Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(3):417-433. doi:10.7150/jca.80097 This issue Cite
Review
Potential clinical treatment prospects behind the molecular mechanism of alternative lengthening of telomeres (ALT)
Haolu Sun2 ![]() *, Guijuan Chen1*, Baochang Guo3, Shushu Lv4, Guojun Yuan1
*, Guijuan Chen1*, Baochang Guo3, Shushu Lv4, Guojun Yuan1 ![]()
1. School of Environment and Chemical Engineering, Anhui Vocational and Technical College, Hefei, 230011, China.
2. Department of Pathophysiology, School of Basic Medical Sciences, Anhui Medical University, Hefei, 230011, China.
3. Rehabilitation Department of Traditional Chinese Medicine, 969 Hospital of the Joint Support Force of the Chinese People's Liberation Army, Hohhot, 010000, China.
4. Department of Pathology, The First Affiliated Hospital of Huzhou University, Huzhou 313000, China.
*These authors contributed equally to this work.
Received 2022-10-22; Accepted 2022-12-25; Published 2023-1-31
Abstract

Normal somatic cells inevitably experience replicative stress and senescence during proliferation. Somatic cell carcinogenesis can be prevented in part by limiting the reproduction of damaged or old cells and removing them from the cell cycle [1, 2]. However, Cancer cells must overcome the issues of replication pressure and senescence as well as preserve telomere length in order to achieve immortality, in contrast to normal somatic cells [1, 2]. Although telomerase accounts for the bulk of telomere lengthening methods in human cancer cells, there is a non-negligible portion of telomere lengthening pathways that depend on alternative lengthening of telomeres (ALT) [3]. For the selection of novel possible therapeutic targets for ALT-related disorders, a thorough understanding of the molecular biology of these diseases is crucial [4]. The roles of ALT, typical ALT tumor cell traits, the pathophysiology and molecular mechanisms of ALT tumor disorders, such as adrenocortical carcinoma (ACC), are all summarized in this work. Additionally, this research compiles as many of its hypothetically viable but unproven treatment targets as it can (ALT-associated PML bodies (APB), etc.). This review is intended to contribute as much as possible to the development of research, while also trying to provide a partial information for prospective investigations on ALT pathways and associated diseases.
Keywords: Telomerase, ALT, molecular mechanism, diseases, therapeutic targets
Introduction
Telomeres are short sections of DNA-protein complexes found at the ends of linear chromosomes in eukaryotic cells. Telomeres are shielded by complex proteins, which prevents them from being identified as DNA double-strand breaks (DSBs) [4]. Telomeres shorten with each cell cycle as a result of lagging replication fork strands' inability to duplicate telomere ends during somatic cell division [5]. We can anticipate that telomere attrition will eventually result in cellular senescence, which is a barrier to carcinogenesis, if cells continue to proliferate [1, 2]. Cancer is characterized by genomic instability, and DNA replication is the biological mechanism most likely to cause instability. Replication stress, a cause of genomic instability and a trait of precancerous and cancerous cells, is caused by any circumstance that produces a lot of DNA damage [6]. Under normal circumstances, the body restricts replication stress cells reproduction and halts the cell cycle [6]. Therefore, to obtain eternal life by preserving telomere length during proliferation, cancer cells must overcome replication stress and senescence [2, 5]. A small number of human cancers, such as adrenocortical carcinoma, neuroblastoma cell tumor, osteosarcoma, and astrocytoma, rely on the alternative lengthening of telomerase (ALT) pathway to cause telomere shortening, despite the fact that the majority of human malignancies (>85%) use telomerase to lengthen telomeres [3, 7-9].
Some cancer types have been reported to contain ALT+, and ALT may offer new possibilities for the next clinical cancer studies. The development of new clinical medicines is based on molecular biological research on ALT tumors, which is also required for the diagnosis and management of ALT tumors and other related disorders. This article seeks to provide information for researchers to investigate ALT tumors and associated diseases by summarizing the primary mechanisms of telomere lengthening and ALT tumor diseases as they are now understood.
What is the ALT mechanism?
Many of the most resistant cancer subtypes exhibit ALT, a telomere preservation mechanism [10]. As we discuss ALT, we have to introduce telomeres. Telomeres become important in preserving the stability of the human DNA [3]. First of all, telomeres shield linear chromosomal ends from being identified as DNADSBs [11]. Then, through the start of cellular senescence, the increasing wear of telomeres that results from each round of cell division has the power to suppress malignancies and restrict cell division [3, 12]. Activating the telomere maintenance mechanism (TMM) allows tumor cells to multiply quickly and become immortal [3]. To achieve TMM, 85-90% of these tumor cells activate telomerase [13]. However, the others 10%-15% of tumor cells through the ALT mechanism [3].
Telomeres structures and ALT tumor cells' typical property
Telomeres structures
Telomeres are conserved nucleoprotein structures localized at the ends of eukaryotic linear chromosomes. (1) Double-stranded TTAGGG repeats can be found in the genetic material [14]. (2) Telomere-associated proteins, such as the Shelterin complex [14]. Telomere repeat binding factor 1 (TRF1), telomere repeat binding factor 2 (TRF2), TRF1-interacting nuclear protein 1 (TIN1), repressor-activator protein 1 homolog (RAP1), tripeptidyl-peptididase 1 (TPP1), and protection of telomere 1 (POT1) are the components of shelterin [15].
ALT cells exhibit a variety of typical traits
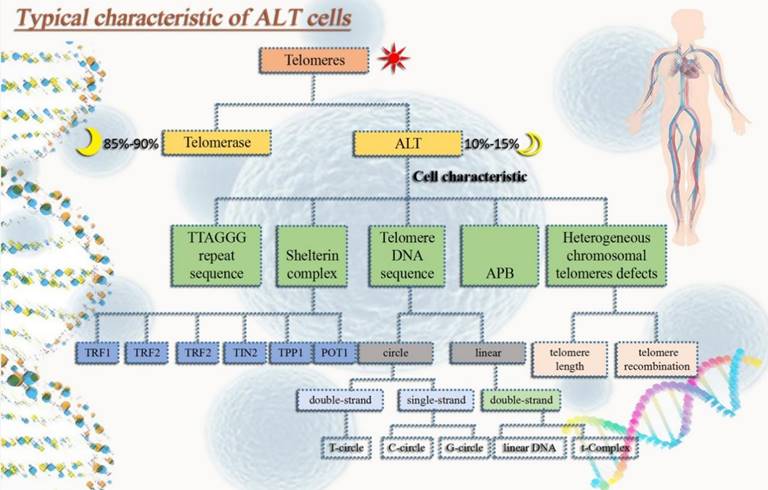
(1) Extrachromosomal telomeric repeats DNA (ECTRs). Mostly telomere circle (t-circle) are included [16], single-stranded circles in part (called C-circle or G-circle depending on whether it is rich in C or G) [17, 18], T-Complex DNA with a very high molecular weight and linear double-stranded DNA [18]. (2) One distinguishing characteristic of ALT cells is the promyelocytic leukemia nucleosome (PML nucleosome), which contains telomere chromatin. It is known as the ALT-related PML body (APB) as a result [19]. (3) Heterogeneous telomere length. Chromosomal telomeres with a high degree of heterogeneity can lead to stability problems. Highly heterogeneous chromosomal telomeres provide for rapid changes in telomere length [20] and a dramatic increase in telomere recombination rates [19, 21]. (4) High level of telomere-sister chromatid exchanges (T-SCEs) [22] (Fig. 1).
Typical characteristic of ALT tumor cells. In order to avoid aging and gain immortality, tumor cells usually extend telomeres in two pathways, one is telomerase and the other one is ALT. ALT tumor cells have typical properties that not only help us identify whether ALT mechanisms occur in tumor cells, but also in some ways help maintain ALT tumor cell homeostasis.
Markers and methods for the identification of ALT
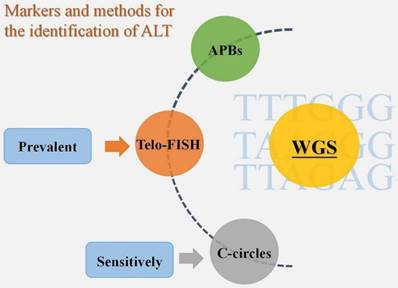
According to the review of earlier studies, there is currently no unambiguous agreement on ALT identification tactics. APBs, Telo-FISH, and C-circles have all been employed extensively in the past as biomarkers for ALT detection [23]. There is proof that the most sensitive ALT biomarker among them is C-circles [17]. Additionally, Telo-FISH was the most often published technique for ALT detection in cohort studies using sizable tumor sample sets [23]. In reality, to guarantee the identification of ALT, we need to apply two or more biomarker identification techniques, regardless of the method we select - Telo-FISH, C-circles, or APBs [23]. A solid option for TMM identification technique is also accessible, based on the varied phenotype of ALT, and it may entail the use of a number of biomarkers, such as CCA, APB, and TIF. Compared to conventional identification methods, the introduction of whole genome sequencing (WGS) has led to new concepts for the identification of TMM [24]. According to one study, WGS may accurately predict tumor TMM by validating the frequency of telomere variant repeats, particularly TTTGGG, TAAGGG, and TTAGAG sequences [24]. The accuracy of WGS in identifying tumor TMM was determined to be 91.6% based on the validation data [24]. The variable repeat content was also found to be sufficient to identify whether a tumor was ALT-positive or ALT-negative based on sequencing a sizable sample of 21 tumor subtypes (n = 821 samples), and this technique may develop into a novel and promising one for ALT+ tumor detection [23] (Fig. 2).
Markers and methods for the identification of ALT. APBs, Telo-FISH, and C-circles are the three biomarkers that have so far been used to diagnose ALT+ tumors, with C-circles being the most sensitive and Telo-FISH being the most popular. WGS, however, is a new technology that may offer greater convenience and accuracy.
Molecular mechanism of ALT cells
On one hand, current studies have shown that at least two types of ALT mechanisms exist. the RAD51-dependent type I and the RAD59-dependent (or RAD51-independent) type II [25]. However, RAD59 is not present in mammals [26]. However, RAD59 is involved in the RAD51- and RAD54-independent break-induced replication (BIR) pathways, suggesting that type II telomerase deficient yeast survivors may use the RAD51-independent BIR pathway for telomere lengthening [27]. It is now widely believed that the RAD51-independent BIR pathway may be more central in mediating ALT [25]. Similar to the yeast type I ALT system, the homologous recombination-dependent ALT pathway in human cancer is a RAD51-mediated mechanism that needs the MRE11-RAD50-NBS1 (MRN) recombination complex to be intact [28-30]. On the other hand, within the nucleus of eukaryotes, the genome is a dynamic, non-randomly ordered structure. Organized into active/inactive regions, membrane-free bodies, thin-layered associated domains, protein- or RNA-mediated loops, enhancer-promoter contacts, and chromatin regions with varying accessibility, chromatin is organized into a complex, highly hierarchical three-dimensional structural network [31-33]. To maintain and sustain the viability of cellular processes, epigenetic and transcriptional mechanisms that are carefully regulated in location and time create this complex chromatin structure [34]. The chromatin structure also divides the genome into constitutive heterochromatin regions, which are characterized by cohesive chromatin fibers, high levels of DNA and histone methylation, and transcriptional repression of the underlying DNA sequence [34]. These regions are large repetitive and gene-poor regions [34]. Telomeres are repeated structures found at the ends of chromosomes that are epistemically kept in a repressed heterochromatic state. This prevents DSBs from being recognized, prevents DNA damage repair, and promotes cell growth [35-37]. The production of Telomere-repeat-containing RNA (TERRA) and a low density of histone methylation are two characteristics of the non-classical, relaxed epigenetic state that telomeres adopt in some cancer cells [35-37]. TERRA is connected to telomere stability, telomere heterochromatin development, and telomerase regulation [38, 39]. According to earlier research, TERRA attaches to extra telomere chromatin and influences the transcription of neighboring genes. TERRA is also associated with a proteome that is engaged in a number of processes, including chromatin remodeling and transcription [40, 41]. Telomeres in yeast and human cells are vulnerable to DSB and homology-directed repair (HDR) because of the TERRA R-loop established there [36, 37, 42]. Without telomerase, HDR may in some circumstances be able to cause telomere lengthening [43, 44]. As a result, it is now thought that TERRA could cause ALT to initiate.
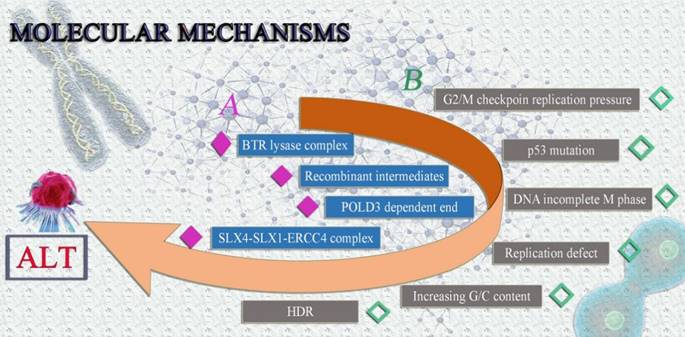
The following are some potential ALT mechanisms: (1) The BLM-TOP3A-RMI(BTR) lysase complex is necessary for ALT-mediated telomere extent [28]. During the chain invasion, the creation of recombinant intermediates is suggested [28]. Similar to this, chain invasion causes the fast and expansive POLD3-dependent end to form [45]. Finally, the SLX4-SLX1-ERCC4 complex uses granular synthesis to speed up resolving recombination intermediates, leading to telomere exchange without telomere extension [46]. (2) Telomere mitotic DNA synthesis (MiDAS) is a conservative DNA synthesis method that builds up replication pressure at G2/M checkpoints in malfunctioning cells [47]. The majority of ALT tumor cells frequently harbor p53 and G2/M checkpoint mutations [48, 49]. Checkpoint and p53 mutation may be the primary causes of the genomic instability of ALT tumor cells, according to conjecture [49, 50]. Because the ALT tumor cells' genomes are unstable, cellular DNA is easily triggered to enter the incomplete M phase, build up synthetic replication pressure, and result in more replication defects [28, 51]. Increased G/C content is found in telomere sequences and secondary structures, such as the creation of G-quadruplexes and R-loops, which induce HDR and set off ALT, as a result of replication errors and persistent DNA damage responses [28] (Fig. 3).
Possible molecular mechanisms of ALT tumor cells. It is currently believed that the formation of ALT involves two possible molecular mechanisms, both of which can lead to the extension of telomeres in tumor cells in the absence of telomerase, avoiding senescence and prolonging survival.
The relationship between the ALT mechanism and medical conditions
ALT incidence in various cancer types
Even though 80-85% of TMM positive cancers show telomerase activity, but, according to many excellent earlier studies, tumors originating in mesenchymal tissues like bone, soft tissue, the neuroendocrine system, the peripheral nervous system, and the central nervous system are typically marked by ALT activity [52]. According to certain data, the prevalence of osteosarcoma ALT+ might reach 64% and 9% in synovial sarcoma [53, 54]. Additionally, ALT+ rates in soft tissue tumors were higher than we had anticipated, at 62%, 58%, and 25% for malignant fibrous histiocytoma, leiomyosarcoma, and liposarcoma, respectively [53-56]. However, more than 50% of tumor cells in the neuroendocrine system employed ALT to prolong telomeres. PanNET made up 53% of them, Paraganglioma 13%, and Carcinoid tumor 6% [53, 55, 57, 58]. The central nervous system and the peripheral nervous system should both be given substantial consideration because they are the "hardest hit" by ALT+ [8, 53, 55]. With 34% of ALT+ in the peripheral nerve system, neuroblastoma takes the top spot, followed by ganglioneuroblastoma (14%), and adrenocortical carcinoma (12%) [8, 53, 55]. Astrocytoma and glioblastoma both exhibit high ALT+ rates in the central nervous system—42% and 28%, respectively [53, 55, 59]. The ALT+ in the gastrointestinal system is also of value, with MSI-H Gastric Carcinoma and Non-MSI-H Gastric Carcinoma having 57% and 19% positive rates, respectively, and Gastric Carcinoma having a 19% ALT+ rate [53, 60]. Additionally, 6% of intestinal cancers exhibited ALT+ [57]. We discovered that ALT has been detected in many different tumor forms and has been positive in more than half of some cancers based on the investigation of ALT incidence in different cancer types. Therefore, it is key for clinical practice to investigate ALT's mechanism.
ALT incidence in various cancer type
| Number | Cancer types | ALT+ rate | References |
|---|---|---|---|
| No.1 | Osteosarcoma | 64% | [53, 54] |
| No.2 | Synovial sarcoma | 9% | [53, 54] |
| No.3 | Malignant fibrous histiocytoma | 62% | [53-56] |
| No.4 | Leiomyosarcoma | 58% | [53-56] |
| No.5 | Liposarcoma | 25% | [53-56] |
| No.6 | PanNET | 53% | [53, 55, 57, 58] |
| No.7 | Paraganglioma | 13% | [53, 55, 57, 58] |
| No.8 | Carcinoid tumor | 6% | [53, 55, 57, 58] |
| No.9 | Neuroblastoma | 34% | [8, 53, 55] |
| No.10 | Ganglioneuroblastoma | 14% | [8, 53, 55] |
| No.11 | Adrenocortical carcinoma | 12% | [8, 53, 55] |
| No.12 | Astrocytoma | 42% | [53, 55, 59] |
| No.13 | Glioblastoma | 28% | [53, 55, 59] |
| No.14 | MSI-H Gastric Carcinoma | 57% | [53, 60] |
| No.15 | Non-MSI-H Gastric Carcinoma | 19% | [53, 60] |
| No.16 | Intestinal cancers | 6% | [57] |
Non-small cell lung cancer (NSCLC)
With an estimated 22 million new cases and 17.9 million deaths per year, lung cancer is one of the most common malignancies and the main cause of cancer-related deaths globally [61]. There have been considerable advancements and gains in survival for many patients as a result of the present research of disease biology, the use of prognostic biomarkers, and improvements in treatment [61]. However, public health initiatives to lower smoking rates have dramatically decreased lung cancer incidence in developed nations [62]. Although numerous studies have looked at the disease process in NSCLC, one of the most prevalent types of lung cancer, there are few studies looking into the mechanisms of ALT [63]. Despite this, the number of new lung cancer diagnoses rises every year in low-income nations [63-65]. In one study, 16 human NSCLC cell lines were used to examine the mechanism of telomere stabilization [66]. The results revealed that the majority of the cancer cell lines were TA-positive [66]. The TA-negative NSCLC cell line SK-LU-1, on the other hand, exhibited all ALT characteristics, including APB and typical TRF length heterogeneity [66]. Surprisingly, we were unable to identify ALT characteristics in two other NSCLC cell lines, SK-LU-1 and hTERT mRNA-negative TA and hTERT [67, 68]. It is common knowledge that the ALT machinery is believed to involve recombination processes that result in heterogeneous and extra-long telomeres that are comparable to those reported in yeast telomerase negative type II survivors [69, 70]. Additionally, it was discovered that TA deficiency was not related with decreased proliferation and tumorigenicity if the cells displayed ALT features during in vitro tests of NSCLC cell lines [66]. These findings imply that telomerase directly contributes to the NSCLC cells' rapid development [66]. The researchers also discovered that SCID mice and TA-negative NSCLC cell lines [3 of 16 (19%)] showed significantly reduced invasive growth [66]. In vitro and animal evidence, which all demonstrated a noticeably worse prognosis in lung cancer patients expressing TA and/or hTERT, particularly in stage I NSCLC, supported the conclusions of multiple clinical trials [71-74]. Furthermore, TA and/or hTERT expression were linked to improved clinical staging, according to clinical investigations [75]. A much worse prognosis based on a combination of recombination events involving extremely long telomeres and enhanced genomic instability based on recombination events involving very short telomeres was blamed for the good prognosis of patients with ALT-positive malignancies [76].
Breast cancer
Breast cancer accounts for 18% of all female cancer cases and is the most frequent malignancy in women worldwide [77]. The most frequent histologic type of breast cancer, accounting for about 68% of cases, is invasive ductal carcinoma [78]. Four primary breast cancer subgroups have been identified as a result of a new classification of breast cancers based on DNA microarray gene expression profiles [79]. These subgroups appear to originate from different cell types with various biological activities. These include subtype A (defined as significant ER and ductal epithelium-related gene expression and low histologic grade), subtype B (defined as lower ER expression and higher histologic grade), subtype C HER-2 positive (defined as low ER and HER-2 expression and high HER-2 expression), and subtype D basal-like (defined as low ER and HER-2 expression and high myoepithelial expression) [79]. Transmembrane tyrosine kinase growth factor receptor ERBB2 is a proto-oncogene that is located on chromosome 17q and that, in 15-20% of invasive breast tumors, amplifies, leading to overexpression of the HER-2 receptor [80]. The tumor suppressor genes p53 and BRCA1 as well as the topoisomerase IIα gene are situated on chromosome 17, along with a number of other genes involved in the development of breast cancer [81]. The ALT phenotype is uncommon in breast cancer but nearly never seen in other malignancies, according to this tiny study [82]. However, it does appear preferentially in a subset of HER-2 overexpressed breast tumors [81]. High levels of gene amplification are a well-known feature of HER-2 positive breast tumors, which may indicate a higher level of genomic complexity and change [81]. The replication and recombination events caused by breaks appear to be how the ALT mechanism works [82, 83]. These activities could result in free chromosome ends that engage in end-to-end association and trigger break-fusion bridge cycles, which would increase the frequency of complicated chromosomal rearrangements [82, 83]. A link between SLX4-interacting protein (SLX4IP) and TMMs has also been identified [83]. It has been demonstrated that SLX4IP is a key regulator of metastatic recurrence in breast cancer metastasis and recurrence. When SLX4IP is inactive, ALT is inhibited, and telomerase is activated simultaneously [83]. In individuals with genetically different breast cancer subtypes, TMMs selection has a significant impact on metastatic progression and survival [83, 84]. In particular, TMM was pharmacologically and genetically modulated in disseminated tumor cells, causing telomere-dependent cell death and preventing disease recurrence [84].
Prostate cancer
With more than 160,000 cases and 26,000 fatalities per year in the United States, prostate cancer is the second most frequent cancer in males [85]. Prostate cancer cells, like those of other cancers, must avoid replicative senescence and the potentially fatal chromosomal instability brought on by telomere malfunction [69]. Recurrent cancer-specific somatic inactivating mutations in the ATRX-DAXX chromatin remodeling complex are strongly correlated with the presence of ALT [86, 87]. It has been demonstrated that deletion of ATRX is sufficient to cause several ALT-related characteristics, including the existence of ALT, in LAPC-4 cell lines [88]. ATRX deletion is crucial for ALT activation, as shown by the development of genuine ALT in adenocarcinoma cells after ATRX inactivation and subsequent loss of telomerase activity [88]. That is, the inactivation of important ALT suppressors like ATRX can change telomere maintenance from telomerase to ALT under the selection pressure of loss of telomerase activity and in the appropriate genetic and/or epigenetic background [89]. Additionally, it has been documented in the literature that TRF2 is crucial to the pathological development of prostate tumors and that prostate tumors with low levels of TRF2 expression are categorized as high-grade androgen receptor-negative tumors [85, 90]. High tumorigenicity and telomere maintenance are provided by prostate cancer stem cells with diminished TRF2 expression via telomerase and ALT [85]. However, the study also found that prostate cancers that survived in the presence of TRF2 and Terc deficiency lacked both telomerase activity and the ALT marker APB [85]. As a result, the researchers hypothesize that prostate cancers may have telomere maintenance mechanisms other than telomerase and ALT processes [85].
Adrenocortical cancer (ACC)
Telomerase activity (TA) and alpha-thymidine kinase are two distinct TMM processes in ACC, according to the high-quality ACC tissue assessment findings gathered by the University of Michigan Health System [8]. Alternative telomere lengthening methods were assessed by telomere restriction fragment analysis (TRF) [91]. To some extent, telomere length is a representation of TA [8]. The "gold standard" for measuring telomere length is TRF analysis, which was created based on the understanding that TTAGGG sequences are highly conserved [92-94]. By assessing the intensity of telomere smears, TRF analysis calculates the average telomere length [92-94]. The repeating telomere sequences (TTAGGG), which do not contain the sites of the restriction enzymes utilized, do not break down when genomic DNA is digested [91]. Consequently, depending on where the restriction enzyme digested submonomeric region is located, each TRF will have a specific number of non-monomeric sequences (referred to as X-regions) [93]. Following DNA digestion, gel electrophoresis is carried out, and Southern blot analysis can be used to identify telomere sequences [93]. The findings demonstrated that ACC involves activation of the ALT mechanism [8].
Neuroblastoma
An embryonal tumor of the autonomic nervous system called neuroblastoma. The neural crest tissue's growing precursor cells and partially committed precursor cells are assumed to be the source of primordial cells [95]. High polymer content, low telomerase reverse transcriptase (TERT) expression, and enhanced TERRA expression are all hallmarks of ALT cells [96, 97]. One of the ALT tumors is the neuroblastoma. More than 50% of ALT-positive neuroblastoma cells had an ATRX-linked mutation, which is associated with -thalassemia. The telomeres of ALT-positive malignancies are also enriched with the heterochromatin histone marker H3K9me3 that ATRX recognizes [7]. It is generally accepted that ATRX mutations or lower expression levels of ATRX/death domain-associated proteins (ATRX/DAXX) in cells are responsible for the phenotypic shift in ALT cells [7]. The reduction of ATRX level in ALT ATRX wild-type neuroblastoma can be accomplished by lowering DAXX level [7]. Reduced DAXX levels cause orphan ATRX protein molecules to degrade and prevent the formation of the ATRX/DAXX complex [7]. The ALT phenotype in neuroblastoma has been linked to ATRX genome changes in several studies, however the linkage between ALT and ATRX genome changes has not been clearly shown (using particular markers) [98]. 60% of ALT cancers had ATRX structural variations, framework fusions, and single nucleotide mutations [98]. Regardless of their ATRX status, patients with the ALT phenotype have poor overall survival in practice, despite these associations [99, 100].
Truly high-risk tumors are those that are TMM-positive, TERT-high-ALT, or both [98]. According to the findings [100], TERT activation in high-risk neuroblastoma is not just present in TERTSV+ and MYCN-amplified tumors. Interestingly, some tumor cells that did not undergo the aforementioned transformation also expressed TERT highly [100]. APB, C-circle, and TERT expression levels were measured by a series of studies. The findings revealed that 12% to 26% of high-risk neuroblastoma tumors, including several MYCN-Amp tumors, had low TERT expression and no ALT activation [101]. The patient's overall survival is also considerably higher than that of high-TERT patients [98].
Osteosarcoma
The most prevalent primary solid malignant bone tumor, osteosarcoma generates malignant bone mesenchymal cells and/or immature bone [102, 103]. According to statistics, there are between 2 and 3 million new cases of osteosarcoma each year, with youth having the highest incidence [104, 105]. The incidence of people aged 15 to 19 could reach 8 to 11 million annually during the peak period [104, 105]. According to historical data, men are 1.4 times more likely than women to develop osteosarcoma [103]. Approximately 60% of osteosarcoma sample contain the ALT mechanism [106]. Recent genomic investigations have found that Osteosarcoma tumors exhibit a high level of structural diversity, including somatic mutations, copy number changes, and different single nucleotide variants or recurring point mutations [107, 108]. Recurrent somatic changes are also common in other putative driver genes, such as ATRX, while TP53 and RB1 genes are the most frequently affected in osteosarcoma [109]. ATRX was one of the most frequently mutated genes among the 288 osteosarcoma patients examined by the American Association for Cancer Research Genomics Project, second only to TP53 [110-112]. Importantly, because current next-generation sequencing tools cannot detect many complicated variations with short sequence reads, the true incidence of ATRX mutations may be underestimated [113]. ATRX is really among a number of oncogenic driver genes with frequent somatic complex differentials in a variety of cancer types of tumors [113]. The relationship between the chromatin remodeling gene ATRX, the histone chaperone DAXX, and the histone variation H3.3 and the ALT status is well known [101, 114]. The promotion of the histone variation H3.3 by ATRX and DAXX in heterochromatin regions raises the possibility that abnormalities in the stability of telomere heterochromatin can result from the loss of ATRX, DAXX, and/or H3.3 [87, 115, 116]. ATRX, DAXX, and H3.3 gene alterations were discovered in ALT-positive cancer samples, according to prior research, and some of these ALT-positive samples also displayed loss of ATRX or DAXX expression or localization [101, 114, 115]. Here, we identified a new gene fusion event between the kinesin motor protein KIFC3 and DAXX using next-generation sequencing, which resulted in the translation of the chimeric DAXX-KIFC3 fusion protein [106]. The expression of the DAXX-KIFC3 fusion protein is activated by the translocation between the untranslated region of the DAXX gene and the central intron of KIFC3, a kinesin family member, which results in functional deficiencies in the DAXX protein and helps to activate the ALT pathway [106]. Other investigations have revealed that the DAXX gene locus on chromosome 6p21, the KIFC3 gene locus on chromosome 16q21, and cells production on chromosome 6p21 have all been identified as common fragile locations in the genome [117, 118]. This finding suggests that the aforementioned areas are unstable and may play a significant role in the final genomic rearrangement and DSB [117, 118].
We looked for molecular proof of TA and ALT in 62 patient osteosarcoma samples. In this cohort of osteosarcoma patients, Kaplan-Meier analysis revealed that the absence of both TA and ALT (18%) was more strongly related with increased survival (P = 0.05) than staging (P = 0.16) or treatment response (P 0.18). However, Kaplan-Meier analysis also revealed that there was no discernible difference in survival between individuals with ALT+ and ALT- osteosarcoma. The survival rate of patients with ALT+ osteosarcoma was the same as or worse than ALT- group [54, 119], despite the fact that the median follow-up time (28 months) was brief. Analyzing whether the ALT status of the osteosarcoma was connected to treatment response concurrently. Chemotherapy had a 35% ALT+ response rate and a 33% ALT- response rate, but the difference was not statistically significant [54].
Astrocytoma
The most prevalent tumors of the central nervous system (CNS) are astrocytomas, which are primarily made up of astrocyte-like cells) [120]. Astrocytomas are categorized into grades II, III, and IV using the World Health Organization (WHO) system. Glioblastoma (GBM) is another name for grade IV cancer [120, 121]. Grade II to IV astrocytomas invade the brain widely and are physiologically malignant [122]. The median survival period for GBM is less than one year, making it one of the most aggressive tumors in human cancers [120, 122]. Unfortunately, they will eventually experience neurological malfunction and pass away despite advancements in neurosurgery, radiation, and chemotherapy [123, 124].
Clinically, it is common practice to employ TERTp and Isocitrate dehydrogenase (IDH) mutations to categorize 80% of GBM into genetic subgroups with various clinical trajectories to aid in diagnosis [125-127]. The strategies used for telomere maintenance vary according on the subgroup of GBM cellular molecules [54]. The development of the initiating transcription factor binding site led to an increase in TERT expression, telomerase activation in the TERTp mutant GBM, and ALT in the IDH mutant GBM as a result of the concurrent loss of ATRX function or mutation [55, 128]. According to these models, genetic modifications to preserve telomeres may be a crucial stage in the development of glioma cells [9]. ATRX (particularly without IDH or TP53 mutations) or GBM with mutations in the SWItch/Sucrose Non-Fermentable (SWI/SNF)-related, matrix associated, actin-dependent regulator of chromatin subfamily A-like 1 (SMARCAL1) include an ALT-positive TERTpWT-IDHWTGBM subgroup known as IDHWT-ALT [9]. An adenosine triphosphate (ATP)-dependent annealing helicase known as SMARCAL1, a new potential driver, is involved in catalyzing the recombination of DNA at the junction of the halted replication fork. Replication Protein A (RPA) [129]. It is now known to play a role in easing the replication stress caused by telomere recombination. RPA attracts SMARCAL1 to DNA [9, 130]. DNA deterioration and replication at the location where the fork has halted encourage fork repair and restart, preserving the stability of the genome [9]. The study's findings suggest that the intact SMARCAL1 helicase domain is crucial in preventing the production of C-circle and other markers in SMARCAL1 mutants and cancer cells that are ALT-positive [9]. By looking up H3.3G34R andIDH1R132H in the public cancer genomes database, other studies have demonstrated [131] that they are thought of as collaborating genes of ATRX. Together, they have an impact on the ALT mutations that may occur in glioblastoma patients who are still young. Studies have also discovered that [73] H3.3G34R increases ALT by preventing telomere KDM4B histone demethylase, and that ATRX mutations are necessary for ALT activation. It has been demonstrated through in vivo research that Kdm4b-/- and H3.3G34R serve the same purpose in activating the ALT mechanism, and IDH1R132H is most likely to act along the same pathway. Additionally, ALT-positive cells with ectopic KDM4B expression suffer from telomere degradation. Therefore, the loss of KDM4B is required for ALT activation, which can be achieved in the primary cell model by manipulating the four specified variables. Telomerase inactivation, TP53 checkpoint activation, ATRX, and KDM4B (H3.3G34R or Kdm4b-/-) are the four variables that have been identified.
Pancreatic neuroendocrine tumors (PanNETs)
With a 60% death rate, panNETs are the second most prevalent pancreatic epithelial tumors [132, 133]. PanNET is divided into three groups by the WHO: low-grade (G1), intermediate-grade (G2), and high-grade (G3) based on the classification and evaluation of the proliferative rate of tumor cells [133]. Although 90% of PanNETs are G1 or G2 tumors, G3 tumors have a higher mortality rate than G1 or G2 tumors [133]. PanNETs are mostly sporadic but can possibly be linked to three genetic syndromes: von Hippel-Lindau syndrome, tuberous sclerosis complex, and multiple endocrine neoplasia type 1 (MEN-1) [134].
In this investigation, primary PanNETs that were ALT-positive had larger tumor sizes and a higher pT classification than primary PanNETs that were ALT-negative [135]. ALT activation is a late occurrence in PanNET tumors, as evidenced by the observational finding that the prevalence of ALT increased considerably with tumor size [135]. Similar outcomes were attained in several experiments, and only PanNET subsets larger than 3 cm showed a decrease of nuclear ATRX or DAXX expression [128]. Only 14.2% of tumors with a diameter of less than 2 cm tested positive for ALT [58]. There are notable instances, nevertheless, where nuclear ATRX and DAXX expression is still present in Alt positive PanNETs [135]. These alterations might result from the genetically altered ATRX or DAXX losing their function, but not from the loss of nuclear protein expression [135]. In a similar vein, Wenzel M. Hacking et al. investigation's revealed that the proportion of patients with non-functional PanNETs (NF-PanNETs) rose sharply [136]. ATRX/DAXX loss and the presence of ALT are intimately related to the known poor prognostic characteristics of NF-PanNETs, including large tumors, high WHO grade, lymphatic vascular infiltration, peripheral nerves Invasive, advanced pathological T stage, and regional lymph node metastasis, according to a significant international multi-agency NF-PanNETs cohort study [137]. In patients with NF-PanNETs, loss of ATRX/DAXX and ALT are independent predictive indicators of shorter relapse-free survival [137].
Leiomyosarcoma
Leiomyosarcoma, which makes up 5% to 10% of all sarcomas [138] is a malignant tumor that differentiates smooth muscle. The 40% survival rate of leiomyosarcoma, an aggressive tumor, is low [139]. Myogenic differentiation, which includes smooth and skeletal muscle as well as myofibroblasts, makes sarcomas more aggressive than non-myogenic sarcomas [140, 141].
A highly complex cytogenetic tumor without recurrent chromosomal abnormalities is leiomyosarcoma [139]. The survival advantage of chemotherapy for metastatic leiomyosarcoma has not been established, and the majority of patients eventually pass away from the condition [142, 143]. The outcomes are particularly bad for deep big tumors and tumors connected to massive blood vessels [144]. According to the available data, ALT is a more significant telomere maintenance mechanism in sarcoma than telomerase activation [139]. 31 of of 59 leiomyosarcoma cases (or 53%) tested positive for ALT in previous large-scale studies utilizing telomere-specific fish [139]. The results of the other two studies were similar; 59% and 62% of the positive samples for leiomyosarcoma were from ALT leiomyosarcoma, respectively [54, 55]. Further research has revealed that ALT-positive leiomyosarcoma is linked to tumor necrosis, poor differentiation, epithelioid/polymorphic cell shape, and a high FNCLCC (Federation of the French Cancer Centres) grade [139]. One aggressive smooth muscle tumor linked to clinical outcomes is uterine leiomyomas (ULMSs) [145, 146]. Several studies that used in vitro sequencing to evaluate the genetic mutations of ULMSs. Tumor protein p53 (6/19; 33%), ATRX (5/19; 26%), and mediator complex subunit 12 (MED12; 4/19; 21%) are the most frequently altered genes [90]. Changes in TP53 and MED12, as opposed to ATRX mutations, have frequently been linked to ULMSs [147].
Malignant fibroushistiocytoma (MFH)
The most prevalent kind of soft tissue sarcoma is MFH. Pleomorphic rhabdomyosarcoma (RM) was the previous name for it [148, 149]. It is a relatively uncommon form of cancer that often affects the extremities and infrequently the peritoneum [140, 148]. Particularly, MFH growths can occur in the liver or heart on occasion, as well as in the skin, head and neck regions, posterior space, or abdominal cavity [148, 150]. MFH is typically detected after it has spread locally or is aggressive [149]. Complete resection followed by chemotherapy appears to be a successful course of treatment [150]. Although there are currently comparable treatments available, individuals with this condition have a 12-month median survival time [150].
Existing data show that 14.3% (32.6%) of the 43 soft tissue malignant fibrous histiocytoma samples tested positive for ALT [151]. Complicated karyotypes with particular translocations that exhibit monotonous cell shape as well as complex genetic and chromosomal instability characteristics might be used to categorize sarcoma subtypes [151, 152]. Soft tissue tumors with complicated karyotypes are somewhat related with ALT [152]. A typical complex karyosarcoma exhibits a low frequency of ALT, demonstrating the varied characteristics of sarcoma, despite research showing that ALT is not the main source of chromosomal instability [54]. Although the Meier curve indicates that patients with ALT-positive telomerase-positive tumors have a worse prognosis than those with ALT-negative telomerase-positive tumors (five-year survival rates of 0% and 71.6%, respectively), and that ALT-negative patients have higher average survival rates than ALT-positive patient [151, 153]. In the meantime, it was noticed that hTERT expression was found in 90.7% of tumor samples, telomerase activity was found in 79.1% of 43 soft tissue malignant fibrous histiocytoma specimens, and ALT involvement in telomere length maintenance mechanisms was seen in 32.6% of tumor samples [154]. The only independent predictive predictor for patient death among the factors examined was the presence of ALT positivity (hazard ratio, 0.275; 95% confidence range, 0.104 to 0.724; p = 0.0089) [154]. In a clinical study of a small sample of bone MFH, a rare primary malignancy, telomerase activity and telomere length were measured in 10 MFH specimens using PCR, gel hybridization, and other techniques [155]. Similar results were shown. According to the assay analysis, telomerase activity, hTERT expression, and evidence of an ALT mechanism were all found in 100% of the tumor samples, and ALT was a significant predictive risk factor for bone MFH (p=0.0316) [155]. The current clinical cohort research on MFH focus more on examining the relationship between the disease's ALT and survival and its course, leaving the investigation of its mechanism in state of obscurity [151, 153]. We anticipate that more researchers will pay attention to the MFH and ALT mechanism in the future, investigate its targets, and offer fresh concepts and approaches for the creation of MFH medications (Fig. 4).
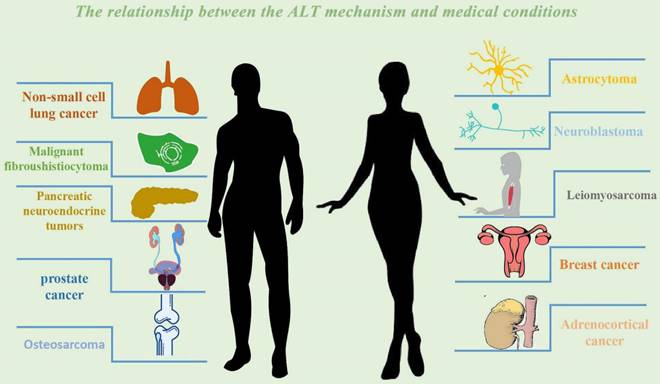
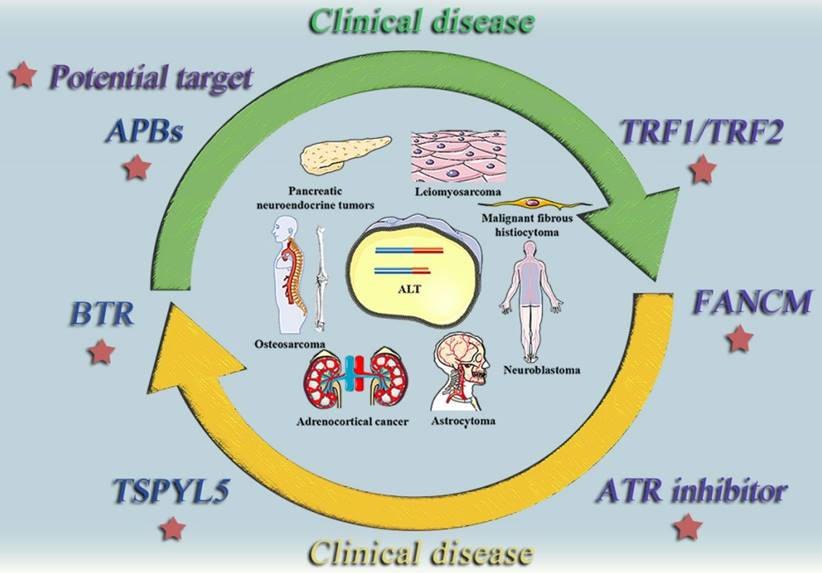
The relationship between the ALT mechanism and medical conditions. The following cancer classes are listed together with the ALT mechanism: Non-small cell lung cancer, Breast cancer, Prostate cancer, Adrenocortical cancer, Neuroblastoma, Osteosarcoma, Astrocytoma. Malignant fibrosarcoma, pancreatic neuroendocrine tumors (PanNETs), and leiomyosarcoma.
ALT tumor inhibitors and possible therapeutic targets in clinical practice
Telomere shortening of ALT+ cells
APBs
APBs are PML organisms with ALT ties. The synthesis of APB and ALT telomeres is stopped by the loss of PML, an essential component of APB. APB and ALT telomere synthesis will also be disrupted by BLM helicase loss, whereas BLM overexpression will promote telomere extension and APB expression [156-158]. In order to determine if PML and its associated APB are crucial to the ALT pathway and to further investigate the long-term impact of ALT on the preservation of telomere length [159], studies have used the PML null cell line. The findings demonstrated that, despite not being a necessary component for long-term cell viability preservation, PML is necessary for ALT+ cell telomere maintenance [159]. In addition, PML is required for the development of the C-cicle, per research findings from PML null cells [159]. PML, which includes BLM, is necessary for the positioning of APB components at the ends of telomeres [159]. By attracting BTR complexes to the ends of telomeres, PML plays a crucial function in ALT [159].
In human cells, PML-I to PML-VI splicing variations have been identified [160]. The ALT+ osteosarcoma cell line U2OS was treated with CRISPR-Cas9 technology to eradicate PML, and a U2OS derivative line devoid of all PML variants was produced [161]. This was done in order to establish which PML variants can sustain ALT. Then, in PML KO cells, all six PML variants were expressed [161]. All cells that had the PML gene knocked out had significantly reduced telomere production when compared to wild-type cells [161]. Only PML-IV among the PML variations may bring back normal levels of telomere synthesis [161]. The primary PML variant that functionally promotes ALT is PML-IV, which is the only PML variant that returns telomere clustering to wild-type levels [161].
The topic of whether PML can be a target site for ALT-positive malignancies is therefore deserving of additional research, especially in light of the significant roles that APBs and PML play in telomere extension and the ALT pathway.
HDR
In ALT cancer cells, telomeres display a distinct nuclear protein structure that results from improper chromatin control, which encourages a cycle of DNA damage and replication stress that activates HDR [162]. It is generally known that the non-homologous end joining (NHEJ) or HDR pathway is used in mammalian cells to repair the majority of DSBs [163]. Of course, there are some rarely used or alternative mechanisms that also aid in DSB repair, such as single strand annealing (SSA) and alternative NHEJ (alt-NHEJ) [164-166]. 80% of DSBs are swiftly repaired by the traditional or classical NHEJ pathway, which predominates in human cells and is active throughout the cell cycle [167, 168]. However, due to the requirement for homologous DNA sequences or the availability of sister chromatids as repair templates, HDR is a significantly slower DSB repair process that is restricted to the S and G2 phases of the cell cycle [169]. Even though NHEJ makes up a larger portion of DSB repair than HDR does, HDR nonetheless becomes a prominent characteristic of ALT+ cancer cells. The HDR pathway can be summed up as follows [163]: (1) the MRN complex binds to each damaged dsDNA end; (2) end excision is carried out by the MRN complex, CtIP, EXO1, BLM, and stabilization of the ssDNA overhang is achieved by binding to RPA; (3) RPA is replaced by RAD51 and Holliday junctions with homologous sequences are formed; and (4) the H End excision generates ssDNA tails, which are then replaced by RPA to create nuclear filaments, which are then replaced by RAD51. The MRN complex (MRE11-RAD50-NBS1), which is necessary for recognizing homologous sequences, is then replaced by RAD51 [170]. Additionally, BRCA1 and BRCA2 aid in the development of RAD51 nuclear filaments. Once RAD51 has been recruited, a homology search can be carried out [171, 172]. If it is successful, the uncut strand can then be allowed to invade the homologous template, allowing the displace template strand to form a displacement-loop(D-loop) [173]. Based on the research that have been discussed thus far, we may infer that the HDR pathway plays a significant role in the development of ALT, and that blocking HDR may be a key mechanism to encourage the death of ALT+ cancer cells.
Inducing ALT+ cell synthesis and lethality
Ataxia Telangiectasia and Rad3-related (ATR) inhibitor
Recombination plays a role in ALT [14, 174]. ALT in cancer is associated with the loss of the chromatin remodeling protein ATRX [101, 175]. Loss of ATRX causes DNA RPA occurs, which generates a recombinant nuclear protein structure and throws off the cell cycle regulation of the telomere non-coding RNA TERRA [176, 177]. A crucial stage in DNA replication and homologous recombination (HR) involves single-stranded DNA (ssDNA) covered with RPA [178]. During DNA replication, RPA momentarily attaches to telomeres before being released following the S phase. In addition to being an HR intermediate, RPA-ssDNA also attracts ATR nucleoprotein [176, 179]. ATR is a crucial protein kinase regulator of an HR, and blocking ATR is essential to preventing RPA recruitment from recombinating [176, 179]. Once recruitment and recombination are suppressed, ALT will be greatly reduced, leading to chromosomal division and ALT tumor cells dying [176]. As ALT-dependent cancer cells are very selectively killed by ATR inhibitors, this inhibitor may be helpful in the treatment of ALT-positive cancers [176].
The Fanconi anemia, complementation group M (FANCM)
The general view is that a certain level of physiological damage to the DNA is required to sustain the ALT mechanism in order to facilitate telomere extension [180]. This indicates that the degree of telomere damage is still below a critical level, where it is sufficient to cause DNA synthesis-based repair but not so great as to cause cell death [180]. The ribonuclease RNaseH1 tightly regulates the level of the telomere R-loop, which is created by TERRA and the telomere DNA and can trigger the replication pressure of ALT [41, 181]. However, its mechanism is still unclear. According to certain mechanistic studies, the Fanconi Anemia (FA) complex's FANCM ATPase/translocase facilitates effective FA complementation group D2 protein (FANCD2) ubiquitination upon replication fork stalling due to physical barriers like DNA crosslinks [182]. The biomarker FANCD2, which is thought to be the most precise and quantitative for ALT and encourages BLM recruitment, is necessary for the development of the C-circle. ALT telomerogenesis is induced by replicative stress [3, 183]. In addition to altering the replication fork, FANCM also suppresses meiotic crossover, attracts DNA repair components to the site of damage, and encourages the activation of ATR checkpoints [180]. Depletion of FANCM can result in telomere replication pressure because it helps replication forks move through telomere bundles in ALT tumor cells efficiently [180, 184]. Additionally, significant replication pressure can also be produced by consuming FANCM complex (FANCM-FAAP24-MHF1&2) in addition to FANCM alone [183]. In other words, Fanconi anemia-associated protein 24 kDa (FAAP24) or FANCM deficiency causes a marked increase in C-circle formation [180]. Insufficient levels of FANCM in ALT cells can result in cell death, activation of the ATR signal, high telomere replication stress, and damage [185]. In FANCM deficient ALT cells, the TERRA R-loop builds up on the telomeres, and downregulating it lowers APBs, replication stress, and C-circle formation [183, 186]. Therefore, by limiting the R-loop, FANCM enables controlling the activity of ALT and the proliferation of ALT cells [180, 183]. Future clinical interventions may target FANCM as a possible target.
TRF1/TRF2
The production of APB, a distinctive characteristic of ALT cells, depends on sumoylation TRF1 and TRF2, as well as a number of PML-related proteins, such as PML, the MRN complex, RAD52, and RPA [68, 187, 188]. The telomere integrity in stem cells and cancer cells may be regulated by a protein called nucleostemini [189, 190]. In ALT cells, the reduction of NS enhanced TA and the quantity of telomere damage lesions while decreasing the percentage of telomere damage caused by APB and the quantity of APB [191]. DNA damage may increase PML-IV and sulfurized TRF1 recruitment by NS in ALT cells, which is one potential molecular mechanism [190]. Previous research has shown that NS knockdown impairs the recruitment of RAD51 and causes spontaneous telomere damage [190]. Similar to this, TRF2ΔBΔM (a TRF2 mutant that causes telomere damage by impairing the stability of the telomere complex), telomere length, low telomere signal, and sister chromatid fusion frequency are all increased by NS depletion [157]. When NS is overexpressed, TRF2ΔBΔM-induced telomere damage in ALT and TA+ cells can be avoided [190]. Other research has demonstrated that the SMC family of proteins (SMC1to SMC6) regulate chromosome dynamics by forming three multi-subunit protein complexes [192, 193]. The study's findings demonstrate the significance of the Structural Maintenance of Chromosome 5/6 (SMC5/6) Complex in ALT cell telomere maintenance [192, 193]. TRF1 and TRF2 are two of the telomere binding proteins that are translated by the SMC5/6 complex SUMO's MMS21 small ubiquitin-like modifier (SUMO) ligase [194, 195]. The synthesis of APB requires the sulfidation of many shelterbelt complex subunits, which is stimulated by the MMS21 SUMO ligase [194, 195]. Telomere shortening and senescence in ALT cells are caused by the inhibition of telomere HR, which is based on the depletion of SMC5/6 subunits by RNA interference [187]. In particular, the SMC5/6 complex promotes the lengthening of telomere HR and ALT cells and the development of APB by vulcanizing telomere binding proteins [187].
Testis-specific Y-encoded-like protein 5 (TSPYL5)
TSPYL5 is a part of the PML body that co-localizes with ALT telomeres and is essential for the survival of ALT+ cells [196]. TSPYL5's ability to combat ubiquitin-specific protease 7 (USP7)-dependent may play a part in its ability to maintain the level of POT1 in ALT+ cells [196, 197]. TSPYL5, which is a part of the PML body, can shield POT1 from USP7 and PML-dependent poly-ubiquitination when it is recruited to ALT telomeres [196]. When TSPYL5 levels are low, the associated ubiquitin ligase may be activated, leading to the ubiquitination and destruction of POT1 via the proteasome [198]. The N- and C-terminal regions of USP7 may directly interact with PML proteins I and IV to explain this mechanism [199]. Without TSPYL5, the interaction of PML and USP7 may aid in the recruitment of PML and E3 ligases to the telomeres and the degradation of POT1 [200]. These ligases include RING finger protein 4 (RNF4), ubiquitin-like with PHD and RING finger domains 1 (UHRF1), mouse double minute 2 (MDM2), and tripartite motif-containing 27 (TRIM27) [200]. Early research has suggested that USP7-dependent poly-ubiquitination may be related to the loss of ATRX function in ALT+ cells, and that alterations in POT1 brought on by degradation interact with DAXX [196]. In summary, preventing the connection between TSPYL5 and USP7 may present novel therapeutic chances to specifically cause cell death in ALT+ malignancies with little adverse effects on healthy normal tissues [196].
BTR
The presence of PML nucleosomes that are tailored for the APB is one of the characteristics of ALT cells [159]. APB collects with telomeres and DNA damage factors at the ends of telomeres. According to several research, PML is necessary for the ALT mechanism [159]. This is required because APB is involved in directing the BTR complex to the end of ALT telomeres [159]. Surprisingly, the BTR complex is recruited to telomeres without the assistance of PML, demonstrating that BTR localization to telomeres is sufficient to maintain ALT activity [159]. In fact, cells lose important ALT signals such telomere length heterogeneity, extrachromosomal C-circle formation, and telomere synthesis in G2/M when PML is absent, which eventually causes telomeres to gradually shorten [161, 201]. The study's findings indicate that the process of APB production in ALT cells encourages the buildup of BTR, which in turn encourages the break-induced replication-mediated telomere elongation [159]. The SUMOization of PML is widely regarded as being essential to its function in ALT [202, 203]. Telomeres are moved to the nuclear pore by SUMOylation in order to encourage telomere elongation [202, 203]. Nuclear polySUMO peptides elicit ALT-like properties in a BLM-dependent way when telomeres are artificially aggregated. The BTR complex at the telomeres is sufficient to induce telomere ALT, increasing the likelihood of BTR recruitment, and it is sufficient to maintain ALT activity without the need for APB components or epigenetic changes [159]. A quick and targeted death of ALT cells can be brought on by the erosion of the BTR complex [159] (Fig. 5).
ALT positive tumor diseases and potential therapeutic targets. This diagram summarizes some typically clinical tumor diseases that are currently believed to be regulated by ALT mechanism and potential therapeutic targets that may be effective in the future.
Conclusion
Telomeres are well known to be crucial in controlling cell development, aging, and death. The growth of tumor cells is also influenced by the length of telomeres in tumor cells. Telomerase lengthens telomeres, one of the two currently acknowledged methods of telomere lengthening; the other is the ALT mechanism. Naturally, ALT tumor cells exhibit certain common traits. These biomarkers aid in determining if the ALT process occurs and, in some cases, maintain its stability. According to earlier research, activation of the ALT pathway has been noted in various tumor disorders. These tumor patients frequently have complex diseases, refractory illnesses, and low overall survival times. Some genes or proteins have been identified as playing a crucial part in the molecular mechanism of ALT tumor cells, and as a result, they may one day serve as viable therapeutic targets for ALT tumor cells.
The definition of ALT, typical ALT tumor cell traits, putative molecular causes of ALT, ALT-related tumor disorders, and potential clinical treatment targets were the main topics of this review. This review aims to deliver partial theoretical information for future ALT investigations by combining the findings of outstanding preliminary studies.
Acknowledgements
The authors would like to thank all the funding and journal staff who contributed to this review.
Funding
This work is supported by Natural Science research project of Higher education in Anhui Province [grant numbe rKJ2020A1037]; the Quality Engineering (MOOK) project of Anhui Province Education Department [grant number 2018mook0134]; Anhui Provincial Educational Research Project [grant number JKZ22012]. Anhui Vocational Technology College Talent Research Fund [grant numberBS2022001]. Scientific research project of Anhui Higher Education Institutions [grant number 2022AH040283].
Author contributions
YGJ provided direction and guidance throughout the preparation of this manuscript. SHL wrote and edited the manuscript. YGJ, CGJ and SHL reviewed and made significant revisions to the manuscript. GBC, LSS revised the manuscript and gave fresh thoughts to it. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Velicescu M, Yu J, Herbert BS, Shay JW, Granada E, Dubeau L. Aneuploidy and telomere attrition are independent determinants of crisis in SV40-transformed epithelial cells. Cancer Res. 2003;63:5813-20
2. Turner KJ, Vasu V, Griffin DK. Telomere Biology and Human Phenotype. Cells. 2019 8
3. Alexander P Sobinoff HAP. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017;33:921-3
4. Zhang JM, Zou L. Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020;10:30
5. Liu J, Wang L, Wang Z, Liu JP. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells. 2019 8
6. Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276-89
7. Hartlieb SA, Sieverling L, Nadler-Holly M, Ziehm M, Toprak UH, Herrmann C. et al. Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun. 2021;12:1269
8. Tobias Else TJG, Gary D Hammer. Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J Clin Endocrinol Metab. 2008;93:1442-9
9. Diplas BH, He X, Brosnan-Cashman JA, Liu H, Chen LH, Wang Z. et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat Commun. 2018;9:2087
10. Song My Hoang RJOS. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends Cancer. 2020;6:247-60
11. Eros Lazzerini Denchi TdL. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1608-71
12. C B Harley ABF, C W Greider. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458-60
13. N W Kim MAP, K R Prowse, C B Harley, M D West, P L Ho, G M Coviello, W E Wright, S L Weinrich, J W Shay. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011-5
14. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319-30
15. Lange Td. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100-10
16. Richard C Wang AS, Titia de Lange. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119:355-68
17. Jeremy D Henson YC, Lily I Huschtscha, Andy C Chang, Amy Y M Au, Hilda A Pickett, Roger R Reddel. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27:1181-5
18. Akira Nabetani FI. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol. 2009;29:703-13
19. Anthony J Cesare RRR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319-30
20. T M Bryan1 AE, J Gupta, S Bacchetti, R R Reddel. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995;14:4240-8
21. J P Murnane LS, B A Marder, W F Morgan. Telomere dynamics in an immortal human cell line. EMBO J. 1994;13:4953-62
22. Londono-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004;64:2324-7
23. MacKenzie D Jr, Watters AK, To JT, Young MW, Muratori J, Wilkoff MH. et al. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers (Basel). 2021 13
24. Lee M, Teber ET, Holmes O, Nones K, Patch AM, Dagg RA. et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018;46:4903-18
25. Lee JJ, Lee J, Lee H. Alternative paths to telomere elongation. Semin Cell Dev Biol. 2021;113:88-96
26. Chen Q, Ijpma A, Greider CW. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol. 2001;21:1819-27
27. Signon L, Malkova A, Naylor ML, Klein H, Haber JE. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol. 2001;21:2048-56
28. Pompili L, Leonetti C, Biroccio A, Salvati E. Diagnosis and treatment of ALT tumors: is Trabectedin a new therapeutic option? J Exp Clin Cancer Res. 2017;36:189
29. Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820-3
30. Teng SC, Chang J, McCowan B, Zakian VA. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell. 2000;6:947-52
31. Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T. et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. 2007;178:925-36
32. Garcia-Cao M, O'Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004;36:94-9
33. Bickmore WA, van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270-84
34. Novo CL. A Tale of Two States: Pluripotency Regulation of Telomeres. Front Cell Dev Biol. 2021;9:703466
35. Feuerhahn S, Chen LY, Luke B, Porro A. No DDRama at chromosome ends: TRF2 takes centre stage. Trends Biochem Sci. 2015;40:275-85
36. Tan J, Duan M, Yadav T, Phoon L, Wang X, Zhang JM. et al. An R-loop-initiated CSB-RAD52-POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 2020;48:1285-300
37. Vohhodina J, Goehring LJ, Liu B, Kong Q, Botchkarev VV Jr, Huynh M. et al. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nat Commun. 2021;12:3542
38. Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798-801
39. Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A. et al. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell. 2017;170:72-85 e14
40. Luke B, Lingner J. TERRA: telomeric repeat-containing RNA. EMBO J. 2009;28:2503-10
41. Arora R, Azzalin CM. Telomere elongation chooses TERRA ALTernatives. RNA Biol. 2015;12:938-41
42. Saha A, Gaurav AK, Pandya UM, Afrin M, Sandhu R, Nanavaty V. et al. TbTRF suppresses the TERRA level and regulates the cell cycle-dependent TERRA foci number with a TERRA binding activity in its C-terminal Myb domain. Nucleic Acids Res. 2021;49:5637-53
43. Hoang SM, Kaminski N, Bhargava R, Barroso-Gonzalez J, Lynskey ML, Garcia-Exposito L. et al. Regulation of ALT-associated homology-directed repair by polyADP-ribosylation. Nat Struct Mol Biol. 2020;27:1152-64
44. Barroso-Gonzalez J, Garcia-Exposito L, Hoang SM, Lynskey ML, Roncaioli JL, Ghosh A. et al. RAD51AP1 Is an Essential Mediator of Alternative Lengthening of Telomeres. Mol Cell. 2019;76:11-26 e7
45. Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539:54-8
46. Sobinoff AP, Allen JA, Neumann AA, Yang SF, Walsh ME, Henson JD. et al. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017;36:2907-19
47. Flynn RL, Heaphy CM. Surviving Telomere Attrition with the MiDAS Touch. Trends Genet. 2019;35:783-5
48. Ge Y, Wu S, Zhang Z, Li X, Li F, Yan S. et al. Inhibition of p53 and/or AKT as a new therapeutic approach specifically targeting ALT cancers. Protein Cell. 2019;10:808-24
49. Stagno D'Alcontres M, Mendez-Bermudez A, Foxon JL, Royle NJ, Salomoni P. Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J Cell Biol. 2007;179:855-67
50. Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B. et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur Urol. 2019;76:89-97
51. Min J, Wright WE, Shay JW. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol Cell Biol. 2017 37
52. De Vitis M, Berardinelli F, Sgura A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int J Mol Sci. 2018 19
53. Amorim JP, Santos G, Vinagre J, Soares P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes (Basel). 2016 7
54. Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA. et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217-25
55. Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E. et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608-15
56. Liau JY, Lee JC, Tsai JH, Yang CY, Liu TL, Ke ZL. et al. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod Pathol. 2015;28:1545-54
57. Dilley RL, Greenberg RA. ALTernative Telomere Maintenance and Cancer. Trends Cancer. 2015;1:145-56
58. Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V. et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453-60 e5
59. Nguyen DN, Heaphy CM, de Wilde RF, Orr BA, Odia Y, Eberhart CG. et al. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol. 2013;23:237-43
60. Omori Y, Nakayama F, Li D, Kanemitsu K, Semba S, Ito A. et al. Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer Sci. 2009;100:413-8
61. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398:535-54
62. Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA. et al. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N Engl J Med. 2020;383:640-9
63. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-907
64. Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B. et al. Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. JAMA. 2014;311:183-92
65. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
66. Brachner A, Sasgary S, Pirker C, Rodgarkia C, Mikula M, Mikulits W. et al. Telomerase- and alternative telomere lengthening-independent telomere stabilization in a metastasis-derived human non-small cell lung cancer cell line: effect of ectopic hTERT. Cancer Res. 2006;66:3584-92
67. Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3:1271-4
68. Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59:4175-9
69. Sommer A, Royle NJ. ALT: A Multi-Faceted Phenomenon. Genes (Basel). 2020 11
70. Sobinoff AP, Pickett HA. Mechanisms that drive telomere maintenance and recombination in human cancers. Curr Opin Genet Dev. 2020;60:25-30
71. Fujita Y, Fujikane T, Fujiuchi S, Nishigaki Y, Yamazaki Y, Nagase A. et al. The diagnostic and prognostic relevance of human telomerase reverse transcriptase mRNA expression detected in situ in patients with nonsmall cell lung carcinoma. Cancer. 2003;98:1008-13
72. Wang L, Soria JC, Kemp BL, Liu DD, Mao L, Khuri FR. hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:2883-9
73. Marchetti A, Pellegrini C, Buttitta F, Falleni M, Romagnoli S, Felicioni L. et al. Prediction of survival in stage I lung carcinoma patients by telomerase function evaluation. Lab Invest. 2002;82:729-36
74. Albanell J, Lonardo F, Rusch V, Engelhardt M, Langenfeld J, Han W. et al. High telomerase activity in primary lung cancers: association with increased cell proliferation rates and advanced pathologic stage. J Natl Cancer Inst. 1997;89:1609-15
75. Hara H, Yamashita K, Shinada J, Yoshimura H, Kameya T. Clinicopathologic significance of telomerase activity and hTERT mRNA expression in non-small cell lung cancer. Lung Cancer. 2001;34:219-26
76. Zhu J, Wang H, Bishop JM, Blackburn EH. Telomerase extends the lifespan of virus-transformed human cells without net telomere lengthening. Proc Natl Acad Sci U S A. 1999;96:3723-8
77. McPherson K, Steel CM, Dixon JM. ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ. 2000;321:624-8
78. Berg JW, Hutter RV. Breast cancer. Cancer. 1995;75:257-69
79. Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360:790-800
80. Ross JS, Fletcher JA. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells. 1998;16:413-28
81. Reinholz MM, Bruzek AK, Visscher DW, Lingle WL, Schroeder MJ, Perez EA. et al. Breast cancer and aneusomy 17: implications for carcinogenesis and therapeutic response. Lancet Oncol. 2009;10:267-77
82. Subhawong AP, Heaphy CM, Argani P, Konishi Y, Kouprina N, Nassar H. et al. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod Pathol. 2009;22:1423-31
83. Robinson NJ, Morrison-Smith CD, Gooding AJ, Schiemann BJ, Jackson MW, Taylor DJ. et al. SLX4IP and telomere dynamics dictate breast cancer metastasis and therapeutic responsiveness. Life Sci Alliance. 2020 3
84. Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2017;26:809-15
85. Wu J, Crowe DL. Telomere DNA Damage Signaling Regulates Prostate Cancer Tumorigenesis. Mol Cancer Res. 2020;18:1326-39
86. Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253-65
87. Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678-91
88. Graham MK, Kim J, Da J, Brosnan-Cashman JA, Rizzo A, Baena Del Valle JA. et al. Functional Loss of ATRX and TERC Activates Alternative Lengthening of Telomeres (ALT) in LAPC4 Prostate Cancer Cells. Mol Cancer Res. 2019;17:2480-91
89. Markiewicz-Potoczny M, Lobanova A, Loeb AM, Kirak O, Olbrich T, Ruiz S. et al. TRF2-mediated telomere protection is dispensable in pluripotent stem cells. Nature. 2021;589:110-5
90. El Mai M, Janho Dit Hreich S, Gaggioli C, Roisin A, Wagner N, Ye J. et al. A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers (Basel). 2021 13
91. Lai TP, Wright WE, Shay JW. Comparison of telomere length measurement methods. Philos Trans R Soc Lond B Biol Sci. 2018 373
92. Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X. et al. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc. 2010;5:1596-607
93. Mender I, Shay JW. Telomere Restriction Fragment (TRF) Analysis. Bio Protoc. 2015 5
94. Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW. et al. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685-91
95. Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202-11
96. Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB. et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349-57
97. Sieverling L, Hong C, Koser SD, Ginsbach P, Kleinheinz K, Hutter B. et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat Commun. 2020;11:733
98. Koneru B, Lopez G, Farooqi A, Conkrite KL, Nguyen TH, Macha SJ. et al. Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res. 2020;80:2663-75
99. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK. et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062-71
100. Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A. et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. 2018;362:1165-70
101. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425
102. Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342-52
103. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21(Suppl 7):vii320-5
104. Stiller CA, Craft AW, Corazziari I, Group EW. Survival of children with bone sarcoma in Europe since 1978: results from the EUROCARE study. Eur J Cancer. 2001;37:760-6
105. Stiller CA, Bielack SS, Jundt G, Steliarova-Foucher E. Bone tumours in European children and adolescents, 1978-1997. Report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42:2124-35
106. Mason-Osann E, Dai A, Floro J, Lock YJ, Reiss M, Gali H. et al. Identification of a novel gene fusion in ALT positive osteosarcoma. Oncotarget. 2018;9:32868-80
107. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E. et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104-12
108. Bousquet M, Noirot C, Accadbled F, Sales de Gauzy J, Castex MP, Brousset P. et al. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann Oncol. 2016;27:738-44
109. Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F. et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun. 2015;6:8940
110. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
111. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
112. Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818-31
113. Ye K, Wang J, Jayasinghe R, Lameijer EW, McMichael JF, Ning J. et al. Systematic discovery of complex insertions and deletions in human cancers. Nat Med. 2016;22:97-104
114. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226-31
115. Elsasser SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature. 2012;491:560-5
116. Elsasser SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature. 2015;522:240-4
117. Burrow AA, Williams LE, Pierce LC, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59
118. Debacker K, Kooy RF. Fragile sites and human disease. Hum Mol Genet. 2007 16 Spec No. 2: R150-8
119. Ulaner GA, Huang HY, Otero J, Zhao Z, Ben-Porat L, Satagopan JM. et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res. 2003;63:1759-63
120. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP. et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119-30
121. Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455-65
122. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J. et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93-8
123. Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120-9
124. Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK. et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311-33
125. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-73
126. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H. et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372:2499-508
127. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-12
128. de Wilde RF, Heaphy CM, Maitra A, Meeker AK, Edil BH, Wolfgang CL. et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol. 2012;25:1033-9
129. Cox KE, Marechal A, Flynn RL. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016;14:1032-40
130. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
131. Udugama M, Hii L, Garvie A, Cervini M, Vinod B, Chan FL. et al. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat Commun. 2021;12:2584
132. Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M. et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245-55
133. Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P. et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65-71
134. Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S. et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17:771-83
135. Kim JY, Brosnan-Cashman JA, An S, Kim SJ, Song KB, Kim MS. et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin Cancer Res. 2017;23:1598-606
136. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y. et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017;3:1335-42
137. Hackeng WM, Brosens LAA, Kim JY, O'Sullivan R, Sung YN, Liu TC. et al. Non-functional pancreatic neuroendocrine tumours: ATRX/DAXX and alternative lengthening of telomeres (ALT) are prognostically independent from ARX/PDX1 expression and tumour size. Gut. 2021
138. Gustafson P, Willen H, Baldetorp B, Ferno M, Akerman M, Rydholm A. Soft tissue leiomyosarcoma. A population-based epidemiologic and prognostic study of 48 patients, including cellular DNA content. Cancer. 1992;70:114-9
139. Liau JY, Tsai JH, Jeng YM, Lee JC, Hsu HH, Yang CY. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am J Surg Pathol. 2015;39:236-44
140. Fletcher CD, Gustafson P, Rydholm A, Willen H, Akerman M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol. 2001;19:3045-50
141. Deyrup AT, Haydon RC, Huo D, Ishikawa A, Peabody TD, He TC. et al. Myoid differentiation and prognosis in adult pleomorphic sarcomas of the extremity: an analysis of 92 cases. Cancer. 2003;98:805-13
142. Hensley ML, Maki R, Venkatraman E, Geller G, Lovegren M, Aghajanian C. et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol. 2002;20:2824-31
143. Hensley ML, Wathen JK, Maki RG, Araujo DM, Sutton G, Priebat DA. et al. Adjuvant therapy for high-grade, uterus-limited leiomyosarcoma: results of a phase 2 trial (SARC 005). Cancer. 2013;119:1555-61
144. Ricci S, Giuntoli RL 2nd, Eisenhauer E, Lopez MA, Krill L, Tanner EJ 3rd. et al. Does adjuvant chemotherapy improve survival for women with early-stage uterine leiomyosarcoma? Gynecol Oncol. 2013;131:629-33
145. Reed NS, Mangioni C, Malmstrom H, Scarfone G, Poveda A, Pecorelli S. et al. Phase III randomised study to evaluate the role of adjuvant pelvic radiotherapy in the treatment of uterine sarcomas stages I and II: an European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group Study (protocol 55874). Eur J Cancer. 2008;44:808-18
146. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119:2922-30
147. Makinen N, Aavikko M, Heikkinen T, Taipale M, Taipale J, Koivisto-Korander R. et al. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016;12:e1005850
148. Katsuda S, Kawahara E, Matsui Y, Ohyama S, Nakanishi I. Malignant fibrous histiocytoma of the liver: a case report and review of the literature. Am J Gastroenterol. 1988;83:1278-82
149. Rothman AE, Lowitt MH, Pfau RG. Pediatric cutaneous malignant fibrous histiocytoma. J Am Acad Dermatol. 2000;42:371-3
150. Schena S, Caniglia A, Agnino A, Caruso G, Ferlan G. Survival following treatment of a cardiac malignant fibrous histiocytoma. Chest. 2000;118:271-3
151. Montgomery E, Argani P, Hicks JL, DeMarzo AM, Meeker AK. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am J Pathol. 2004;164:1523-9
152. Borden EC, Baker LH, Bell RS, Bramwell V, Demetri GD, Eisenberg BL. et al. Soft tissue sarcomas of adults: state of the translational science. Clin Cancer Res. 2003;9:1941-56
153. Terasaki T, Kyo S, Takakura M, Maida Y, Tsuchiya H, Tomita K. et al. Analysis of telomerase activity and telomere length in bone and soft tissue tumors. Oncol Rep. 2004;11:1307-11
154. Matsuo T, Shay JW, Wright WE, Hiyama E, Shimose S, Kubo T. et al. Telomere-maintenance mechanisms in soft-tissue malignant fibrous histiocytomas. J Bone Joint Surg Am. 2009;91:928-37
155. Matsuo T, Shimose S, Kubo T, Fujimori J, Yasunaga Y, Ochi M. Alternative lengthening of telomeres as a prognostic factor in malignant fibrous histiocytomas of bone. Anticancer Res. 2010;30:4959-62
156. Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA. et al. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 2009;16:1244-51
157. Grobelny JV, Godwin AK, Broccoli D. ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci. 2000 113 Pt 24: 4577-85
158. Min J, Shay JW. Telomere clustering drives ALT. Aging (Albany NY). 2019;11:8046-7
159. Loe TK, Li JSZ, Zhang Y, Azeroglu B, Boddy MN, Denchi EL. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020;34:650-62
160. Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 2001;20:7223-33
161. Zhang JM, Genois MM, Ouyang J, Lan L, Zou L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol Cell. 2021;81:1027-42 e4
162. Hoang SM, O'Sullivan RJ. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends Cancer. 2020;6:247-60
163. Kelm JM, Samarbakhsh A, Pillai A, VanderVere-Carozza PS, Aruri H, Pandey DS. et al. Recent Advances in the Development of Non-PIKKs Targeting Small Molecule Inhibitors of DNA Double-Strand Break Repair. Front Oncol. 2022;12:850883
164. Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20:698-714
165. Sfeir A, Symington LS. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci. 2015;40:701-14
166. Bhargava R, Onyango DO, Stark JM. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016;32:566-75
167. Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320-9
168. Stinson BM, Loparo JJ. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu Rev Biochem. 2021;90:137-64
169. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497-510
170. Aasland D, Gotzinger L, Hauck L, Berte N, Meyer J, Effenberger M. et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019;79:99-113
171. Mesman RLS, Calleja F, Hendriks G, Morolli B, Misovic B, Devilee P. et al. The functional impact of variants of uncertain significance in BRCA2. Genet Med. 2019;21:293-302
172. Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L. et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol. 2020;22:87-96
173. Kwon Y, Sung P. Biochemical Analysis of D-Loop Extension and DNA Strand Displacement Synthesis. Methods Mol Biol. 2021;2153:87-99
174. Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Semin Cancer Biol. 2011;21:349-53
175. Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T. et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8:e1002772
176. Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ. et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347:273-7
177. Flynn RL, Centore RC, O'Sullivan RJ, Rai R, Tse A, Songyang Z. et al. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature. 2011;471:532-6
178. Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61-92
179. Schramke V, Luciano P, Brevet V, Guillot S, Corda Y, Longhese MP. et al. RPA regulates telomerase action by providing Est1p access to chromosome ends. Nat Genet. 2004;36:46-54
180. Silva B, Pentz R, Figueira AM, Arora R, Lee YW, Hodson C. et al. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat Commun. 2019;10:2253
181. Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun. 2014;5:5220
182. Xue Y, Li Y, Guo R, Ling C, Wang W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum Mol Genet. 2008;17:1641-52
183. Pan X, Chen Y, Biju B, Ahmed N, Kong J, Goldenberg M. et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci Rep. 2019;9:19110
184. Lu R, O'Rourke JJ, Sobinoff AP, Allen JAM, Nelson CB, Tomlinson CG. et al. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat Commun. 2019;10:2252
185. Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, Zhang D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A. 2017;114:E5940-E9
186. Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC. et al. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell. 2015;60:351-61
187. Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007;14:581-90
188. Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347-52
189. Tsai RY, McKay RD. A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 2002;16:2991-3003
190. Hsu JK, Lin T, Tsai RY. Nucleostemin prevents telomere damage by promoting PML-IV recruitment to SUMOylated TRF1. J Cell Biol. 2012;197:613-24
191. Jiang WQ, Zhong ZH, Henson JD, Reddel RR. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene. 2007;26:4635-47
192. Hirano T. SMC proteins and chromosome mechanics: from bacteria to humans. Philos Trans R Soc Lond B Biol Sci. 2005;360:507-14
193. Koshland DE, Guacci V. Sister chromatid cohesion: the beginning of a long and beautiful relationship. Curr Opin Cell Biol. 2000;12:297-301
194. Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell Biol. 2001;2:202-10
195. Zhao X, Blobel G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A. 2005;102:4777-82
196. Episkopou H, Diman A, Claude E, Viceconte N, Decottignies A. TSPYL5 Depletion Induces Specific Death of ALT Cells through USP7-Dependent Proteasomal Degradation of POT1. Mol Cell. 2019;75:469-82 e6
197. Epping MT, Meijer LA, Krijgsman O, Bos JL, Pandolfi PP, Bernards R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat Cell Biol. 2011;13:102-8
198. Garcia-Exposito L, Bournique E, Bergoglio V, Bose A, Barroso-Gonzalez J, Zhang S. et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016;17:1858-71
199. Sarkari F, Wang X, Nguyen T, Frappier L. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One. 2011;6:e16598
200. Cheng X, Kao HY. Post-translational modifications of PML: consequences and implications. Front Oncol. 2012;2:210
201. Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londono-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 2009;106:15726-31
202. Keiten-Schmitz J, Wagner K, Piller T, Kaulich M, Alberti S, Muller S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol Cell. 2020;79:54-67 e7
203. Gartner A, Muller S. PML, SUMO, and RNF4: guardians of nuclear protein quality. Mol Cell. 2014;55:1-3
Author contact
![]() Corresponding authors: Guojun Yuan, School of Environment and Chemical Engineering, Anhui Vocational and Technical College,Hefei 230011, China. E-mail address: yuangjedu.cn; Haolu Sun, School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui 230032, People's Republic of China. E-mail address: 869375016com.
Corresponding authors: Guojun Yuan, School of Environment and Chemical Engineering, Anhui Vocational and Technical College,Hefei 230011, China. E-mail address: yuangjedu.cn; Haolu Sun, School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui 230032, People's Republic of China. E-mail address: 869375016com.