Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(11):3349-3356. doi:10.7150/jca.38391 This issue Cite
Review
Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers
Rui Chen, Wei-qing Zhao, Cheng Fang, Xin Yang ![]() , Mei Ji
, Mei Ji ![]()
Department of Oncology, the Third Affiliated Hospital of Soochow University, The First People's Hospital of Changzhou, No. 185 Juqian Road, Tianning District, Changzhou 213003, China.
Received 2019-7-13; Accepted 2020-2-9; Published 2020-3-5
Abstract
Epigenetic regulation plays an important role in the occurrence, development and treatment of malignant tumors; and a great deal of attention has been paid to the histone methylation level in recent years. As a 230-kD epigenetic regulator, the histone H3 lysine 36 histone (H3K36) methyltransferase SETD2 is a key enzyme of the nuclear receptor SET domain-containing (NSD) family, which is associated with a specific hyperphosphorylated domain, a large subunit of RNA polymerase II (RNAPII), named RNAPII subunit B1 (RPB1), and SETD2 which methylates the ly-36 position of dimethylated histone H3 (H3K36me2) to generate trimethylated H3K36 (H3K36me3). SETD2 is involved in various cellular processes, including transcriptional regulation, DNA damage repair, non-histone protein-related functions and some other processes. Great efforts of high-throughput sequencing have revealed that SETD2 is mutated or its function is lost in a range of solid cancers, including renal cancer, gastrointestinal cancer, lung cancer, pancreatic cancer, osteosarcoma, and so on. Mutation, or functional loss, of the SETD2 gene produces dysfunction in corresponding tumor tissue proteins, leading to tumorigenesis, progression, chemotherapy resistance, and unfavorable prognosis, suggesting that SETD2 possibly acts as a tumor suppressor. However, its underlying mechanism remains largely unexplored. In the present study, we summarized the latest advances of effects of SETD2 expression at the mRNA and protein levels in solid cancers, and its potential molecular and cellular functions as well as clinical applications were also reviewed.
Keywords: SETD2, Mutation, Tumor suppressor, Solid cancers
Introduction
As a process consisting of complex and consecutive changes with high morbidity and mortality, cancer is the leading cause of death in the world [1]. Recent studies have revealed that histone methylation plays a crucial role in regulatory mechanism, histone lysine methyltransferases (KMTs) are associated with cell biosynthesis and its gene mutation or functional loss as well as subsequent downstream signaling pathways facilitates oncogenic processes [2]. Histone methyltransferase SETD2 (also known as HYPB) is first isolated from human hematopoietic stem cells, and it is thought to be associated with Huntington's disease [3]. Previous studies have reported that many other KMTs can catalyze H3K36 to generate monomethylated histone H3 (H3K36me1) or H3K36me2, such as ASH1L (absent small and homeotic disks protein 1 homolog), NSD1, NSD2, NSD3 (nuclear receptor-binding SET domain-containing proteins 1-3) and SMYD2 (SET and MYND domain containing 2). SETD2 is a key member of nuclear receptor SET domain-containing (NSD) family [4], and it is the only methyltransferase which can alter the trimethylation status of H3K36 and regulate protein structure as well as its function [5,6]. SETD2 is mutated or its function is lost in various solid tumors [7,8], leading to imbalance in methylation, demethylation and epimutation, which eventually causes tumorigenesis. Loss of SETD2 affects the progress of the transcriptional elongation, resulting in failure of DNA damage repair. SETD2 deficiency has also been linked to p53, downstream signaling pathway and non-histone protein process. All of these data suggest that mutation of SETD2 gene or its functional deficiency exists in tumors, and it may function as a tumor suppressor (Table 1).
Overview of SETD2 mutation in a selection of solid tumors based on the COSMIC database (Mar. 21, 2019)
| Tissue/tumor subtype | Number and percentage of samples with mutation (%) | Total mutated samples (%) | Total samples tested | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nonsense substitution | Missense substitution | Synonymous substitution | Inframe insertion | Frameshift insertion | Inframe deletion | Frameshift deletion | Complex mutation | Other | |||
| Kidney | 83 (26.52%) | 95 (30.35%) | 5 (1.60%) | 2 (0.64%) | 20 (6.39%) | 4 (1.28%) | 77 (24.60%) | 1 (0.32%) | 4 (1.28%) | 313 (9.19%) | 3407 |
| Skin | 18 (19.15%) | 70 (74.47%) | 8 (8.51%) | 0 (0.00%) | 1 (1.06%) | 0 (0.00%) | 1 (1.06%) | 1 (1.06%) | 0 (0.00%) | 94 (5.39%) | 1744 |
| Not specified (NS) | 8 (26.67%) | 17 (56.67%) | 2 (6.67%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 1 (3.33%) | 0 (0.00%) | 1 (3.33%) | 30 (5.35%) | 561 |
| Pleura | 10 (40.00%) | 6 (24.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 9 (36.00%) | 0 (0.00%) | 0 (0.00%) | 25 (5.35%) | 467 |
| Large intestine | 13 (8.12%) | 107(66.88%) | 23 (14.37%) | 0(0.00%) | 3(1.88%) | 0(0.00%) | 39(24.38%) | 0(0.00%) | 1(0.62%) | 160(4.46%) | 3590 |
| Urinary tract | 1 (2.08%) | 41 (85.42%) | 4 (8.33%) | 0 (0.00%) | 0 (0.00%) | 0(0.00%) | 3 (6.25%) | 0 (0.00%) | 0 (0.00%) | 48 (3.98%) | 1206 |
| Endometrium | 5 (14.29%) | 31 (88.57%) | 5 (14.29%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 2 (5.71%) | 0 (0.00%) | 0 (0.00%) | 35 (3.69%) | 948 |
| Lung | 36 (23.53%) | 90 (58.82%) | 5 (3.27%) | 0 (0.00%) | 8 (5.23%) | 1 (0.65%) | 15 (9.80%) | 3 (1.96%) | 0 (0.00%) | 153 (3.58%) | 4268 |
| Liver | 11 (16.18%) | 43 (63.24%) | 9 (13.24%) | 0 (0.00%) | 3 (4.41%) | 0 (0.00%) | 5 (7.35%) | 0 (0.00%) | 0 (0.00%) | 68 (2.95%) | 2307 |
| Soft tissue | 6 (18.18%) | 23 (69.70%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 3 (9.09%) | 0 (0.00%) | 1 (3.03%) | 33 (2.5%) | 1320 |
| Breast | 22(22.92%) | 49(51.04%) | 7(7.29%) | 0(0.00%) | 2(2.08%) | 6(6.25%) | 19(19.79%) | 1(1.04%) | 1(1.04%) | 96(2.24%) | 4278 |
| Stomach | 0(0.00%) | 17(65.38%) | 4(15.38%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 5(19.23%) | 0(0.00%) | 0(0.00%) | 26(2.12%) | 1225 |
| Cervix | 1(12.50%) | 8(100.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) | 8(2.01%) | 398 |
*Tumor subtypes with a sample size less than 100 cases and mutation frequencies less than 2% have been excluded.
Protein structure of SETD2
We denominated SETD2 with three genes, Su (var)3-9, enhancer of zeste and trithorax. SETD2 is located at cytogentic band p21.31 of chromosome 3, and SETD2 protein consists of three main functional domains as follows: (1) the methyltransferase activity domains: AWS (associated with SET), SET and PS (post-SET); (2) protein-binding domains: WW (tryptophan-tryptophan), CC (Coiled-Coiled) [9,10] and SRI (Set2-Rpb1 interacting); and (3) other unclear domains.
The biological function and potential mechanism of SETD2
Previous studies have identified that SETD2 and its dependent H3K36me3 both participate in a series of cellular processes [11]. Mutation of SETD2 gene and dysfunction of downstream signaling pathways affect biological functions in many different ways, eventually causing tumorigenesis. However, the underlying mechanism remains unknown. In the present study, we summarized the latest research advances in terms of potential cellular and molecular mechanisms.
Transcriptional regulation
Transcription is a highly regulated and congenitally stochastic biochemical process [12], and such process is carried out by binding the specific regions of DNA and RNA [13]. Transcriptional regulation is achieved by varying the rates of different transcription procedures [14]. The methyltransferase Set2 in yeast, which has homology similarity to human SETD2 [9], is responsible for histone methylation, and both of Set2 and H3K36me3 are involved in transcription [15-17]. H3K36me3 can prevent initiation of spurious transcription by recruiting histone deacetylase complexes, and depletion of Set2 results in increased level of this adverse transcription [16, 18-20]. Set2 also plays a role in elongation through directly associating with RNAPII via WW domain [21, 22]. Consistently, SETD2 and H3K36me3 participate in transcriptional program in humans as well [15, 23-25]. SETD2 binds to the hyperphosphorylated carboxy-terminal domain (CTD) of RNAPII via the SRI domain, and then recruits MORF-related gene on chromosome 15 (MRG15) and polypyrimidine tract binding protein (PTB) during transcriptional process [26].
DNA damage repair
DNA is a complex molecule that stores genetic information packaged inside histones. DNA damage can be triggered by various causes, and failure of damage repair is lethal for genome stability, leading to the occurrence of malignant diseases. DNA double-strand break (DSB) is one form of DNA damage, and its misrepair is a feature of tumors [27]. SETD2 and H3K36me3 are mainly related to human homologous recombination (HR) [28], and the latter is a critical repair as well as tolerance pathway [29], which facilitates DNA DSB repair [30]. SETD2 medicates the binding of H3K36me3 to LEDGF (lens epithelium-derived growth factor)/CtIP (C-terminal binding protein interacting protein) complex [31,32] by promoting DSB resection, recruiting DNA-repair members, including RAD51 and replication protein A (RPA) to damaged sites, and then completing repair-related events [33]. In renal cancer, SETD2 mutation leads to unrepairable DNA damage, by which the key suppressor p53-mediated checkpoint can't be activated [30]. DNA mismatch repair (MMR) is known as a biological pathway which ensures genome stability [34]. SETD2 and H3K36me3 participate in human MMR in vivo [35, 36], H3K36me3 is required to recruit MMR protein hMutSα (hMSH2-hMSH6) (a significant player in MMR-associated processes) by direct combination with the specific MSH6's conserved domain PWWP (proline-tryptophan-tryptophan-proline) [37-39], and then hMutSα is located to chromatin. Further studies have found that SETD2 and H3K36me3 neither physically modulate MMR nor alter the expression level or function of MMR gene [36].
Other functions
Interestingly, the biological function of SETD2 is more than just that. SETD2 has been described to be involved in the process of tumorigenesis through combining tumor suppressor p53 and regulating its downstream genes [40], the WW and SET domains are necessary during this process [41], p53 is a key transcriptional activator which regulates cell cycle and apoptosis, and SETD2 down-regulates the expression of downstream hdm2, which is a p53 target, leading to enhanced p53 protein stability [41]. SETD2 also has some functions with regard to non-histone targets, and its levels are significantly increased during mitosis, wherein cytoplasmic methylation of tubulin by SETD2 maintains the genome stability by promoting proper chromosome segregation [42]. Recently, SETD2 protein has been identified to interact with STAT1 (signal transducers and activators of transcription), finally causing dysfunction of cell signaling pathway [43]. Besides, SETD2 and H3K36me3 are implicated in alternative splicing (AS) [44,45], and this event is related to histone modification [46-49]. H3K36me3 is also related to crosstalk [11] and genomic integrity (Figure 1) [33].
Biological Function of SETD2
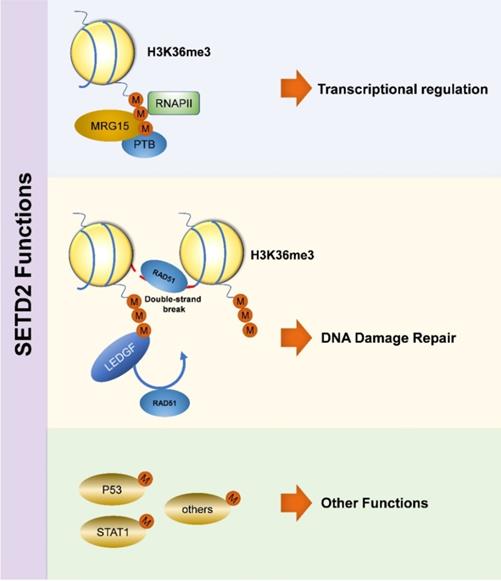
The medical application of SETD2 in solid cancers
SETD2 in reproductive and urinary system cancers
SETD2 in renal cancer
Studies have identified that 3P chromosome loss occurs in almost all cases of clear cell renal cell cancer (ccRCC) [50-53], causing mutation of multifarious tumor suppressor genes either independently or simultaneously [54]. Some diseases are emerging with poor prognosis [55-57], and the therapeutic resistance has gradually developed. SETD2 mutation fluently occurs in renal cancer [11, 58-62], and the absence of SETD2 protein is more evident than deficiency of the SETD2 gene itself. The mutation of SETD2 gene in ccRCC has a proportion of 34.07%, with such a mutation being related to producing an aggressive cancer phenotype [63]. The expression levels of SETD2 and H3K36me3 is associated with the tumor size, clinical stage and risk of carcinoma-related death, suggesting the worse prognosis in ccRCC patients with or without metastasis [57,64,65]. Besides, a comprehendsive evaluation of SETD2 and H3K36me3 expressions may be a factor in predicting preoperative risk stratification and guiding future treatment in patients with early-stage ccRCC [57]. Mechanically, SETD2 in renal cancer plays a role in epigenetic dysfunction and metabolism regulation. H3K36me3 works with H4K16ac to promote transcriptional activation [11], and a lower level of H3K36me3 fails to accomplish DNA repair and AS [45], and activate p53 [30] in renal cancer. SETD2 deficiency is associated with metabolism of three major substances, including carbohydrate, glycosaminoglycan and creatine [11], and SETD2-inactive cells are likely to enhance the PGC1α expression at the protein (PGC1α has been known as a stimulator in tumor of digestive system[66]) level and undergo a higher level of tricarboxylic acid cycle process with released energy. The dysfunction of SETD2-PGC1α metabolic pathway may act as a stimulating factor in ccRCC and provide a potential based on metabolomics of targeted treatment [67]. SETD2 is a leading cause of sunitinib resistance and may be related to MCL-1 protein expression [68]. All of these studies have indicated that SETD2 acts as a tumor suppressor in renal cancer [69].
SETD2 in prostate cancer
Global cancer statistics have shown that prostate cancer is the second most commonly diagnosed malignancy in male patients [1,70,71]. The most appropriate treatment approach is chosen according to the pathological type, clinical stage and molecular subtype. In combination with The Cancer Genome Atlas (TCGA), correlation and regression analysis show that histone methyltransferase plays an important role in prostate cancer and may be linked to genesis and progression of disease. SETD2 mutation does exist in human prostate cancer. Surprisingly, it is significantly clustered in prostate cancer samples over-expressing androgen receptors, indicating that functional loss of SETD2 may lead to resistance to surgical and drug castration in prostate patients and the specific underlying mechanism needs to be further explored [72].
SETD2 in digestive system cancers
SETD2 in gastric cancer (GC)
Studies have identified that mutation of SETD2 gene or its functional loss also occurs in advanced GC [73-75]. The SETD2 expression at the mRNA and protein levels has been respectively investigated by immunohistochemistry, quantitative PCR (qPCR), real-time PCR, and immunoblotting. The results have shown that the SETD2 expression at the mRNA and protein levels is remarkably lower in tumor tissue compared with adjacent normal tissue, and the SETD2 expression at the mRNA level is declined in nearly 80% tumor tissue samples. Besides, the expression level of SETD2 is significantly linked to tumor size, TNM stage, and lymph node metastasis, and down-regulated SETD2 expression is significantly associated with lower overall survival and lower 5-year survival in GC patients. All of these data have identified that SETD2 may be one negative prognosis factor of advanced GC patients [73,74]. A univariate analysis has discovered that SETD2 mutation or its functional loss in GC patients in Singapore, especially in high-risk gastrointestinal stromal tumor (GIST) [75]. Further studies have identified that in GC cell lines, over-expression of SETD2 can significantly inhibit the biological functions, including cell proliferation, migration and invasion [73]. SETD2 may act as a tumor suppressor gene, and the specific mechanism is divided into two aspects. SETD2 binds to p53 to form a co-immunoprecipitate, and this combination can occur under various conditions. Moreover, SETD2 can affect p53 and then participate in regulating activity of its transcriptional factors [73]. Loss of SETD2 subsequently leads to a lower level of SETD2-dependent H3K36me3, and SETD2 silenced cells show a tendency to promote DNA damage. Therefore, SETD2 may act as a suppressor gene and have the potential to serve as a molecular marker for prognosis and treatment in GC and predict the GIST patient risk stratification.
SETD2 in colorectal cancer (CRC)
Recently, SETD2 has been clarified to play a suppressive role in CRC [76], and inactivation of SETD2 induces tumor malignant potential and increases susceptibility to tumorigenesis. Further studies on the mechanism of SETD2 have shown that SETD2 protects the intestinal cells via fine-tune Wnt signaling pathway and some other pathways, such as SIRT7, JAG1 and PKM2. SETD2 deficiency may promote the expression of dishevelled segment polarity protein 2 (DVL2), resulting in increased stabilization and transcriptional activity of β-catenin, leading to promoted oncogenesis, proliferation and metastasis. In addition, functional loss of SETD2 disturbs SETD2-H3K36me3 expression and eventually affects AS in CRC [77], and such dysfunction is also linked to intestinal tumorigenesis.
SETD in pancreatic cancer
Recent studies have found that epigenetic mechanisms also play a role in pancreatic cancer. SETD2 gene alters or mutates in around 10% patients with pancreatic cancer, and such data are consistent with TCGA [8,78]. Studies have analyzed various clinical stages and pathological types of pancreatic cancer, mainly composed of normal pancreatic tissues, pancreatic intraepithelial neoplasia (PanIN), pancreatic ductal adenocarcinoma (PDA), intraductal papillary mucinous neoplasm (IPMN) and advanced PDA, and the results show that the expression of SETD2 in pancreatic cancer tissues is significantly decreased compared with the normal tissues. No matter it is precancerous lesions or advanced metastatic cancer tissue, the difference is statistically significant. More interestingly, the lowest SETD2 expression is found in PanIN lesions, supporting that SETD2 is associated with the early development and metastasis in pancreatic cancer. In addition, unlike other tumors, bioinformatic analysis indicates that there is no significant correlation between the SETD2 expression and prognosis in pancreatic cancer patients [79].
SETD2 in lung cancer
Lung cancer is one of the world's most pressing diseases, which is characterized by high morbidity and poor survival [1,80]. With the development of targeted therapy, anti-angiogenenic therapy and immunotherapy have gradually been matured, leading to effectively improved survival rate. Mutation of SETD2 gene or its functional loss in lung cancer [81-86] has a higher frequency in metastatic sites compared with primary sites, which is linked to poor prognosis [82,83]. SETD2-deficient cells display a robust tumor-driver property [41], functional loss of SETD2 and lower expression of H3K36me3 accelerate the progression of both early- and late-stage lung adenocarcinoma (LUAD), and such situation may be associated with deficiency of SWI/SNF complex [87]. Previous reports have identified that SETD2 deficiency is involved in resistance of chemotherapy drugs in solid tumors [68,72], and SETD2 also plays a key role in the treatment of lung cancer. Cisplatin constitutes a mainstay drug in combination chemotherapy regiments with other drugs in treatment of advanced non-small cell lung cancer (NSCLC), SETD2 mutation facilitates acquired cisplatin resistance due to decreased apoptosis of cancer cells, and such apoptosis occurs with abnormal H3K36me3 expression and ERK signaling pathway [82], finally contributing to worse relapse-free survival [88, 89]. In conclusion, SETD2 may function as a tumor suppressor gene in the development of lung cancer.
SETD2 in other tumors
Previous research has shown that mutations of SETD2 gene frequently occur in high-grade gliomas and only occur in the area named cerebral hemispheres, and such situation is more common in children and adolescents than elderly populations. Functional loss of SETD2 and deficiency of H3K36me3 may be linked to tumorigenesis [90]. Interestingly, SETD2 mutation also occurs in central nervous system tumors, including low-grade gliomas and non-glial tumors, and this gene mutation also occurs in people over 55 years of age [91, 95, 96]. According to the TCGA and METABRIC databases, SETD2 is mutated in all subtypes of breast cancer with a proportion of 2.62%, and its incidence in triple-negative breast cancer is 1.2%. However, it remains unclear whether SETD2 is mutated in Luminal B subtype, Luminal A and Her+. Further studies have shown that in all types of breast cancer, the expression level of SETD2 is significantly related to the prognosis of patients and that the higher the expression, the better the prognosis. Such correlation also exists in the triple-negative breast cancer, and the difference is more obvious, indicating that SETD2 mutation is linked to poor prognosis. However, the SETD mutation has few influences on patient outcome with chemotherapy [94]. Another study, which is the first one in the literature, also identified that the expression of SETD2 at the mRNA level in malignant tissue is markedly reduced compared to normal tissue. Further detection has shown that the expression of SETD2 is negatively correlated with tumor grade, stages and lymph node metastasis. These two studies have confirmed that SETD2 may play a role in tumor suppression, and it can be used as a prognostic marker in breast cancer due to its association with abnormal activity of p53 [40]. SETD2 mutation in osteosarcoma can inhibit the growth of tumor cells. In addition, it significantly affects cisplatin-induced apoptosis, resulting in inactivation of Wnt/β-catenin signaling, and sequentially leading to dysfunction of downstream c-myc, cyclin D1 and CD133 [93]. Malignant peritoneal mesothelioma is a rare tumor with limited treatment [92,97], gene sequencing of 13 patients with malignant mesothelioma has shown SETD2 mutation in malignant peritoneal mesothelioma, and it may be linked to PI3K-mTOR signaling pathway, which is expected to become a new target for the treatment of this type of tumor [92,97] (Table 2).
Overview of SETD2 in a selection of solid tumors based on this work
| Tissue/tumor subtype | Function | Mechanism | Gene change | Reference/s |
|---|---|---|---|---|
| Clear cell renal cell carcinoma | tumorigenesis | SETD2-PGC1α metabolic pathway | loss | [67] |
| treatment | SETD2-MCL-1 axis | mutation | [68] | |
| Prognosis | unknown | mutation | [57] | |
| Lung cancer | Tumorigenesis/progression | SWI/SNF complex | loss | [83, 87] |
| therapy | ERK signaling pathway | mutation | [82] | |
| Gastric cancer | proliferation, migration, invasion/ prognosis | p53 | Mutation/loss | [73-75]. |
| Colorectal cancer | Tumorigenesis/proliferation/metastasis | alternative splicing/ wnt signal pathway | mutation | [76, 77] |
| Central nervous system tumors | Tumorigenesis | unknown | mutation | [90, 91] |
| Pancreatic cancer | Tumorigenesis | unknown | mutation | [8],[79] |
| prostate cancer | Treatment | unknown | mutation | [72] |
| malignant peritoneal mesothelioma | Tumorigenesis | PI3K-mTOR signal pathway | mutation | [92] |
| osteosarcoma | tumorigenesis, treatment | Wnt/β-catenin signal pathway | mutation | [93] |
| Breast cancer | Prognosis | p53 | mutation | [40, 94] |
Conclusions
In the present review, we summarized all recent findings on SETD2 mutation or its functional loss in a variety of solid tumors. SETD2 plays an important role in the initiation, progression, prognosis and treatment of solid tumors. SETD2 participates in transcriptional regulation, DNA MMR, human DNA HR and some other processes, leading to progression of malignant tumor. Mutation of SETD2 gene or its functional loss causes dysfunction of downstream signaling pathways, including wnt signaling pathway, PGC1α metabolic pathway and PI3K-mTOR signaling pathway, and such dysfunction is related to tumor occurrence. In addition, there are some signaling pathways, which are linked to drug resistance, such as Wnt/β-catenin signaling pathway and ERK signaling pathway. Taken together, SETD2 may be a regulator of tumor suppressor and can be used as a novel prognostic biomarker and therapeutic target. However, the underlying mechanism remains unknown, and there is still a lack of relevant research, especially in vitro studies. Therefore, it is necessary to clarify how SETD2 is involved in oncogenesis, progression and prognosis of cancers, which may help select the most appropriate and cost-effective therapeutic drugs, and monitor recurrence and drug resistance, and assess prognosis for each patient.
Abbreviations
SETD2: SET domain-containing 2; H3K36: histone H3 lysine 36; H3K36me1: monomethylated histone H3; H3K36me2: dimethylated histone H3; H3K36me3: trimethylated H3K36; RNAPII: RNA polymerase II; RPB1: RNAPII subunit B1; KMTs: histone lysine methyltransferases; NSD: nuclear receptor SET domain-containing; ASH1L: Absent small and homeotic disks protein 1 homolog; NSD1, NSD2, NSD3: Nuclear receptor-binding SET domain-containing proteins 1-3; SMYD2: SET and MYND domain containing 2; AWS: associated with SET; PS: post-SET; CC: Coiled-Coiled; SRI: Set2-Rpb1 interacting; CTD: carboxy-terminal domain; MRG15: MORF-related gene on chromosome 15; PTB: polypyrimidine tract binding protein; DSB: DNA double-strand break; HR: DNA homologous recombination repair; RPA: Replication Protein A; PWWP: Proline-tryptophan-tryptophan-Proline; MMR: mismatch repair; LEDGF: lens epithelium-derived growth factor; CtIP: C-terminal binding protein interacting protein; STAT1: signal transducers and activators of transcription; ccRCC: clear cell renal cell cancer; AS: Alternative splicing; GC: gastric cancer; LUAD: lung adenocarcinoma; NSCLC: non-small cell lung cancer; CRC: colorectal cancer; DVL2: dishevelled segment polarity protein 2.
Acknowledgements
This work was supported by grants from the Natural Science Youth Foundation of China (81501971), Project funded by China Postdoctoral Science Foundation (2018M630603), the Natural Science Youth Foundation of Jiangsu Province (BK20150252), the Human Resource Summit Grant of Jiangsu Province (WSW-142) and the Youth Medical Professionals Foundation of Jiangsu Province (QNRC2016279).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Skucha A, Ebner J, Grebien F. Roles of SETD2 in Leukemia-Transcription, DNA-Damage, and Beyond. Int J Mol Sci. 2019;20:1029
3. Faber PW, Barnes GT, Srinidhi J. et al. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998;7:1463-74
4. Wang S, Yuan X, Liu Y. et al. Genetic polymorphisms of histone methyltransferase SETD2 predicts prognosis and chemotherapy response in Chinese acute myeloid leukemia patients. J Transl Med. 2019;17:101
5. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature reviews Molecular cell biology. 2012;13:115-26
6. Xu Q, Xiang Y, Wang Q. et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet. 2019;51:844-56
7. Kandoth C, Mclellan MD, Vandin F. et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-9
8. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
9. Hacker KE, Fahey CC, Shinsky SA. et al. Structure/Function Analysis of Recurrent Mutations in SETD2 Protein Reveals a Critical and Conserved Role for a SET Domain Residue in Maintaining Protein Stability and Histone H3 Lys-36 Trimethylation. The Journal of biological chemistry. 2016;291:21283-95
10. Mcdaniel SL, Strahl BD. Shaping the cellular landscape with Set2/SETD2 methylation. Cellular and molecular life sciences: CMLS. 2017;74:3317-34
11. Li L, Miao W, Huang M. et al. Integrated Genomic and Proteomic Analyses Reveal Novel Mechanisms of the Methyltransferase SETD2 in Renal Cell Carcinoma Development. Molecular & cellular proteomics: MCP. 2019;18:437-47
12. Rullan M, Benzinger D, Schmidt GW. et al. An Optogenetic Platform for Real-Time, Single-Cell Interrogation of Stochastic Transcriptional Regulation. Mol Cell. 2018;70:745-56.e6
13. Payne JL, Khalid F, Wagner A. RNA-mediated gene regulation is less evolvable than transcriptional regulation. Proc Natl Acad Sci U S A. 2018;115:E3481-E90
14. Bartman CR, Hamagami N, Keller CA. et al. Transcriptional Burst Initiation and Polymerase Pause Release Are Key Control Points of Transcriptional Regulation. Mol Cell. 2019;73:519-32.e4
15. Strahl BD, Grant PA, Briggs SD. et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298-306
16. Kizer KO, Phatnani HP, Shibata Y. et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305-16
17. Fuchs SM, Kizer KO, Braberg H. et al. RNA polymerase II carboxyl-terminal domain phosphorylation regulates protein stability of the Set2 methyltransferase and histone H3 di- and trimethylation at lysine 36. The Journal of biological chemistry. 2012;287:3249-56
18. Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971-8
19. Keogh MC, Kurdistani SK, Morris SA. et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593-605
20. Rechtsteiner A, Ercan S, Takasaki T. et al. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010;6:e1001091
21. Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. The Journal of biological chemistry. 2002;277:49383-8
22. Sudol M, Sliwa K, Russo T. Functions of WW domains in the nucleus. FEBS Lett. 2001;490:190-5
23. Schaft D, Roguev A, Kotovic KM. et al. The histone 3 lysine 36 methyltransferase, SET2, is involved in transcriptional elongation. Nucleic Acids Res. 2003;31:2475-82
24. Carvalho S, Raposo AC, Martins FB. et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013;41:2881-93
25. Wen H, Li Y, Xi Y. et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263-8
26. Sanidas I, Polytarchou C, Hatziapostolou M. et al. Phosphoproteomics screen reveals akt isoform-specific signals linking RNA processing to lung cancer. Mol Cell. 2014;53:577-90
27. Jeggo P, Lavin MF. Cellular radiosensitivity: how much better do we understand it? Int J Radiat Biol. 2009;85:1061-81
28. Pfister SX, Ahrabi S, Zalmas LP. et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006-18
29. Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113-39
30. Carvalho S, Vítor AC, Sridhara SC. et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife. 2014;3:e02482
31. Pradeepa MM, Sutherland HG, Ule J. et al. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717
32. Daugaard M, Baude A, Fugger K. et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol. 2012;19:803-10
33. Kanu N, Grönroos E, Martinez P. et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. 2015;34:5699-708
34. Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85-98
35. Fang J, Huang Y, Mao G. et al. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc Natl Acad Sci U S A. 2018;115:9598-603
36. Li F, Mao G, Tong D. et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153:590-600
37. Laguri C, Duband-Goulet I, Friedrich N. et al. Human mismatch repair protein MSH6 contains a PWWP domain that targets double stranded DNA. Biochemistry. 2008;47:6199-207
38. Dhayalan A, Rajavelu A, Rathert P. et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. The Journal of biological chemistry. 2010;285:26114-20
39. Vezzoli A, Bonadies N, Allen MD. et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617-9
40. Sarakbi WA, Sasi W, Jiang WG. et al. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC Cancer. 2009;9:290
41. Xie P, Tian C, An L. et al. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell Signal. 2008;20:1671-8
42. Park IY, Powell RT, Tripathi DN. et al. Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell. 2016;166:950-62
43. Chen K, Liu J, Liu S. et al. Methyltransferase SETD2-Mediated Methylation of STAT1 Is Critical for Interferon Antiviral Activity. Cell. 2017;170:492-506.e14
44. Luco RF, Pan Q, Tominaga K. et al. Regulation of alternative splicing by histone modifications. Science (New York, NY). 2010;327:996-1000
45. Ho TH, Park IY, Zhao H. et al. High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene. 2016;35:1565-74
46. Zhou HL, Luo G, Wise JA. et al. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014;42:701-13
47. Xu Y, Zhao W, Olson SD. et al. Alternative splicing links histone modifications to stem cell fate decision. Genome Biol. 2018;19:133
48. Naftelberg S, Schor IE, Ast G. et al. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165-98
49. He R, Kidder BL. H3K4 demethylase KDM5B regulates global dynamics of transcription elongation and alternative splicing in embryonic stem cells. Nucleic Acids Res. 2017;45:6427-41
50. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43-9
51. Bihr S, Ohashi R, Moore AL. et al. Expression and Mutation Patterns of PBRM1, BAP1 and SETD2 Mirror Specific Evolutionary Subtypes in Clear Cell Renal Cell Carcinoma. Neoplasia (New York, NY). 2019;21:247-56
52. Hakimi AA, Ostrovnaya I, Reva B. et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3259-67
53. Morris MR, Latif F. The epigenetic landscape of renal cancer. Nature reviews Nephrology. 2017;13:47-60
54. Gossage L, Murtaza M, Slatter AF. et al. Clinical and pathological impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in clear cell renal cell carcinoma. Genes Chromosomes Cancer. 2014;53:38-51
55. Li J, Kluiver J, Osinga J. et al. Functional Studies on Primary Tubular Epithelial Cells Indicate a Tumor Suppressor Role of SETD2 in Clear Cell Renal Cell Carcinoma. Neoplasia (New York, NY). 2016;18:339-46
56. Hsieh JJ, Le VH, Oyama T. et al. Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2018;36:JCO2018792549
57. Liu W, Fu Q, An H. et al. Decreased Expression of SETD2 Predicts Unfavorable Prognosis in Patients With Nonmetastatic Clear-Cell Renal Cell Carcinoma. Medicine. 2015;94:e2004
58. Dalgliesh GL, Furge K, Greenman C. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360-3
59. Hakimi AA, Chen YB, Wren J. et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol. 2013;63:848-54
60. Varela I, Tarpey P, Raine K. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539-42
61. Guo G, Gui Y, Gao S. et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2011;44:17-9
62. Su X, Zhang J, Mouawad R. et al. Inactivation and Mutation Drive a Convergence toward Loss of Function of H3K36 Writers in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017;77:4835-45
63. Liu L, Guo R, Zhang X. et al. Loss of SETD2, but not H3K36me3, correlates with aggressive clinicopathological features of clear cell renal cell carcinoma patients. Biosci Trends. 2017;11:214-20
64. Wang J, Liu L, Qu Y. et al. Prognostic Value of SETD2 Expression in Patients with Metastatic Renal Cell Carcinoma Treated with Tyrosine Kinase Inhibitors. The Journal of urology. 2016;196:1363-70
65. Ho TH, Kapur P, Joseph RW. et al. Loss of histone H3 lysine 36 trimethylation is associated with an increased risk of renal cell carcinoma-specific death. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2016;29:34-42
66. Bhalla K, Hwang BJ, Dewi RE. et al. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011;71:6888-98
67. Liu J, Hanavan PD, Kras K. et al. Loss of SETD2 Induces a Metabolic Switch in Renal Cell Carcinoma Cell Lines toward Enhanced Oxidative Phosphorylation. J Proteome Res. 2019;18:331-40
68. Elgendy M, Fusco JP, Segura V. et al. Identification of mutations associated with acquired resistance to sunitinib in renal cell cancer. Int J Cancer. 2019;145:1991-2001
69. Duns G, Van DBE, Van DI. et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70:4287-91
70. Ferlay J, Soerjomataram I, Dikshit R. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
71. Lozano R, Naghavi M, Foreman K. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (London, England). 2012;380:2095-128
72. Zhang Y, Yan L, Yao W. et al. Integrated Analysis of Genetic Abnormalities of the Histone Lysine Methyltransferases in Prostate Cancer. Medical science monitor: international medical journal of experimental and clinical research. 2019;25:193-239
73. Chen Z, Raghoonundun C, Chen W. et al. SETD2 indicates favourable prognosis in gastric cancer and suppresses cancer cell proliferation, migration, and invasion. Biochem Biophys Res Commun. 2018;498:579-85
74. Liu X, Chu KM. Molecular biomarkers for prognosis of gastrointestinal stromal tumor. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2019;21:145-51
75. Huang KK, Mcpherson JR, Tay ST. et al. SETD2 histone modifier loss in aggressive GI stromal tumours. Gut. 2016;65:1960-72
76. Feodorova Y, Tashkova D, Koev I. et al. Novel insights into transcriptional dysregulation in colorectal cancer. Neoplasma. 2018;65:415-24
77. Yuan H, Li N, Fu D. et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. The Journal of clinical investigation. 2017;127:3375-91
78. Gao J, Aksoy BA, Dogrusoz U. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
79. Ettel M, Zhao L, Schechter S. et al. Expression and prognostic value of NSD1 and SETD2 in pancreatic ductal adenocarcinoma and its precursor lesions. Pathology. 2019;51:392-8
80. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
81. Imielinski M, Berger AH, Hammerman PS. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107-20
82. Kim IK, Mccutcheon JN, Rao G. et al. Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene. 2019;38:180-93
83. Kadara H, Choi M, Zhang J. et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Annals of oncology: official journal of the European Society for Medical Oncology. 2017;28:75-82
84. Lee JJ, Park S, Park H. et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell. 2019;177:1842-1857.e21
85. Hao C, Wang L, Peng S. et al. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett. 2015;357:179-85
86. Hwang DH, Sholl LM, Rojas-Rudilla V. et al. KRAS and NKX2-1 Mutations in Invasive Mucinous Adenocarcinoma of the Lung. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2016;11:496-503
87. Walter David M, Venancio et al. Systematic Inactivation of Chromatin-Regulating Enzymes Identifies Setd2 as a Potent Tumor Suppressor in Lung Adenocarcinoma. Cancer Res. 2017;77:1719-29
88. Yang R, Piperdi S, Gorlick R. Activation of the RAF/mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway mediates apoptosis induced by chelerythrine in osteosarcoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6396-404
89. Mizumoto Y, Kyo S, Mori N. et al. Activation of ERK1/2 occurs independently of KRAS or BRAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci. 2007;98:652-8
90. Viaene AN, Santi M, Rosenbaum J. et al. SETD2 mutations in primary central nervous system tumors. Acta neuropathologica communications. 2018;6:123
91. Fontebasso AM, Schwartzentruber J, Khuong-Quang DA. et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125:659-69
92. Boussios S, Moschetta M, Karathanasi A. et al. Malignant peritoneal mesothelioma: clinical aspects, and therapeutic perspectives. Annals of gastroenterology. 2018;31:659-69
93. Jiang C, He C, Wu Z. et al. Histone methyltransferase SETD2 regulates osteosarcoma cell growth and chemosensitivity by suppressing Wnt/β-catenin signaling. Biochem Biophys Res Commun. 2018;502:382-8
94. Morcillo-Garcia S, Noblejas-Lopez MDM, Nieto-Jimenez C. et al. Genetic mutational status of genes regulating epigenetics: Role of the histone methyltransferase KMT2D in triple negative breast tumors. PLoS One. 2019;14:e0209134
95. Brennan CW, Verhaak RG, Mckenna A. et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462-77
96. Wu G, Diaz AK, Paugh BS. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444-50
97. Joseph NM, Chen YY, Nasr A. et al. Genomic profiling of malignant peritoneal mesothelioma reveals recurrent alterations in epigenetic regulatory genes BAP1, SETD2, and DDX3X. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2017;30:246-54
Author contact
![]() Corresponding authors: Xin Yang, Department of Oncology, The Third Affiliated Hospital of Soochow University, The First People's Hospital of Changzhou, Changzhou 213003, China, E-mail: yangxindoctorcom. Tel.: +8618861235566; Mei Ji, Department of Oncology, the Third Affiliated Hospital of Soochow University, The First People's Hospital of Changzhou, Changzhou 213003, China, E-mail: zlkjimeicom, Tel.: +8613775181579.
Corresponding authors: Xin Yang, Department of Oncology, The Third Affiliated Hospital of Soochow University, The First People's Hospital of Changzhou, Changzhou 213003, China, E-mail: yangxindoctorcom. Tel.: +8618861235566; Mei Ji, Department of Oncology, the Third Affiliated Hospital of Soochow University, The First People's Hospital of Changzhou, Changzhou 213003, China, E-mail: zlkjimeicom, Tel.: +8613775181579.