Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(18):5385-5393. doi:10.7150/jca.57463 This issue Cite
Research Paper
Mutation SVCT2 promotes cell proliferation, invasion and migration in colorectal cancer
Sang-Soo Park1,2,*, Yea Seong Ryu1,2,*, Dong-In Koh1, Seung-Woo Hong1, Jai-Hee Moon1, Jae-Sik Shin1, Mi Jin Kim1,2, Do Yeon Kim1,2, Jun Ki Hong1,2, Eun Ho Kim1,2, Hong-Rae Jeong1,2, Yoon Sun Park1,2, Joseph Kim1,2, Dong Min Kim1,2, Hyeseon Yun1,2, Joo-Yeon Shin1,2, Dong-Hoon Jin1,2,3 ![]()
1. Asan Institute for Life Science, Asan Medical Center, Seoul, Republic of Korea
2. Department of Medical Science, Asan Medical Institute of Convergence Science and Technology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
3. Department of Convergence Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
* These authors contributed equally to this work.
Received 2020-12-23; Accepted 2021-6-4; Published 2021-7-6
Abstract

The sodium-dependent vitamin C transporter 2 (SVCT2) surface glycoprotein regulates ascorbate accumulation in the plasma, often resulting in the induction of cancer cell death. Therefore, high expression of this gene associates with increased overall survival in several cancers. However, in colorectal cancer (CRC), high (likely mutated) SVCT2 expression relates to poor overall survival, and its functional significance has not been studied. Thus, we hypothesize that mutant SVCT2 expression could affect CRC patient survival. According to biological databases, SVCT2 has been found to be mutated frequently, and SVCT2 E264K has a particularly high pathogenic score (0.98), compared to other SVCT2 mutant sites, in CRC patients. Interestingly, our results reveal expression of SVCT2 E264K in many CRC tissues and cells. Also, we found wild-type SVCT2 expression to be largely localized to the cytoplasm and membrane, while SVCT2 E264K was restricted to the cytoplasm. We further found that SVCT2 E264K overexpression increases cell growth. By contrast, SVCT2 E264K knockdown significantly reduced cell proliferation and promoted cell apoptosis, resulting in inhibition of cell invasion and migration. Taken together, SVCT2 E264K plays a critical role in proliferation in CRC. Our results suggest that SVCT2 E264K could be a promising novel therapeutic target in CRC.
Keywords: SVCT2, colorectal cancer, cell proliferation, invasion, migration
Introduction
Colorectal cancer (CRC) is the third-most common cancer worldwide, and its incidence and mortality are steadily rising in Asia, Europe, and North and South America [1, 2]. According to GLOBOCAN, CRC is the second-leading cause of global mortality, accounting for an estimated 9.6 million annual deaths [2]. Accumulating evidence has shown that early cancer stage, i.e., stage 1 CRC has a 5-year survival rate of over 90%; in stage 4, however, such survival drops to ~10% [3]. Furthermore, about 25% of total CRC patients still develop symptoms from the disease or its metastasis [4-6].
Genetic alterations in the colonic epithelium drive from transformation of normal epithelium to an adenoma, progressing to in situ carcinoma, and eventually, to an invasive and metastatic tumor [7]. CRC is a genetically heterogeneous disease involving a vast array of mutations, suggesting that targeted therapy, for specific molecular aberrations, is likely to be effective for patients [8]. Importantly, it has become obvious that developments in molecular staging add a source of prognostic and predictive information, to enhance the classic staging system, in which CRC patients are classified into four different prognostic groups, based on the extent of the primary tumor, the involvement of regional lymph nodes, and the presence/absence of metastasis [8]. Therefore, it is of significant importance to identify clinical biomarkers responsible for monitoring CRC treatment, which may further lead to the development of novel therapeutic targets, ultimately decreasing the risk of death in CRC patients.
Sodium-dependent vitamin C transporters (SVCTs) are surface glycoproteins, encoded by two different genes, having very similar structures, while also showing distinct functional properties, depending on their cellular distribution [9-11]. SVCT1 is primarily expressed in epithelial tissues such as the intestine, liver, and kidney, while SVCT2 is ubiquitously expressed in a wide variety of tissues, including the placenta, liver, brain, heart, lung, intestine, and eye [12-18]. With regard to function, SVCT1 is a low affinity and high capacity vitamin C transporter that responds to most ascorbate transporters, whereas SVCT2 is a high affinity and low capacity vitamin C transporter that regulates ascorbate accumulation in the plasma [19-21]. Although most animals can synthesize their own vitamin C in the liver, homozygous SVCT2 knockout mice die within a few minutes after birth, due to brain hemorrhage and respiratory failure [22] indicating that this transporter is required for maintaining vitamin C homeostasis.
According to Kaplan-Meier survival analysis, high SVCT2 expression associates with good prognosis in several cancers, including liver, pancreatic, and urothelial cancer, but not in CRC. Consequently, we hypothesize that mutant SVCT2 expression could be overexpressed and even affect survival in CRC. Mutations or DNA methylation in a tumor suppressor gene results in a loss or reduction in its function and often upregulation and activation of oncoproteins [23]. According to the Catalogue of Somatic Mutations in Cancer (COSMIC) database, SVCT2 has been found to be mutated frequently and SVCT2 E264K, specifically, has a pathogenic score of 0.98, distinctly higher than other SVCT2 mutant sites, in CRC patients. However, the functional significance of SVCT2 E264K has not been studied. This finding suggests that previous studies showing SVCT2 overexpression, in tumors, could have actually been high expression of a mutant SVCT2 gene (similar to the case of the tumor suppressor p53) (PMID: 2137806). It was also reported that the missense mutant SVCT2 results in decreased amino acid uptake, and consequent inhibition of osteoblast-like differentiation in murine calvarial MC3T3-E1 cells [24]. Furthermore, genetic variations in SVCT2 associate with increased risk of numerous malignancies, including gastric cancer, lymphoma, and head and neck squamous cell carcinomas [25-27]. Therefore, it is important to consider the molecular characterization of mutant SVCT2 E264K for CRC patient prognostic and therapeutic assessment.
In this study, we examined functional differences between wild-type SVCT2 and SVCT2 E264K in CRC. Moreover, we investigated the mechanisms underlying mutant form SVCT2-induced cell proliferation, thus assessing its importance as a novel therapeutic target for CRC treatment.
Materials and methods
Cell culture
Human CRC HCT 116, HCT-15, COLO 320DM, HT-29, DLD-1, Caco2, SW480, SW480E, SW620, and RKO cell lines were purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea). Human embryonic kidney 293T cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were cultured in RPMI 1640 (Welgene, Daegu, Korea) or DMEM (Welgene) medium containing 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C in an incubator with a humidified atmosphere of 5% CO2.
Patients and specimens
Tumor samples from a total of 64 patients were used in this study. Clinical samples were provided by ProteoGenex, Inc. (Inglewood, CA, USA).
Kaplan-Meier Plotter tool analysis
The Kaplan-Meier Plotter database (GEPIA) was used to determine the association between SVCT2 and overall survival in CRC patients. SVCT2 mRNA expression was classified as 'high group' and 'low group' according to the gene expression values with preestablished cutoffs. The Kaplan-Meier survival plot, hazards ratio (HR), 95% confidence interval (CI), and log rank p were directly determined and exhibited on the web page. p value less than 0.05 was considered statistically significant.
Transfection of plasmids and short hairpin RNAs (shRNAs)
Cells were transiently transfected with wild-type SVCT2-, and SVCT2 E264K-expressing plasmids, and shRNA against both mRNAs using jetPRIME reagent (Polyplus-transfection SA, Illkirch-Graffenstaden, France), according to the manufacturer's instructions. The integrity of the constructs was confirmed by sequencing. Targeting the SVCT2-coding region with shRNA was used to silence gene expression. Negative control scrambled shRNA, with no significant homology to human gene sequences, was used to control for nonspecific effects.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and 1 μg RNA was reversed transcribed using the AccuPower® RT PreMix (Bioneer, Daejeon, Korea). PCR was then performed using AccuPower® Gold Multiplex PCR PreMix (Bioneer) in a ABI 9902 thermal cycler (Applied Biosystems, Foster City, CA, USA). The primers for PCR products were as follows: using the forward primer 5'-CATCGGTCCCTTGACCATTAC-3', and the reverse primer 5'-GTTGGGCTGATGGGTAAGTAG-3', for amplification of fragment #1; the forward primer 5'-CCAAGTTACCTCAGACAGAACC-3', and the reverse primer 5'-CCAGCAATGGACACTCTCAA-3', for the amplification of fragment #2; the forward primer 5'-CATCGGTCCCTTGACCATTAC-3', and the reverse primer 5'-ACCGTGAAGATGAAGCAGAG-3', for the amplification of fragment #3. PCR for all four amplicons were as follows: 5 min at 95°C, for initial denaturation; 35 cycles of 30 sec at 95°C, 30 sec at 58°C, and 45 sec at 72°C, and final elongation for 5 min, at 72°C. The PCR products were then resolved by 2% agarose gel electrophoresis.
Subcellular fractionation
Cytoplasmic and membrane fractions were isolated using Subcellular Protein Fractionation Kits for Cultured Cells (Thermo Fisher Scientific), following the manufacturer's instructions. In brief, cell pellet was mixed with ice-cold CEB buffer including protease inhibitors and incubated at 4°C for 10 min. Insoluble material was sedimented at 500 × g for 5 min and the resulting supernatant, the cytoplasmic extract, was transferred to a clean pre-chilled tube on ice. The pellet was mixed with ice-cold MEB buffer containing protease inhibitors and incubated at 4°C for 10 min. The insoluble component was sedimented at 3,000 × g for 5 min. The supernatant (membrane extract) was transferred into a clean pre-chilled tube on ice. Fractions were stored at -80°C until further use.
Western blotting
Total proteins were extracted, separated by 10-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to 0.45 μm polyvinylidine difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were incubated with 5% skim milk to block nonspecific binding at room temperature for 1 h, and then incubated with primary antibodies overnight at 4°C, including anti-SVCT2 from Abcam (Cambridge, MA, USA). Antibodies against Myc-tag, phospho-Akt, Ak, Survivin, phospho-Erk, Erk, and E-cadherin were from Cell Signaling Technology (Danvers, MA, USA), while antibodies against α-Tubulin, β-Actin and Gapdh were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The membranes with bound primary antibodies were then reacted with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz) for 2 h at room temperature, and the protein bands then detected by chemiluminescence (Amersham, Little Chalfont, Buckinghamshire, UK). Antibodies against α-Tubulin, β-Actin and Gapdh were used as loading controls. The intensity of the bands was quantified by Image J software.
Proliferation assay
Following cell transfection, 1 × 105 cells were seeded in 6-well plates, for the indicated times. First, the cultured cells were prepared into single cell suspensions by trypsin solution. The cells were then pipetted up and down to obtain single cell suspensions and mixed with an equal amount of 0.4% trypan blue stain (Gibco-BRL, Thermo Fisher Scientific). Since viable cells maintain membrane integrity and do not take up trypan blue, stained (dead) cells were counted under a light microscope [28]. These assays were repeated at least three times.
Colony formation assay
Following cell transfection, cells were seeded in 6-well plates (500 cells/well) for a ≤ 2-week incubation, which was terminated when white clone spots were observed by the naked eye. The colonies were then fixed with 100% methyl alcohol, and stained with 0.05% crystal violet for 20 minutes at room temperature. Colonies containing 50 or more cells were considered viable.
Microscopic examination
To examine morphological changes, 1 × 105 cells were seeded in 6-well plates and transfected with SVCT2 E264K. The cells were rinsed twice with PBS and an inverted phase-contrast microscope (EVOS® FL Cell Imaging System, Thermo Fisher Scientific) was used to observe cell changes. Using a conventional digital camera and adaptor, the cell status for each group was recorded at × 200 magnifications.
Cell apoptosis analysis
Flow cytometry analysis was performed to detect apoptosis using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit Ι, following the manufacturer's instructions (BD Biosciences PharmingenTM, Franklin Lakes, NJ, USA). Cells were seeded in 6-well plates (3 × 105 cells /well) for 24 h. The cells were then trypsinized, and the single-cell suspension was added with FITC-Annexin V and propidium idodide (PI) for 15 min in the dark. Data was analyzed with FlowJo software (BD Biosciences). Stained cells were detected by a flow cytometer (Canto II, BD Biosciences). Early apoptosis is FITC Annexin V-positive and PI-negative, whereas late apoptosis is FITC Annexin V and PI double-positive [29].
Migration and Invasion assay
Cell migration and invasion assays were performed using transwell chambers (8 μm pore size; BD Falcon, Bedford, MA, USA), which were coated without (migration assay) or with (invasion assay) Matrigel™ (Corning, Corning, NY, USA). Knocked-down SVCT2 E264K-transfected HCT-15 cells (1 × 105 cells) were suspended in 200 μl serum-free RPMI 1640 medium, and then added to the upper chambers. The lower chambers were filled with FBS-supplemented RPMI 1640 medium. After incubation for 18 h at 37°C in a 5% CO2 humidified incubator, cells were fixed with methanol, stained with hematoxylin and eosin (H&E), and then rinsed in distilled water, until the water was colorless. The cells in the inner chamber were removed with a cotton swab and cells attached to the bottom side of the membrane were counted in five random fields, per well, at × 200x magnification. Each experiment was performed in triplicate.
Statistical analysis
Statistical analyses were performed using SigmaStat software v12 (Systat Software Inc., San Jose, CA, USA). Data were presented as means ± standard deviations (SDs) from 3 independent experiments. Significance of statistical analysis was analyzed using the Student's t-test. p<0.05 was considered statistically significant.
Results
SVCT2 E264K is overexpressed in CRC
Kaplan-Meier Plotter survival curves showed that the overall survival was significantly longer in patients with lowly expressed SVCT2, compared to patients with highly expressed SVCT2 (Fig. 1A). To identify target molecules for CRC patients, we performed RT-PCR using SVCT2 E264K sequences and specific primers. We confirmed SVCT2 E264K expression by DNA sequence analysis, in 58.5% of CRC patient tissues (Fig. 1B), suggesting that mutant SVCT2 E264K is a potential novel target for CRC. Therefore, to investigate the biological roles of SVCT2 in the initiation and progression of CRC, we first examined its expression in human CRC cell lines HCT 116, HCT-15, COLO 320DM, HT-29, DLD-1, Caco2, SW480, SW480E, SW620 and RKO (Fig. 1C). As a result of sequencing analysis of the cell lines, it was found that SW620 was the SVCT2 wild-type and HCT-15 was the SVCT2 E264K type (Fig. 1D). In the immunohistochemistry data from the Human Protein Atlas, CRC patient's samples showed that SVCT2 expression is located in the cell membrane and cytoplasm (Supple. 1). Isolation of cytoplasmic and membrane fractions, from SW620 cells, showed SVCT2 to be present in both the cytosolic and membrane components, whereas SVCT2 E264K, in HCT-15 cells, was restricted to the cytoplasm, rather than the membrane fraction (Fig. 1E). These results suggest that wild-type SVCT2 and SVCT2 E264K could serve different functions.
Overexpression of SVCT2 E264K in CRC. (A) Overall survival analysis showed that SVCT2 low expression (n=81) has a better prognosis than SVCT2 high expression(n=81). p=0.038, HR(high)=1.8, p(HR)=0.041. (B) Expression of SVCT2 E264K in 64 CRC patients using RT-PCR methods. (C) SVCT2 levels were confirmed in 10 human CRC cell lines. (D) Expression of SVCT2 E264K was analyzed by sequencing in SW620 and HCT-15. (E) SVCT2 localized to the membrane and cytosol in wild-type SVCT2-expressing SW620 cells , while HCT15 cells expressed SVCT2 E264K only in the cytosol.
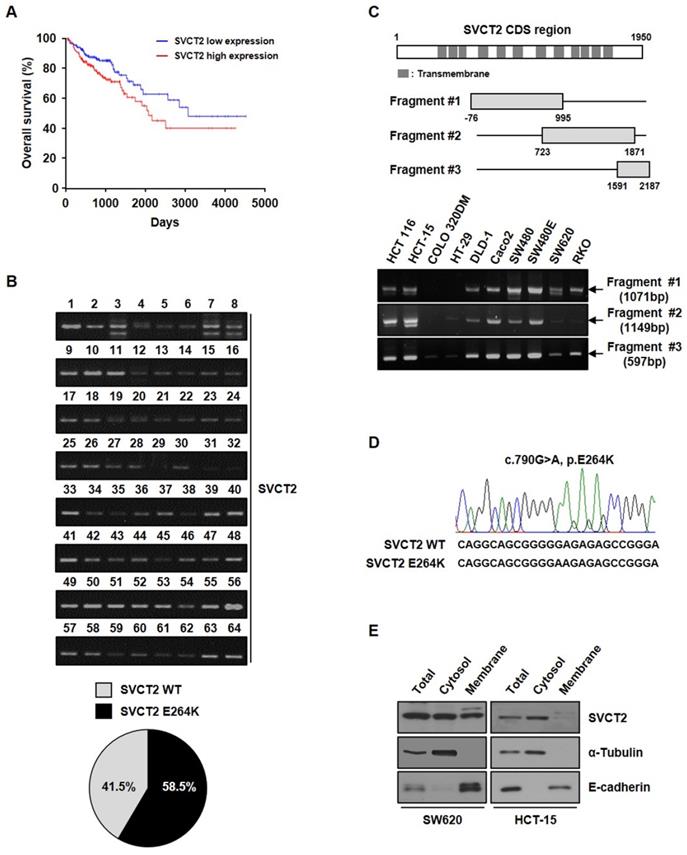
SVCT2 E264K promotes cell proliferation by upregulating p-Akt
To determine whether SVCT2 E264K expression can accelerate the growth rate of cells, we confirmed the expression of a cell proliferation related marker. As a result, it was confirmed the expression of proto-oncoprotein p-Akt and Survivin was increased by SVCT2 E264K (Fig. 2A). Also, SVCT2 E264K showed greatly increased cell proliferation by trypan blue staining and colony forming assays, compared to wild-type SVCT2 (Fig. 2B and 2C). These results suggest that SVCT2 E264K associates with pathogenic capacity in CRC.
The effects of SVCT2 E264K on cell proliferation. (A) Expression of p-Akt and Survivin was evaluated by western blotting in HEK293T cells. Cellular proliferation of SVCT2 WT and SVCT2 E264K-overexpressing cells analyzed using (B) trypan blue assay staining and (C) colony formation assay. *p<0.05 indicates significantly different from control group.
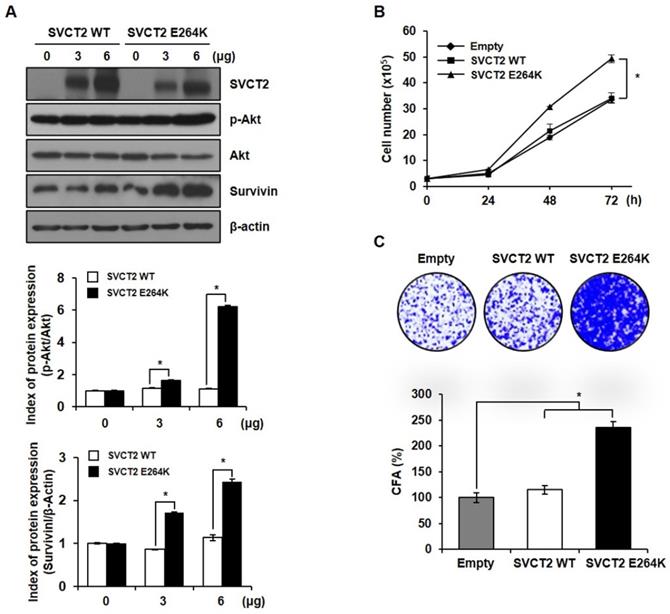
Knockdown of SVCT2 E264K suppresses CRC proliferation
To study the role of SVCT2 E264K, we investigated morphology changes following SVCT2 E264K knockdown. Under microscopic observation, control HCT-15 cells displayed a round or polygonal shapes, and close cell-cell proximity, reminiscent of cellular tight-junctions. SVCT2 E264K-knocked down HCT-15 cells, by contrast, dissociated from one another (Fig. 3A). Next, trypan blue staining revealed that SVCT2 E264K deficiency significantly increased cell death, implying that upregulation of the SVCT2 E264K facilitates growth or survival of cancer cells (Fig. 3B). In addition, we analyzed the type of cell death in SVCT2-depleted cells by flow cytometry. FITC-conjugated Annexin V and PI double staining assay both showed that SVCT2 E264K knockdown increases both early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptosis (Fig. 3C). And caspase3/7 activity was significantly increased in SVCT2 E264K knockdowned-cells (Fig. 3D). Erk 1/2 and Akt pathway are also important for cell proliferation [30, 31], and consistently, we observed that Akt activation (i.e., self-phosphorylation) was not influenced by SVCT2 E264K knockdown. However, Erk 1/2 phosphorylation was definitively downregulated in these cells (Fig. 3E). These data imply that SVCT2 E264K is strongly involved in CRC cell progression via inhibition of cell growth pathways.
The effects of SVCT2 E264K knockdown on cell apoptosis. (A) Following SVCT2 E264K knockdown, as achieved by shRNA antagonism, HCT-15 cells were photographed under a light microscope. Scale bar, 100 μm. (B) Cell death after SVCT2 E264K knockdown was confirmed by (B) trypan blue staining and (C) FITC-annexin V and PI staining (D) caspase 3/7 activity. (E) Expression of p-Erk and p-Akt was shown by western blotting in HCT-15 cells. *p<0.05; significantly different from control group.
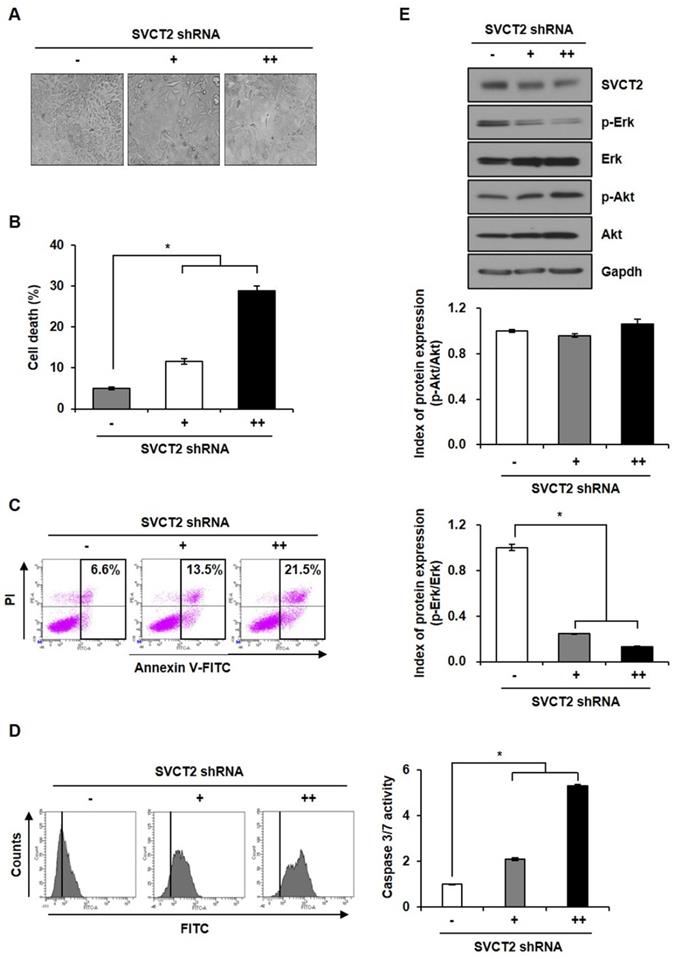
Knockdown of SVCT2 E264K inhibits cell migration and invasion
To evaluate whether knockdown of SVCT2 modulated cell motility, we performed transwell invasion assays on SVCT2 E264K-expressing HCT-15 cells. After knockdown of SVCT2 E264K, cell migration ability was significantly inhibited (Fig. 4A). Consistently, invasion assays likewise indicated that knockdown of SVCT2 E264K decreased cell invasiveness (Fig. 4B). These results strongly suggest that knockdown of SVCT2 E264K inhibits CRC tumor progression.
The effects of SVCT2 E264K knockdown on cell migration and invasion. (A) Migration of SVCT2 E264K knockdown HCT-15 cells was assessed using uncoated transwell chambers. (B) Invasion of SVCT2 E264K knockdown HCT-15 cells was confirmed with Matrigel™-coated transwell chambers. Scale bar: 100 μm. *p<0.05 indicate significantly different from control group.
Discussion
SVCT2 is a high-affinity vitamin C transporter that regulates plasma-to-tissue accumulation of ascorbate, with SVCT2 expressed ubiquitously in cells. Moreover, the SVCT2 transporter is highly expressed in the brain, where it is necessary for maintaining the high ascorbate levels required for proper brain function and survival [32, 33]. In the human bronchial epithelium, SVCT2 protein expression inversely correlates with ascorbate concentration, in the respiratory tract-lining fluid [34]. Also, mutant or wild-type SVCT2 protein was readily detected in Lewis lung tumors grown in ascorbate-dependent mice, and as predicted, SVCT2 protein levels varied over time, following a single high-dose ascorbate injection, although its association with tumor ascorbate levels is complex [35]. In cancer, as the key protein responsible for vitamin C uptake in the liver, SVCT-2 plays crucial roles in regulating sensitivity to ascorbate-induced cytotoxicity [36].
In this study, however, we found that high levels of SVCT2 associated with poor survival in CRC, indicating that mutant or wild-type SVCT2 expression could respectively, positively or negatively affect tumor growth. In in silico analysis, SVCT2 is frequently mutated, and has a high pathogenic score (0.98), according to the Functional Analysis through Hidden Markov Models (FATHMM) (PMID: 23033316). It has previously been reported that CRC is a genetically heterogeneous disease by the sequential accumulation of multiple mutations. Specifically, our results showed expression of mutant SVCT2 E264K in many CRC patients and cells. This genetic mutation can alter the subcellular localization of the SVCT2 protein [37], as we observed that wild-type SVCT2 largely localized to the cytoplasm and membrane, while mutant SVCT2 E264K accumulated solely in the cytoplasm.
To further confirm the role of SVCT2 E264K, we assessed whether wild-type SVCT, or its mutant, might modulate cell growth. These results confirmed that SVCT2 E264K upregulates the expression of p-Akt and Survivin proteins, both associated with cell proliferation, compared to SVCT2 wild-type, and that SVCT2 E264K promotes cell proliferation and colony-forming ability. These results indicate that SVCT2 E264K associates with acquired oncogenic potential. It has also been demonstrated that genetic mutations or DNA methylation lead to inactivation of functional tumor suppressor genes [38]. Moreover, SVCT2 E264K knockdown leads to increased cellular apoptosis and altered cell morphology. The SVCT2 E264K-expressing HCT15 cells displayed a round or polygonal shape, and close cell-cell proximity, reminiscent of cellular tight-junctions. By contrast, SVCT2 E264K knockdown cells became dissociated from one another. This change in morphology is suggestive of the epithelial-to-mesenchymal transition (EMT). EMT refers to a global cellular and molecular transition in which polarized epithelial cells gain mesenchymal properties, allowing them to migrate, which plays a vital role in local invasion, and metastatic dissemination, during malignancy [39]. Our results suggest that SVCT2 E264K knockdown suppresses cell migration and invasion.
In summary, SVCT2 E264K can influence tumor progression in CRC, and also is a promising potential biomarker for cancer diagnosis and prognosis. We will investigate further studies on which factors cause SVCT2 mutation. The present study provides novel insight into SVCT2-related cancers, and it is reasonable for us to conclude that SVCT2 E264K knockdown inhibits CRC cell proliferation, invasion and migration. Therefore, this mutant protein could serve as a potential therapeutic target and prognostic predictor, for CRC patients.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arnold M, Sierra MS, Laversanne M. et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:638-91
2. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and motality worldwide for 36 cancers in 185 countires. CA Cancer J Clin. 2018;68:394-424
3. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490-502
4. Garborg K, Holme Ø, Løberg M. et al. Current status of screening for colorectal cancer. Ann Oncol. 2013;24:1963-72
5. Edwards BK, Ward E, Kohler BA. et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factores, screening, and treatment) to reduce future rates. Cancer. 2010;116:544-573
6. Sargent D, Sobrero A, Grothey A. et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20, 898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872-877
7. Hong SN. Genetic and epigenetic alterations of colorectal cancer. Intest Res. 2018;16:327-37
8. Punt CJA, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14:235-246
9. Maulen NP, Henriquez EA, Kempe S. et al. Upregulation and polarized expression of the sodium-ascorbic acid transporter SVCT1 in post-confluent differentiated CaCo-2 cells. J Biol Chem. 2003;278:9035-41
10. MacDonald L, Thumser AE, Sharp P. Decreased expression of the vitamin C transporter SVCT1 by ascorbic acid in a human intestinal epithelial cell line. Br J Nutr. 2002;87:97-100
11. Boyer JC, Campbell CE, Sigurdson WJ. et al. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem Biophys Res Commun. 2005;334:150-156
12. Castro T, Low M, Salazar K. et al. Differential distribution of the Sodium-vitamin C cotransporter-1 along the proximal tubule of the mouse and human kidney. Kidney Int. 2008;74:1278-1786
13. Lv H, Wang C, Fang T. et al. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis Oncol. 2018;2:1
14. Fulzele S, Chothe P, Sangani R. et al. Sodium-dependent vitamin C transporter SVCT2: expression and function in bone marrow stromal cells and in osteogenesis. Stem Cell Res. 2013;10:36-47
15. Rajan DP, Huang W, Dutta B. et al. Human placental sodium-dependent vitamin C transporter (SVCT2): molecular cloning and transport function. Biochem Biophys Res Commun. 1999;262:762-768
16. Tsukaguchi H, Tokui T, Machenzie B. et al. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70-75
17. Chothe PP, Chutkan N, Sangani R. et al. Sodium-coupled vitamin C transporter (SVCT2): expression, function, and regulation in intervertebral disc cells. Spine J. 2013;13:549-557
18. Bürzle M, Suzuki Y, Ackermann D. et al. The sodium-dependent ascorbic acid transporter family SLC23. Mol Aspects Med. 2013;34:436-454
19. Daruwala R, Song J, Koh WS. et al. Cloning and functional characterization of the human sodiumdependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999;460:480-484
20. Tsukaguchi H, Tokui T, Mackenzie B. et al. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature. 1999;399:70-75
21. Wang Y, Mackenzie B, Tsukaguchi H. et al. Human vitamin C (l-ascorbic acid) transporter SVCT1. Biochem Biophys Res Commun. 2000;267:488-494
22. Sotiriou S, Gispert S, Cheng J. et al. Ascorbic acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat med. 2002;8:514-517
23. Li H, Zhang J, Tong JHM. et al. Targeting the oncogenic p53 mutants in colorectal cancer and other solid tumors. Int J Mol Sci. 2019;20:5999
24. Wu X, Zeng LH, Taniguchi T. et al. Activation of PKA and phosphorylation of sodium-dependent vitamin C transporter 2 by prostaglandin E2 promote osteoblast-like differentiation in MC3T3-E1 cells. Cell Death Differ. 2007;14:1792-1801
25. Chen AA, Marsit CJ, Christensen BC. et al. Genetic variation in the vitamin C transporter, SLC23A2, modifies the risk of HPV16-associated head and neck cancer. Carcinogenesis. 2009;30:977-981
26. Wright ME, Andreotti G, Lissowska J. et al. Genetic variation in sodium-dependent ascorbic acid transporters and risk of gastric cancer in Poland. Eur J Cancer. 2009;45:1824-1830
27. Skibola CF, Bracci PM, Halperin E. et al. Polymorphisms in the estrogen receptor 1 and vitamin C and matrix metalloproteinase gene families are associated with susceptibility to lymphoma. PloS One. 2008;3:e2816
28. Weisenthal LM, Marsden JA, Dill PL. et al. A novel dye exclusion method for testing in vitro chemosensitivity of human tumors. Cancer Res. 1983;43:749-757
29. Brauchle E, Thude S, Brucker SY. et al. Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci Rep. 2014;4:4698
30. Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1-to S-phase transition. Oncogene. 2007;26:3227-3239
31. Xu N, Lao Y, Zhang Y. et al. Akt: A double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724
32. Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med. 2009;46:719-730
33. Meredith ME, Harrison FE, May JM. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem Biophys Res Commun. 2011;414:737-742
34. Larsson N, Rankin GD, Bicer EM. et al. Identification of vitamin C transporters in the human airways: a cross-sectional in vivo study. BMJ Open. 2015;5:e006979
35. Campbell EJ, Vissers MC, Wohlrab C. et al. Pharmacokinetic and anti-cancer properties of high dose ascorbate in solid tumours of ascorbate-dependent mice. Free Radic Biol Med. 2016;99:451-462
36. Hong SW, Lee SH, Moon JH. et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene. 2013;32:1508-1517
37. Emr SD, Schwartz M, Silhavy TJ. Mutations altering the cellular localization of the phage lambda receptor, an Escherichia coli outer membrane protein. Proc Natl Acad Sci USA. 1978;75:5802-5806
38. Jia P, Zhao Z. Characterization of tumor-suppressor gene inactivation events in 33 cancer types. Cell Reports. 2019;26:496-506
39. Jung HY, Fattet L, Yang J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21:962-968
Author contact
![]() Corresponding author: Professor Dong-Hoon Jin. Department of Convergence Medicine, Asan Medicdal Center, University of Ulsan College of Medicine, 88 Olympicro-43gil, Songpa-gu, Seoul, Korea 05505. E-mail: inno183seoul.kr; Tel: + 82-2-3010-2643; Fax: + 82-2-3010-4665
Corresponding author: Professor Dong-Hoon Jin. Department of Convergence Medicine, Asan Medicdal Center, University of Ulsan College of Medicine, 88 Olympicro-43gil, Songpa-gu, Seoul, Korea 05505. E-mail: inno183seoul.kr; Tel: + 82-2-3010-2643; Fax: + 82-2-3010-4665