (mechanisms) | References | |
|---|---|---|---|---|---|---|
| PD-1×TIM-3 | PD-1-blocking antibody | TIM-3-blocking antibody | Lung adenocarcinoma | Genetically engineered mouse models of lung cancer: EGFR L858R T790M mutation and CC10 RTTA double-positive mice, KrasG12D mice. | TIM-3 blockade treatment after PD-1 blockade failure significantly improved anti-tumor efficacy and survival. The combination therapy increased IFN-γ production and proliferation of TIM-3+ CD8+ T cells from PD-1-resistant mice, decreased the expression of some tumor-promoting cytokines, such as IL-6 and progranulin. | [91] |
| PD-1×TIM-3 | Anti-PD1 mAb | Anti-TIM-3 mAb | Hepatocellular carcinoma | BALB/c nude mice bearing with HepG2 cells | The combined blocking exhibited a more significant anti-tumor effect than a single blockade in mice with hepatocellular carcinoma, through increased production of T cell effector cytokines (IFN-γ and TNF-α) and TILs, decreased levels of immunosuppressive cytokines (IL-10 and IL-6) and the amounts of PD-1+TIM-3+CD8+ T cells within the TME. | [124] |
| PD-1×TIGIT | Anti-PD-1 mAb | Anti-TIGIT mAb 10D7.G8 | Melanoma | CD8+ T lymphocytes from PBMCs obtained from patients | Dual blockades of TIGIT and PD-1 further increased NY-ESO-1-specific CD8+ T cell counts. PD-1 blockade increased TIGIT expression, but TIGIT blockade could not increase PD-1 expression. | [136] |
| PD-1×TIGIT | Anti-PD-1 (4 H2) | Anti-TIGIT (clone 4B1 mIgG2 a, depleting isotype) | Glioblastoma | C57 BL/6 J mice with intracranial tumor | Both combined blockade and PD-1 single blockade can establish anti-tumor immune memory. The superior efficacy of combination therapy may be due to increased CD8+ and CD4+ T-cell infiltration, higher IFN-γ and TNF-α production, and reduced tumor-infiltrating dendritic cells. | [137] |
The preclinical studies of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Tumor types | Model | Findings (mechanisms) | References |
|---|---|---|---|---|---|---|
| PD-L1×VISTA | Anti-PD-L1 mAb | Anti-VISTA mAb | Colon cancer | C57BL/6 mice with CT26 colon cancer | Combined treatment showed significant anti-tumor effects in mice with colon cancer, through increased cytokine (IFN-γ, TNF-α, and granzyme B) production by tumor-specific CD8+ T cells from tumor-draining lymph nodes more than single blockade or control. | [142] |
| PD-L1×IDO1 | Anti-PD-L1 (clone 10F.9G2) | IDO1 inhibitor (PF-06840003) | Colon carcinoma | BALB/c and C57BL/6 mice with colon carcinoma | IDO1 inhibitor PF-06840003, when used in combination with anti-PD-L1, could induce a higher proportion of T cells secreting IFN-γ and the expression of the cytolytic enzyme granzyme A, thereby enhancing anti-tumor effects. | [151] |
| PD-L1×IDO | Anti-PD-L1 antibody (clone 10 F.9G2) | IDO inhibitor (INCB23843) | Melanoma | C57BL/6 mice bearing with B16-dsRed-SIY cells | Combination therapy increased IL-2-producing, proliferating polyfunctional T cells in the tumor, prolonged peripheral tumor-reactive lymphocytes' duration and frequency later on, but didn't boost early anti-tumor CD8+ T cells in tumor-draining lymph nodes. | [152] |
| PD-L1×IDO | Anti-PD-L1 | NLG-RGD NI | Pancreatic cancer | pancreatic cancer cell line Pan02 | IDO nano-inhibitor enhanced the anti-tumor efficacy of anti-PD-L1 via decreasing the proportion of Tregs and increasing the proportion of the IFN-γ secreting tumor-infiltrating T cells. | [153] |
The preclinical studies of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Tumor types | Model | Findings (mechanisms) | References |
|---|---|---|---|---|---|---|
| PD-L1×CD47 | IBI322 (CD47/PD-L1 bispecific antibody) | Burkitt's lymphoma, melanoma | NOD-SCID mouse bearing with Raji-PDL1 cell or A375 cells | CD47 and PD-L1 bispecific antibody (IBI322) was found to exhibit lower toxicity reactions and synergistic anti-tumor effects via increasing IFN-γ levels and promoting phagocytosis of macrophages. | [193] | |
| PD-L1×CD47 | Anti-PD-L1 mAb | SIRPα-Fc | B-cell lymphoma | BALB/c mice with lymphoma | Dual blockade with anti-CD47 and anti-PD-L1 therapy activated CD8+ T cells, increased the secretion of perforin, granzyme B and IFN-γ, and enhanced macrophage infiltration. | [162] |
| PD-1×A2AR | RMP1-14 (anti-PD-1 mAb) | SCH58261 | Breast carcinoma | C57BL/6 and BALB/C mice bearing with MC38 cells and 4T1.2 cells | Blocking PD-1 upregulated A2AR expression on CD8+ TILs. The combination blockade of PD-1 and A2AR increased the production of IFN-γ by tumor-infiltrating CD8+ T cells both in vitro and in vivo. | [168] |
| PD-1×A2AR | Anti-PD-1 mAb (RMP1-14, Bioxcell) | CPI-444 (A2AR antagonist) | Colon cancer, melanoma | C57BL/6 mice bearing with MC38 cells and B16-OVA cells | CPI-444 moderately suppressed tumor growth, while combined with anti-PD-1 showed significant anti-tumor efficacy in vivo (causing tumor regression and improving survival), which might be related to the inhibition of PD-1 and LAG-3 expression by CPI-444 on CD8+ T cells and Tregs. | [170] |
The clinical trials of the combination of PD-1/PD-L1 blockade with Other ICB in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Clinicaltrial no. | Phase | Tumor types | Findings | References |
|---|---|---|---|---|---|---|---|
| PD-1×CTLA-4 | Nivolumab | Ipilimumab | NCT03033576 | II | Refractory metastatic melanoma | The combination of nivolumab and ipilimumab resulted in a statistically significant improvement in PFS over ipilimumab (hazard ratio, 0.63, p = 0.04). ORR were 28% and 9%, respectively (p = 0.05). | [194] |
| PD-1×CTLA-4 | Nivolumab | Ipilimumab | NCT02716272 | II | Relapsed malignant pleural mesothelioma | In the nivolumab group, 24 (44%) of 54 patients achieved 12-week disease control, compared to 27 (50%) of 54 in the combination group. In the intention-to-treat population, 25 (40%) of 63 in the nivolumab group and 32 (52%) of 62 in the combination group achieved 12-week disease control. | [195] |
| PD-1×CTLA-4 | Nivolumab | Ipilimumab | NCT01844505 | III | Melanoma | Median PFS was 11.5 months for combined treatment (2.9 months for ipilimumab alone, 6.9 months for nivolumab alone). In PD-L1-positive patients, median PFS was 14.0 months for both combination and nivolumab alone groups. In PD-L1-negative patients, combination PFS was 11.2 months vs 5.3 months with nivolumab alone. | [196] |
| PD-L1×CTLA-4 | Durvalumab | Tremelimumab | NCT02592551 | II | Malignant pleural mesothelioma | Patients receiving combination blockades had longer median overall survival compared with those receiving monotherapy. Tumor PR occurred in 6 of 17 patients receiving ICB and thoracotomy (35.3%), among which major PR (>90% tumor regression) occurred in 2 (11.8%). | [197] |
| PD-L1×CTLA-4 | Durvalumab | Tremelimumab | NCT02319044 | II | Recurrent or metastatic HNSCC | Objective response rate was 7.8% in the combination arm (n = 129), 9.2% for durvalumab monotherapy (n = 65), and 1.6% for tremelimumab monotherapy (n = 63); median overall survival for all patients treated was 7.6, 6.0, and 5.5 months, respectively. | [198] |
| PD-1×CTLA-4 | Cadonilimab | NCT04220307 | II | Recurrent or metastatic nasopharyngeal carcinoma | ORR was 26.1 %. The ORR were 44.4 % and 14.3 % in patients with tumor PD-L1 expression ≥50 % and <50 %, respectively. ORR was achieved in 40.0 % of patients with EBV-DNA level <4000 IU/ml and 15.4 % of those with ≥4000 IU/ml. | [199] | |
ICB: immune checkpoint blockades; ORR: objective response rate; DCR: disease control rate; PFS: progression-free survival; PR: partial response; HNSCC: head and neck squamous cell carcinoma.
The clinical trials of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Clinicaltrial no. | Phase | Tumor types | Findings | References |
|---|---|---|---|---|---|---|---|
| PD-L1×TIM-3 | LY3300054 | LY3321367 | NCT02791334 | Ib | Microsatellite instability-high/MMR-deficient tumors | Objective response occurred in 13 patients (32.5%) with monotherapy, 9 (45.0%) in the PD-1/PD-L1 inhibitor-naïve combination cohort, and 1 patient (4.5%) in the PD-1/PD-L1 inhibitor-resistant/refractory combination cohort. | [200] |
| PD-L1×TIM-3 | LY300054 | LY3321367 | NCT03099109 | Ia/b | Advanced solid Tumors | In the NSCLC monotherapy expansion cohort, anti-PD-1/L1 refractory patients (n= 23, ORR 0%, DCR 35%, PFS 1.9 months) versus anti-PD-1/L1 responders (n = 14, ORR 7%, DCR 50%, PFS 7.3 months). In combination expansion cohorts (n = 91), ORR and DCR were 4% and 42%. | [201] |
| PD-1×TIM-3 | Spartalizumab | Sabatolimab | NCT02608268 | I/Ib | Advanced solid Tumors | No response was seen with sabatolimab. 5 patients receiving combination treatment had PR (6%; lasting 12-27 months) in colorectal cancer (n = 2), NSCLC, malignant perianal melanoma, and SCLC. | [202] |
| PD-L1×TIM-3 | LY3415244 | NCT03752177 | I | Advanced solid Tumors | One patient with PD-1 refractory NSCLC had a near partial response (29.6%). | [203] | |
ICB: immune checkpoint blockades; ORR: objective response rate; DCR: disease control rate; PFS: progression-free survival; NSCLC: non-small cell lung cancer; CAR-T: chimeric antigen receptor T-cell immunotherapy.
PD-1/PD-L1 Blockade in Combination with Non-Immunotherapeutic Strategies
Besides, some preclinical studies and clinical trials confirmed that PD-1/PD-L1 blockade achieved synergetic anti-tumor treatment benefits when combined with some classical treatments. Here, we briefly discuss the combination with chemotherapy, radiotherapy, and targeted therapy.
Combination with chemotherapy
Chemotherapy is the classical treatment for cancers, but systematic toxic adverse effects limit its application. It was found that PD-1/PD-L1 blockade achieved synergetic anti-tumor treatment benefits when combined with chemotherapy. For example, in mouse model of pancreatic cancer liver metastasis, the group treated with gemcitabine and anti-PD-1 had the lowest average volume of liver metastatic nodules and prolonged survival, compared to gemcitabine alone or anti-PD-1 alone. The mechanism may be related to the enhancement of immune responses mediated by Th1 lymphocytes and M1 macrophages, as well as CD8+ T cells [173]. In a phase 3 clinical trial involving advanced gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma, the combination of nivolumab with chemotherapy showed superior overall survival (OS) and progression-free survival (PFS) benefits compared to chemotherapy alone, along with acceptable safety [174]. Additionally, a retrospective cohort study on nodular-type oral mucosal melanoma revealed that patients treated with chemotherapy combined with anti-PD-1 showed significant improvements in 2-year OS and PFS, and were safer and better tolerated, when compared to chemotherapy alone [175].
The clinical trials of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Clinicaltrial no. | Phase | Tumor types | Findings | References |
|---|---|---|---|---|---|---|---|
| PD-1×LAG-3 | Nivolumab | Relatlimab | NCT04205552 | II | Resectable NSCLC | Major pathological and objective radiographic responses were achieved in 27% and 10% (nivolumab) and in 30% and 27% (nivolumab and relatlimab) of patients, respectively. With 12 months median duration of follow-up, disease-free survival and overall survival rates at 12 months were 89% and 93% (nivolumab), and 93% and 100% (nivolumab and relatlimab). | [204] |
| PD-1×LAG-3 | Nivolumab | Relatlimab | NCT03470922 | II/III | Untreated advanced melanoma | The median PFS was 10.1 months; PFS at 12 months was 47.7%. | [205] |
| PD-1×LAG-3 | Spartalizumab | Ieramilimab | NCT02460224 | I/II | Advanced malignancies | Anti-tumor activity was observed in the combination arm, with 3 (2%) complete response and 10 (8%) partial response in a mixed population of tumor types. In the combination arm, eight patients (6.6%) experienced stable disease for 6 months or longer versus six patients (4.5%) in the single-agent arm. | [206] |
| PD-1×LAG-3 | Tebotelimab | NCT03219268 | I | Solid tumors and hematologic cancers | There were tumor decreases in 34% (59/172) of response-evaluable patients in the dose-escalation cohorts, with objective response in multiple solid tumor types, including PD-1-refractory disease, and in LAG-3 non-Hodgkin lymphomas, including CAR-T refractory disease. | [207] | |
ICB: immune checkpoint blockades; ORR: objective response rate; DCR: disease control rate; PFS: progression-free survival; NSCLC: non-small cell lung cancer; CAR-T: chimeric antigen receptor T-cell immunotherapy.
The clinical trials of the combination of PD-1/PD-L1 blockade with other ICBs in cancer therapy
| Targets | PD-1/PD-L1 blockade | Other ICB | Clinicaltrial no. | Phase | Tumor types | Findings | References |
|---|---|---|---|---|---|---|---|
| PD-1×TIGIT | Pembrolizumab | Vibostolimab | NCT02964013 | I | Advanced solid tumors, including NSCLC | Part A: confirmed ORR was 0% with monotherapy and 7% with combination therapy. Part B: confirmed ORR was 3% with monotherapy and 3% with combination therapy. | [208] |
| PD-L1×TIGIT | Atezolizumab | Tiragolumab | NCT03563716 | II | PD-L1-positive NSCLC | 21/67 patients (31.3%) in the combined group versus 11/68 patients (16.2%) in the placebo plus atezolizumab group had an objective response. Median PFS was 5.4 months in the combined group versus 3.6 months in the placebo plus atezolizumab group. | [209] |
| PD-1×IDO | Pembrolizumab | Epacadostat | NCT03414229 | II | Advanced sarcoma | The best ORR at 24 weeks was 3.3% (PR, 1/30). The median PFS was 7.6 weeks. Combined treatment was well tolerated and showed limited antitumor activity in sarcoma. | [210] |
| PD-1×IDO | Pembrolizumab | Epacadostat | NCT02752074 | III | Unresectable stage III or IV melanoma | No significant differences were found between the treatment groups for PFS (median 4.7 months for epacadostat plus pembrolizumab vs 4.9 months for placebo plus pembrolizumab) or overall survival. | [211] |
| PD-1×CD47 | Pembrolizumab | Evorpacept | NCT03013218 | I | Advanced solid tumours | Among patients who received evorpacept plus pembrolizumab, overall responses were recorded in 4/20 patients with HNSCC, in 1/ 20 patients with NSCLC, and in 4/19 patients with gastric or gastroesophageal junction cancer. | [212] |
| PD-L1×A2AR | Durvalumab | AZD4635 | NCT02740985 | Ia/b | Solid tumors | In patients with metastatic castration-resistant prostate cancer receiving monotherapy or combination treatment, tumor responses (2/39 and 6/37, respectively) and prostate-specific antigen responses (3/60 and 10/45, respectively) were observed. High versus low blood-based adenosine signature was associated with median PFS of 21 weeks versus 8.7 weeks. | [213] |
ICB: immune checkpoint blockades; ORR: objective response rate; DCR: disease control rate; PFS: progression-free survival; PR: partial response; NSCLC: non-small cell lung cancer; HNSCC: head and neck squamous cell carcinoma.
Combination with radiotherapy
Radiotherapy, as a first-line oncological therapy, plays the role of treatment, prevention of recurrence, and palliative care in different types of tumors. However, it is noted that radiotherapy may lead to the activation of immunosuppressive pathways in the TME by increasing TIL frequency, upregulating PD-L1 and MHC-I expression [176]. Thus, radiotherapy may benefit immunotherapy [177]. For example, in a systematic review and meta-analysis study on NSCLC, it was found that the combination therapy of PD-1/PD-L1 inhibitors and radiotherapy can improve the OS, PFS, and tumor response rate of advanced NSCLC patients without increasing serious adverse events [178]. Similarly, in another study, the combination therapy of radiotherapy and anti-PD-1/PD-L1 treatment had enhanced anti-tumor efficacy. Radiotherapy made nasopharyngeal carcinoma cells sensitive to the cytotoxic effects of NK cells, and upregulated the expression of PD-L1 on nasopharyngeal carcinoma cells and PD-1 on NK cells. Blocking the PD-L1/PD-1 checkpoint further increased the cytotoxicity of NK cells against nasopharyngeal carcinoma cells during radiotherapy [179].
Combination with targeted therapies
Targeted therapies, based on the unique molecular markers or signaling pathways in cancer cells, have achieved therapeutic effects in numerous cancers while minimized adverse effects on normal cells and tissues.
Epidermal growth factor receptor (EGFR)
In EGFR-mutant lung adenocarcinoma, the combination of EGFR inhibitor erlotinib and anti-PD-1 monoclonal antibody significantly inhibited tumor growth. However, this synergistic anti-tumor effect was not observed in EGFR wild-type tumors [180]. In another research, it was found that after acquiring resistance to the EGFR inhibitor gefitinib, the expression of PD-L1 in the specimens increased from less than 1% to 50% or more. This suggested that patients would be more likely to benefit from anti-PD-1 after EGFR inhibitor treatment, providing a rationale for the combination of EGFR-targeted drugs and PD-1/PD-L1 blockades [181].
Human epidermal growth factor receptor-2 (HER2)
HER2-positive is identified by the overexpression of the HER2 receptor due to HER2/ERBB2 gene amplification. It was confirmed that HER2-targeted therapy significantly improved post-treatment disease-free survival of HER2-positive breast cancer patients [182]. However, longtime treatment showed unsatisfactory response rate, development of drug resistance, and disease recurrence [183]. Combination therapy of anti-PD-1/PD-L1 (BMS-202) and trastuzumab (a drug targeting HER2) significantly reduced the survival rate and invasiveness of breast cancer cells [184]. In a clinical trial on HER2-positive gastric cancer, the addition of pembrolizumab to the standard treatment (trastuzumab and chemotherapy) achieved significant improvements, reducing tumor size and increasing the objective response rate [185].
Vascular endothelial growth factor (VEGF)
The abnormal tumor vasculature system may mediate immune suppression in the TME. The combination of VEGF-target drugs and anti-PD-1/PD-L1 had shown promise and potential effects [186]. In mouse model of small cell lung cancer, it was found that the combination of anti-VEGF and anti-PD-L1 had a synergistic effect, characterized by improved PFS, OS, and enhanced CD4+ T cell infiltration in the tumor [187]. In another study, the dual blockade of VEGFR-2/PD-1 significantly delayed tumor growth in mice with liver cancer. The potential mechanisms of combination therapy include reprogramming of the TME (such as a significant increase in the number of tumor-infiltrating CTLs, and upregulation of PD-L1 and PD-1 expression following VEGFR-2 blockade [188].
Poly-ADP-ribose polymerase (PARP)
PARP inhibitors, a class of drugs targeting poly-ADP-ribose polymerase, are often used to treat cancer patients with BRCA1 and BRCA2 gene mutations. In breast cancer, the PARP inhibitor significantly upregulated the expression of PD-L1 in cancer cells and in mouse model through the inactivation of GSK3β. Administering anti-PD-L1 treatment could restore the reduction of tumor-infiltrating cytotoxic CD8+ T cells after PARP inhibitor treatment [189]. In ovarian cancer, the PARP inhibitor significantly upregulated PD-L1 expression through the Chk1 pathway in vitro, and treatment with anti-PD-L1 could reverse the suppression of CD8+ T cells caused by PARP inhibitor treatment. Furthermore, combined anti-PD-L1 with PARP inhibitor treatment showed a synergistic anti-tumor efficacy in vivo [190].
Based on the above, anti-PD-1/PD-L1 therapy showed better response to some extent when combined with chemotherapy, radiotherapy, or targeted therapies. However, the combinations couldn't lead to a reduction in the incidence of treatment-related adverse events. In a phase 3 trial clinical study on gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma, it was found that 59% of the nivolumab-plus-chemotherapy group experienced grade 3-4 treatment-related adverse events, compared with 44% of the chemotherapy group [174]. In another clinical trial (phase 3), pembrolizumab plus chemotherapy was superior to placebo plus chemotherapy for progression-free survival in patients with oesophageal squamous cell carcinoma (6.3 months vs 5.8 months). However, 266 (72%) patients in the pembrolizumab plus chemotherapy group experienced treatment-related adverse events of grade 3 or higher, while 250 (68%) patients in the placebo plus chemotherapy group [191]. In addition, in a phase 2 clinical trial of platinum-resistant recurrent or metastatic nasopharyngeal carcinoma, the objective response rate was significantly higher in the bevacizumab and pembrolizumab group (58.3% [95% confidence interval: 36.6-77.9] than that in the pembrolizumab group (12.5% [2.7-32.4], and the grade 3 treatment-related adverse events occurred in 29% (7/24) of the combination group compared to 8% (2/24) in the pembrolizumab group [192]. It can be seen that the therapeutic potential of PD-1/PD-L1 blockade in combination with non-immunotherapeutic strategies. However, the treatment-related adverse events need to be addressed, such as irAEs induced by PD-1/PD-L1 blockade, systematic toxicity induced by chemotherapy, local dermatitis induced by radiotherapy, etc.
Conclusion and Prospects
In recent years, despite the rapid development of ICBs in the field of tumor treatment, their clinical use is limited by low response rates and the potential problem of drug resistance. Therefore, it is urgent to expand the population that can benefit from ICBs and overcome resistance. Among them, the PD-1/PD-L1 is the most widely studied and promising immune checkpoint in cancer immunotherapy research and clinical application. Thus, it is important and meaningful to investigate the complex mechanisms of PD-1/PD-L1 blockade resistance. In this review, we summarized the vital aspects. However, it should be recognized that resistance mechanisms are dynamic and complex due to the high heterogeneity of patients and tumors. In the current review, we focused on the strategies of combining PD-1/PD-L1 blockade with other ICBs, which showed synergetic anti-tumor effects in preclinical and clinical studies.
For the combination therapy of PD-1/PD-L1 and other ICBs, some issues still need to be resolved and studied: (1) To uncover more targets and more effective immune checkpoint combinations based on preclinical and clinical research; (2) Personalized cancer immunotherapy treatment plans need to be formulated based on tumor heterogeneities, drug resistance issues, and individual immune status; (3) Further research on biomarkers for combination therapy is needed, such as discovering more effective biomarkers, combining multiple biomarkers, developing non-invasive biomarker detection, etc., to predict treatment response and adverse reactions more predictively; (4) Preclinical research related to combination therapy needs more precise research for clinical translation, such as tumor organoid culture can simulate the TME and thus better predict drug sensitivity and assess prognosis; (5) Combination therapy plans still need to be optimized from multiple aspects such as efficacy, safety, avoiding and handling immune-related adverse events.
Abbreviations
PD-1: programmed death-1; PD-L1: programmed death-ligand 1; TME: tumor microenvironment; ICBs: immune checkpoints blockades; ITIM: immune receptor tyrosine-based inhibitory motif; ITSM: immune receptor inhibitory tyrosine-based switch motif; TCR: T cell receptor; irAEs: immune-related adverse events; SD: stable disease period; TILs: tumor-infiltrating lymphocytes; CAFs: cancer-associated fibroblasts; MDSCs: myeloid-derived suppressor cells; TAMs: tumor-associated macrophages; CA-MSCs: cancer-associated mesenchymal stem cells; MHC: major histocompatibility complex; ECM: extracellular matrix; LSD1: lysine-specific demethylase 1; NSCLC: non-small cell lung cancer; APCs: antigen-presenting cells; TMB: tumor mutation burden; MMR: mismatch repair; MSI: microsatellite instability; Tregs: regulatory T cells; CTLs: cytotoxic T lymphocytes; FMT: fecal microbiota transplantation; CTLA-4: cytotoxic T-lymphocyte associated antigen-4; TIM-3: T cell immunoglobulin-3; LAG-3: lymphocyte activation gene 3; TALs: tumor-associated lymphocytes; TIGIT: T cell immunoreceptor with immunoglobulin and ITIM domain; VISTA: V-domain Ig suppressor of T cell activation; IDO: indoleamine 2,3-dioxygenase; A2AR: adenosine A2A receptor; TSP-1: thrombospondin-1; OS: overall survival; PFS: progression-free survival; EGFR: epidermal growth factor receptor; HER2: human epidermal growth factor receptor-2; PARP: poly-ADP-ribose polymerase; VEGF: vascular endothelial growth factor.
Acknowledgements
We thank Professor Zhaowu Ma for the critical expert review of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81602303), and the Start Fund of First Affiliated Hospital of Yangtze University (2022DIF01).
Author contributions
LDL and XY initiated the topic and designed the review. LDL and XHY collected the related literature, wrote and edited the manuscript. ZD made the figures and tables. WYF collected the related literature. XY and ZWJ revised the manuscript, and provided feedback and guidance. All authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350-5
2. Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2018;175:313-26
3. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
4. Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW. et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients with First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1571-80
5. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD. et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J Clin Oncol. 2022;40:127-37
6. André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C. et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383:2207-18
7. Eggermont AMM, Kicinski M, Blank CU, Mandala M, Long GV, Atkinson V. et al. Association Between Immune-Related Adverse Events and Recurrence-Free Survival Among Patients with Stage III Melanoma Randomized to Receive Pembrolizumab or Placebo: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2020;6:519-27
8. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X. et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129
9. Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J. et al. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021;11:838-57
10. Kim TK, Herbst RS, Chen L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol. 2018;39:624-31
11. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo j. 1992;11:3887-95
12. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704
13. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515-48
14. Jiang Y, Chen M, Nie H, Yuan Y. PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother. 2019;15:1111-22
15. Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J. et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin Cancer Res. 2017;23:3158-67
16. Fu S, Li J, You J, Liu S, Dong Q, Fu Y. et al. Baicalin attenuates PD-1/PD-L1 axis-induced immunosuppression in piglets challenged with Glaesserella parasuis by inhibiting the PI3K/Akt/mTOR and RAS/MEK/ERK signalling pathways. Vet Res. 2024;55:95
17. Thompson RH, Dong H, Kwon ED. Implications of B7-H1 expression in clear cell carcinoma of the kidney for prognostication and therapy. Clin Cancer Res. 2007;13:709s-15s
18. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104:3360-5
19. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC. et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170:1120-33.e17
20. Mojic M, Takeda K, Hayakawa Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int J Mol Sci. 2017;19:e89
21. Sanmamed MF, Chen L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014;20:256-61
22. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800
23. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT. et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116
24. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel). 2020;12:738
25. Rodrigues M, Vanoni G, Loap P, Dubot C, Timperi E, Minsat M. et al. Nivolumab plus chemoradiotherapy in locally-advanced cervical cancer: the NICOL phase 1 trial. Nat Commun. 2023;14:3698
26. Balar AV, Kamat AM, Kulkarni GS, Uchio EM, Boormans JL, Roumiguié M. et al. Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unresponsive to BCG (KEYNOTE-057): an open-label, single-arm, multicentre, phase 2 study. Lancet Oncol. 2021;22:919-30
27. Wang FH, Wei XL, Feng J, Li Q, Xu N, Hu XC. et al. Efficacy, Safety, and Correlative Biomarkers of Toripalimab in Previously Treated Recurrent or Metastatic Nasopharyngeal Carcinoma: A Phase II Clinical Trial (POLARIS-02). J Clin Oncol. 2021;39:704-12
28. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73:6900-12
29. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60-5
30. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K. et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447-52
31. Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A. et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114:4993-8
32. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
33. Das S, Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer. 2019;7:306
34. Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. 2017;28:2377-85
35. Wang SJ, Dougan SK, Dougan M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer. 2023;9:543-53
36. de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm Metab Res. 2019;51:145-56
37. Axelrod ML, Meijers WC, Screever EM, Qin J, Carroll MG, Sun X. et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature. 2022;611:818-26
38. Sonpavde GP, Grivas P, Lin Y, Hennessy D, Hunt JD. Immune-related adverse events with PD-1 versus PD-L1 inhibitors: a meta-analysis of 8730 patients from clinical trials. Future Oncol. 2021;17:2545-58
39. Poto R, Troiani T, Criscuolo G, Marone G, Ciardiello F, Tocchetti CG. et al. Holistic Approach to Immune Checkpoint Inhibitor-Related Adverse Events. Front Immunol. 2022;13:804597
40. Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I. et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol. 2019;30:1080-7
41. Kluger HM, Tawbi HA, Ascierto ML, Bowden M, Callahan MK. et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer. 2020;8:e00089
42. Vesely MD, Zhang T, Chen L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu Rev Immunol. 2022;40:45-74
43. Zhang T, Yu W, Cheng X, Yeung J, Ahumada V, Norris PC. et al. Up-regulated PLA2G10 in cancer impairs T cell infiltration to dampen immunity. Sci Immunol. 2024;9:eadh2334
44. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74-80
45. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203-13
46. Cascio S, Chandler C, Zhang L, Sinno S, Gao B, Onkar S. et al. Cancer-associated MSC drive tumor immune exclusion and resistance to immunotherapy, which can be overcome by Hedgehog inhibition. Sci Adv. 2021;7:eabi5790
47. Desbois M, Udyavar AR, Ryner L, Kozlowski C, Guan Y, Dürrbaum M. et al. Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nat Commun. 2020;11:5583
48. Li L, Wei JR, Dong J, Lin QG, Tang H, Jia YX. et al. Laminin γ2-mediating T cell exclusion attenuates response to anti-PD-1 therapy. Sci Adv. 2021;7:eabc8346
49. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231-5
50. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265-76
51. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW. et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326-36
52. Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA. et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat Commun. 2020;11:4520
53. Meng Y, Ye F, Nie P, Zhao Q, An L, Wang W. et al. Immunosuppressive CD10(+)ALPL(+) neutrophils promote resistance to anti-PD-1 therapy in HCC by mediating irreversible exhaustion of T cells. J Hepatol. 2023;79:1435-49
54. Liu Y, Debo B, Li M, Shi Z, Sheng W, Shi Y. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat Commun. 2021;12:6831
55. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H. et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol. 2019;4:eaay0555
56. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B. et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10
57. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A. et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188-201
58. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139-45
59. Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med. 2016;213:2835-40
60. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W. et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382-6
61. Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W. et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28:862-4
62. Yin Z, Yu M, Ma T, Zhang C, Huang S, Karimzadeh MR. et al. Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: a key role of exosomal PD-L1. J Immunother Cancer. 2021;9:e001698
63. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479-85
64. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298-312
65. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13
66. Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A. et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat Commun. 2020;11:1897
67. Ziogas DC, Theocharopoulos C, Koutouratsas T, Haanen J, Gogas H. Mechanisms of resistance to immune checkpoint inhibitors in melanoma: What we have to overcome? Cancer Treat Rev. 2023;113:102499
68. Tu J, Xu H, Ma L, Li C, Qin W, Chen X. et al. Nintedanib enhances the efficacy of PD-L1 blockade by upregulating MHC-I and PD-L1 expression in tumor cells. Theranostics. 2022;12:747-66
69. Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res. 2020;26:290-300
70. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med. 2017;377:2500-1
71. Sun JY, Zhang D, Wu S, Xu M, Zhou X, Lu XJ. et al. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms, predictive factors, and future perspectives. Biomark Res. 2020;8:35
72. Ricciuti B, Wang X, Alessi JV, Rizvi H, Mahadevan NR, Li YY. et al. Association of High Tumor Mutation Burden in Non-Small Cell Lung Cancers with Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA Oncol. 2022;8:1160-8
73. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353-65
74. Forschner A, Battke F, Hadaschik D, Schulze M, Weißgraeber S, Han CT. et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J Immunother Cancer. 2019;7:180
75. Pu Y, Ji Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front Immunol. 2022;13:874589
76. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75
77. Wang Y, Shang K, Zhang N, Zhao J, Cao B. Tumor-Associated Macrophage-Derived Exosomes Promote the Progression of Gastric Cancer by Regulating the P38MAPK Signaling Pathway and the Immune Checkpoint PD-L1. Cancer Biother Radiopharm. 2021;36:455-464
78. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C. et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130:5380-96
79. Liu CQ, Xu J, Zhou ZG, Jin LL, Yu XJ, Xiao G. et al. Expression patterns of programmed death ligand 1 correlate with different microenvironments and patient prognosis in hepatocellular carcinoma. Br J Cancer. 2018;119:80-8
80. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS. et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017;9:eaal3604
81. Nixon BG, Kuo F, Ji L, Liu M, Capistrano K, Do M. et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity. 2022;55:2044-58.e5
82. Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y. et al. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22:29
83. Alspach E, Lussier DM, Schreiber RD. Interferon γ and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol. 2019;11(3):a028480
84. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151-67
85. Jeong SK, Yang K, Park YS, Choi YJ, Oh SJ, Lee CW. et al. Interferon gamma induced by resveratrol analog, HS-1793, reverses the properties of tumor associated macrophages. Int Immunopharmacol. 2014;22:303-10
86. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413-8
87. Horn S, Leonardelli S, Sucker A, Schadendorf D, Griewank KG, Paschen A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J Natl Cancer Inst. 2018;110:677-81
88. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544-8
89. Bule P, Aguiar SI, Aires-Da-Silva F, Dias JNR. Chemokine-Directed Tumor Microenvironment Modulation in Cancer Immunotherapy. Int J Mol Sci. 2021;22:9804
90. Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6:e1249561
91. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501
92. Kumar A, Chamoto K. Immune metabolism in PD-1 blockade-based cancer immunotherapy. Int Immunol. 2021;33:17-26
93. Fang X, Zhao P, Gao S, Liu D, Zhang S, Shan M. et al. Lactate induces tumor-associated macrophage polarization independent of mitochondrial pyruvate carrier-mediated metabolism. Int J Biol Macromol. 2023;237:123810
94. Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q. et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723-33
95. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W. et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249-53
96. Ding L, Lu S, Li Y. Regulation of PD-1/PD-L1 Pathway in Cancer by Noncoding RNAs. Pathol Oncol Res. 2020;26:651-63
97. Zhang LX, Gao J, Long X, Zhang PF, Yang X, Zhu SQ. et al. The circular RNA circHMGB2 drives immunosuppression and anti-PD-1 resistance in lung adenocarcinomas and squamous cell carcinomas via the miR-181a-5p/CARM1 axis. Mol Cancer. 2022;21:110
98. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML. et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104-8
99. Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H. et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol Res. 2020;8:1251-61
100. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91-7
101. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459-65
102. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131:58-67
103. Manches O, Fernandez MV, Plumas J, Chaperot L, Bhardwaj N. Activation of the noncanonical NF-κB pathway by HIV controls a dendritic cell immunoregulatory phenotype. Proc Natl Acad Sci U S A. 2012;109:14122-7
104. Lax BM, Palmeri JR, Lutz EA, Sheen A, Stinson JA, Duhamel L. et al. Both intratumoral regulatory T cell depletion and CTLA-4 antagonism are required for maximum efficacy of anti-CTLA-4 antibodies. Proc Natl Acad Sci U S A. 2023;120:e2300895120
105. Kitano S, Tsuji T, Liu C, Hirschhorn-Cymerman D, Kyi C, Mu Z. et al. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol Res. 2013;1:235-44
106. Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P. et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin Cancer Res. 2016;22:2908-18
107. Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M. et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63:675-83
108. Fiegle E, Doleschel D, Koletnik S, Rix A, Weiskirchen R, Borkham-Kamphorst E. et al. Dual CTLA-4 and PD-L1 Blockade Inhibits Tumor Growth and Liver Metastasis in a Highly Aggressive Orthotopic Mouse Model of Colon Cancer. Neoplasia. 2019;21:932-44
109. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541-50
110. Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Domenig C, Habicht A. et al. Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J Immunol. 2005;174:3408-15
111. Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother. 2017;66:1609-17
112. Robert L, Harview C, Emerson R, Wang X, Mok S, Homet B. et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology. 2014;3:e29244
113. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543-53
114. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275-80
115. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591-603
116. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235:172-89
117. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276:97-111
118. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173-85
119. Tomkowicz B, Walsh E, Cotty A, Verona R, Sabins N, Kaplan F. et al. TIM-3 Suppresses Anti-CD3/CD28-Induced TCR Activation and IL-2 Expression through the NFAT Signaling Pathway. PLoS One. 2015;10:e0140694
120. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175-86
121. Kuai W, Xu X, Yan J, Zhao W, Li Y, Wang B. et al. Prognostic Impact of PD-1 and Tim-3 Expression in Tumor Tissue in Stage I-III Colorectal Cancer. Biomed Res Int. 2020;2020:5294043
122. He X, Peng Y, He G, Ye H, Liu L, Zhou Q. et al. Increased co-expression of PD1 and TIM3 is associated with poor prognosis and immune microenvironment heterogeneity in gallbladder cancer. J Transl Med. 2023;21:717
123. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH. et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117:4501-10
124. Zhang XS, Zhou HC, Wei P, Chen L, Ma WH, Ding L. et al. Combined TIM-3 and PD-1 blockade restrains hepatocellular carcinoma development by facilitating CD4+ and CD8+ T cell-mediated antitumor immune responses. World J Gastrointest Oncol. 2023;15:2138-49
125. Lee JB, Ha SJ, Kim HR. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front Pharmacol. 2021;12:681320
126. Hannier S, Triebel F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. Int Immunol. 1999;11:1745-52
127. Guy C, Mitrea DM, Chou PC, Temirov J, Vignali KM, Liu X. et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat Immunol. 2022;23:757-67
128. Okoye I, Namdar A, Xu L, Crux N, Elahi S. Atorvastatin downregulates co-inhibitory receptor expression by targeting Ras-activated mTOR signalling. Oncotarget. 2017;8:98215-32
129. Paik J. Nivolumab Plus Relatlimab: First Approval. Drugs. 2022;82:925-31
130. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359-77
131. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T. et al. Expression Analysis and Significance of PD-1, LAG-3, and TIM-3 in Human Non-Small Cell Lung Cancer Using Spatially Resolved and Multiparametric Single-Cell Analysis. Clin Cancer Res. 2019;25:4663-73
132. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48-57
133. Liu L, Wang A, Liu X, Han S, Sun Y, Zhang J. et al. Blocking TIGIT/CD155 signalling reverses CD8(+) T cell exhaustion and enhances the antitumor activity in cervical cancer. J Transl Med. 2022;20:280
134. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020;8:e000957
135. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344-55
136. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C. et al. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J Clin Invest. 2015;125:2046-58
137. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS. et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7:e1466769
138. Noelle RJ, Lines JL, Lewis LD, Martell RE, Guillaudeux T, Lee SW. et al. Clinical and research updates on the VISTA immune checkpoint: immuno-oncology themes and highlights. Front Oncol. 2023;13:1225081
139. ElTanbouly MA, Zhao Y, Nowak E, Li J, Schaafsma E, Le Mercier I. et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science. 2020;367(6475):eaay0524
140. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A. et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. 2019;574:565-70
141. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C. et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. 2019;156:74-85
142. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD. et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A. 2015;112:6682-7
143. Schaafsma E, Croteau W, ElTanbouly M, Nowak EC, Smits NC, Deng J. et al. VISTA Targeting of T-cell Quiescence and Myeloid Suppression Overcomes Adaptive Resistance. Cancer Immunol Res. 2023;11:38-55
144. Dounay AB, Tuttle JB, Verhoest PR. Challenges and Opportunities in the Discovery of New Therapeutics Targeting the Kynurenine Pathway. J Med Chem. 2015;58:8762-82
145. Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol. 2007;7:817-23
146. Liu X, Yang M, Xu P, Du M, Li S, Shi J. et al. Kynurenine-AhR reduces T-cell infiltration and induces a delayed T-cell immune response by suppressing the STAT1-CXCL9/CXCL10 axis in tuberculosis. Cell Mol Immunol. 2024;21:1426-40
147. Volarevic V, Markovic BS, Jankovic MG, Djokovic B, Jovicic N, Harrell CR. et al. Galectin 3 protects from cisplatin-induced acute kidney injury by promoting TLR-2-dependent activation of IDO1/Kynurenine pathway in renal DCs. Theranostics. 2019;9:5976-6001
148. Théate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC. et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015;3:161-72
149. Zhou QH, Han H, Lu JB, Liu TY, Huang KB, Deng CZ. et al. Up-regulation of indoleamine 2,3-dioxygenase 1 (IDO1) expression and catalytic activity is associated with immunosuppression and poor prognosis in penile squamous cell carcinoma patients. Cancer Commun (Lond). 2020;40:3-15
150. Lin DJ, Ng JCK, Huang L, Robinson M, O'Hara J, Wilson JA. et al. The immunotherapeutic role of indoleamine 2,3-dioxygenase in head and neck squamous cell carcinoma: A systematic review. Clin Otolaryngol. 2021;46:919-34
151. Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S. et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol Cancer Ther. 2018;17:2530-42
152. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3
153. Han X, Cheng K, Xu Y, Wang Y, Min H, Zhang Y. et al. Modularly Designed Peptide Nanoprodrug Augments Antitumor Immunity of PD-L1 Checkpoint Blockade by Targeting Indoleamine 2,3-Dioxygenase. J Am Chem Soc. 2020;142:2490-6
154. Volaric A, Gentzler R, Hall R, Mehaffey JH, Stelow EB, Bullock TN. et al. Indoleamine-2,3-Dioxygenase in Non-Small Cell Lung Cancer: A Targetable Mechanism of Immune Resistance Frequently Coexpressed With PD-L1. Am J Surg Pathol. 2018;42:1216-23
155. Reinhold MI, Lindberg FP, Plas D, Reynolds S, Peters MG, Brown EJ. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47). J Cell Sci. 1995;108( Pt 11):3419-25
156. Jia X, Yan B, Tian X, Liu Q, Jin J, Shi J. et al. CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int J Biol Sci. 2021;17:3281-7
157. Li Y, Zhou H, Liu P, Lv D, Shi Y, Tang B. et al. SHP2 deneddylation mediates tumor immunosuppression in colon cancer via the CD47/SIRPα axis. J Clin Invest. 2023;133:e162870
158. Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X. et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 2019;12:124
159. Miller TW, Kaur S, Ivins-O'Keefe K, Roberts DD. Thrombospondin-1 is a CD47-dependent endogenous inhibitor of hydrogen sulfide signaling in T cell activation. Matrix Biol. 2013;32:316-24
160. Wang R, Zhang C, Cao Y, Wang J, Jiao S, Zhang J. et al. Blockade of dual immune checkpoint inhibitory signals with a CD47/PD-L1 bispecific antibody for cancer treatment. Theranostics. 2023;13:148-60
161. Chen SH, Dominik PK, Stanfield J, Ding S, Yang W, Kurd N. et al. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J Immunother Cancer. 2021;9:e003464
162. Nan Y, Zhang X, Wang S, Xu C, Wang Y, Han L. et al. Targeting CD47 enhanced the antitumor immunity of PD-L1 blockade in B-cell lymphoma. Immunotherapy. 2023;15:175-87
163. Sun C, Wang B, Hao S. Adenosine-A2A Receptor Pathway in Cancer Immunotherapy. Front Immunol. 2022;13:837230
164. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M. et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018;78:1003-16
165. Jenabian MA, Seddiki N, Yatim A, Carriere M, Hulin A, Younas M. et al. Regulatory T cells negatively affect IL-2 production of effector T cells through CD39/adenosine pathway in HIV infection. PLoS Pathog. 2013;9:e1003319
166. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A(2A) Adenosine Receptor Gene Deletion or Synthetic A(2A) Antagonist Liberate Tumor-Reactive CD8(+) T Cells from Tumor-Induced Immunosuppression. J Immunol. 2018;201:782-91
167. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK. et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020;10:40-53
168. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S. et al. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res. 2015;3:506-17
169. Kamai T, Kijima T, Tsuzuki T, Nukui A, Abe H, Arai K. et al. Increased expression of adenosine 2A receptors in metastatic renal cell carcinoma is associated with poorer response to anti-vascular endothelial growth factor agents and anti-PD-1/Anti-CTLA4 antibodies and shorter survival. Cancer Immunol Immunother. 2021;70:2009-21
170. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J. et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother. 2018;67:1271-84
171. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D. et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006-17
172. Kelly RJ, Landon BV, Zaidi AH, Singh D, Canzoniero JV, Balan A. et al. Neoadjuvant nivolumab or nivolumab plus LAG-3 inhibitor relatlimab in resectable esophageal/gastroesophageal junction cancer: a phase Ib trial and ctDNA analyses. Nat Med. 2024;30:1023-34
173. Ho TTB, Nasti A, Seki A, Komura T, Inui H, Kozaka T. et al. Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J Immunother Cancer. 2020;8:e001367
174. Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27-40
175. Wu Y, Wei D, Ren G, Guo W. Chemotherapy in combination with anti-PD-1 agents as adjuvant therapy for high-risk oral mucosal melanoma. J Cancer Res Clin Oncol. 2023;149:2293-300
176. Jarosz-Biej M, Smolarczyk R, Cichoń T, Kułach N. Tumor Microenvironment as A "Game Changer" in Cancer Radiotherapy. Int J Mol Sci. 2019;20:3212
177. Kordbacheh T, Honeychurch J, Blackhall F, Faivre-Finn C, Illidge T. Radiotherapy and anti-PD-1/PD-L1 combinations in lung cancer: building better translational research platforms. Ann Oncol. 2018;29:301-10
178. Geng Y, Zhang Q, Feng S, Li C, Wang L, Zhao X. et al. Safety and Efficacy of PD-1/PD-L1 inhibitors combined with radiotherapy in patients with non-small-cell lung cancer: a systematic review and meta-analysis. Cancer Med. 2021;10:1222-39
179. Makowska A, Lelabi N, Nothbaum C, Shen L, Busson P, Tran TTB. et al. Radiotherapy Combined with PD-1 Inhibition Increases NK Cell Cytotoxicity towards Nasopharyngeal Carcinoma Cells. Cells. 2021;10:2458
180. Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, Kataoka K. et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci Immunol. 2020;5:eaav3937
181. Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K. et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165
182. Puglisi F, Fontanella C, Amoroso V, Bianchi GV, Bisagni G, Falci C. et al. Current challenges in HER2-positive breast cancer. Crit Rev Oncol Hematol. 2016;98:211-21
183. Vernieri C, Milano M, Brambilla M, Mennitto A, Maggi C, Cona MS. et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: Current knowledge, new research directions and therapeutic perspectives. Crit Rev Oncol Hematol. 2019;139:53-66
184. Padmanabhan R, Kheraldine H, Gupta I, Meskin N, Hamad A, Vranic S. et al. Quantification of the growth suppression of HER2+ breast cancer colonies under the effect of trastuzumab and PD-1/PD-L1 inhibitor. Front Oncol. 2022;12:977664
185. Janjigian YY, Kawazoe A, Yañez P, Li N, Lonardi S, Kolesnik O. et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature. 2021;600:727-30
186. Hack SP, Zhu AX, Wang Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front Immunol. 2020;11:598877
187. Meder L, Schuldt P, Thelen M, Schmitt A, Dietlein F, Klein S. et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018;78:4270-81
188. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A. et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology. 2020;71:1247-61
189. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM. et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017;23:3711-20
190. Xue C, Xu Y, Ye W, Xie Q, Gao H, Xu B. et al. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol Oncol. 2020;157:222-33
191. Sun JM, Shen L, Shah MA, Enzinger P, Adenis A, Doi T. et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet. 2021;398:759-71
192. Chong WQ, Low JL, Tay JK, Le TBU, Goh GS, Sooi K. et al. Pembrolizumab with or without bevacizumab in platinum-resistant recurrent or metastatic nasopharyngeal carcinoma: a randomised, open-label, phase 2 trial. Lancet Oncol. 2025;26:175-86
193. Wang Y, Ni H, Zhou S, He K, Gao Y, Wu W. et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother. 2021;70:365-76
194. VanderWalde A, Bellasea SL, Kendra KL, Khushalani NI, Campbell KM, Scumpia PO. et al. Ipilimumab with or without nivolumab in PD-1 or PD-L1 blockade refractory metastatic melanoma: a randomized phase 2 trial. Nat Med. 2023;29:2278-85
195. Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Dô P, Bylicki O. et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019;20:239-53
196. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD. et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23-34
197. Lee HS, Jang HJ, Ramineni M, Wang DY, Ramos D, Choi JM. et al. A Phase II Window of Opportunity Study of Neoadjuvant PD-L1 versus PD-L1 plus CTLA-4 Blockade for Patients with Malignant Pleural Mesothelioma. Clin Cancer Res. 2023;29:548-59
198. Siu LL, Even C, Mesía R, Remenar E, Daste A, Delord JP. et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1-Low/Negative Recurrent or Metastatic HNSCC: The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol. 2019;5:195-203
199. Chen QY, Guo SS, Luo Y, Qu S, Wu DH, Chen XZ. et al. Efficacy and safety of cadonilimab in previously treated recurrent or metastatic nasopharyngeal carcinoma(COMPASSION-06): A phase II multicenter study. Oral Oncol. 2024;151:106723
200. Hollebecque A, Chung HC, de Miguel MJ, Italiano A, Machiels JP, Lin CC. et al. Safety and Antitumor Activity of α-PD-L1 Antibody as Monotherapy or in Combination with α-TIM-3 Antibody in Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Tumors. Clin Cancer Res. 2021;27:6393-404
201. Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J. et al. Blocking TIM-3 in Treatment-refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 Antibody. Clin Cancer Res. 2021;27:2168-78
202. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM. et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin Cancer Res. 2021;27:3620-9
203. Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT. et al. Safety and Immunogenicity of LY3415244, a Bispecific Antibody Against TIM-3 and PD-L1, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2021;27:2773-81
204. Schuler M, Cuppens K, Plönes T, Wiesweg M, Du Pont B, Hegedus B. et al. Neoadjuvant nivolumab with or without relatlimab in resectable non-small-cell lung cancer: a randomized phase 2 trial. Nat Med. 2024;30:1602-11
205. Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E. et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med. 2022;386:24-34
206. Schöffski P, Tan DSW, Martín M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD. et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J Immunother Cancer. 2022;10:e003925
207. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV. et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. 2023;29:2814-24
208. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M. et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann Oncol. 2022;33:169-80
209. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N. et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022;23:781-92
210. Kelly CM, Qin LX, Whiting KA, Richards AL, Avutu V, Chan JE. et al. A Phase II Study of Epacadostat and Pembrolizumab in Patients with Advanced Sarcoma. Clin Cancer Res. 2023;29:2043-51
211. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S. et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083-97
212. Lakhani NJ, Chow LQM, Gainor JF, LoRusso P, Lee KW, Chung HC. et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021;22:1740-51
213. Lim EA, Bendell JC, Falchook GS, Bauer TM, Drake CG, Choe JH. et al. Phase Ia/b, Open-Label, Multicenter Study of AZD4635 (an Adenosine A2A Receptor Antagonist) as Monotherapy or Combined with Durvalumab, in Patients with Solid Tumors. Clin Cancer Res. 2022;28:4871-84
Author contact
![]() Corresponding authors: Ying Xiang, PhD, Associated Professor, email: xying316edu.cn, Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, 1 Nanhuan Road, Jingzhou, Hubei 434023, China. Weijia Zhang, PhD, Associate Chief Physician, email: happyweijia100com, Department of Oncology, First Affiliated Hospital of Yangtze University, No. 8 Hang Kong Road, Shashi District, Jingzhou, Hubei 434023, China.
Corresponding authors: Ying Xiang, PhD, Associated Professor, email: xying316edu.cn, Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, 1 Nanhuan Road, Jingzhou, Hubei 434023, China. Weijia Zhang, PhD, Associate Chief Physician, email: happyweijia100com, Department of Oncology, First Affiliated Hospital of Yangtze University, No. 8 Hang Kong Road, Shashi District, Jingzhou, Hubei 434023, China.