Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(7):1985-1993. doi:10.7150/jca.40002 This issue Cite
Research Paper
Systematic analysis of genetic variants in cancer-testis genes identified two novel lung cancer susceptibility loci in Chinese population
Zhihua Li1*, Jianwei Tang1*, Wei Wen1*, Weibing Wu1, Jun Wang1, Jing Xu1, Yue Yu1, Zhicheng He1, Xianglong Pan1, Haixing Wei1, Yining Zhu1, Shuo Hu1, Jing Cao1, Hongbing Shen2,3, Jun Que1, Wei Wang1, Quan Zhu1 ![]() , Liang Chen1
, Liang Chen1 ![]()
1. Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, China.
2. Department of Epidemiology, Center for Global Health, International Joint Research Center, School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, China.
3. Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Jiangsu Collaborative Innovation Center of Cancer Personalized Medicine, Nanjing Medical University, Nanjing, 211166, China.
*These authors contributed equally to this work.
Received 2019-9-4; Accepted 2019-12-25; Published 2020-2-3
Abstract
Cancer-testis (CT) genes played important roles in the progression of malignant tumors and were recognized as promising therapeutic targets. However, the roles of genetic variants in CT genes in lung cancer susceptibility have not been well depicted. This study aimed to evaluate the associations between genetic variants in CT genes and lung cancer risk in Chinese population. A total of 22,556 qualified SNPs from 268 lung cancer associated CT genes were initially evaluated based on our previous lung cancer GWAS (Genome-wide association studies) with 2,331 cases and 3,077 controls. As a result, 17 candidate SNPs were further genotyped in 1,056 cases and 1,053 controls using Sequenom platform. Two variants (rs6941653, OPRM1, T > C, screening: OR = 1.24, 95%CI: 1.12-1.38, P = 2.40×10-5; validation: OR = 1.18, 95%CI: 1.01-1.37, P = 0.039 and rs402969, NLRP8, C > T, screening: OR = 1.15, 95%CI: 1.04-1.26, P = 0.006; validation: OR = 1.16, 95%CI: 1.02-1.33, P = 0.028) were identified as novel lung cancer susceptibility variants. Stratification analysis indicated that the effect of rs6941653 was stronger in lung squamous cell carcinoma (OR = 1.36) than that in lung adenocarcinoma (OR = 1.15, I2 = 77%, P = 0.04). Finally, functional annotations, differential gene expression analysis, pathway and gene ontology analyses were performed to suggest the potential functions of our identified variants and genes. In conclusion, this study identified two novel lung cancer risk variants in Chinese population and provided deeper insight into the roles of CT genes in lung tumorigenesis.
Keywords: cancer-testis genes, lung cancer susceptibility, single nucleotide polymorphisms, Chinese population, Sequenom platform
Introduction
Lung cancer has been the most frequently diagnosed caner type and the leading cause of cancer-related deaths for decades in China [1]. The tumorigenesis of lung cancer was a multiple-stage process, and both environmental and genetic factors were involved. It was estimated that the heritability of lung cancer was about 15.2% in Chinese population [2]. However, up till now, GWAS (Genome-Wide Association Study)-reported lung cancer associated single nucleotide polymorphisms (SNPs) could only account for limited lung cancer heritability (less than 1%) [2, 3]. Therefore, more effective strategies were wanted to identify novel lung cancer risk loci based on GWAS data.
Epigenetic alterations have been recognized as an important feature of tumorigenesis [4, 5]. Notably, cancer-testis (CT) genes, which were restrictedly expressed in germ cells and malignant tumor cells, were usually activated through epigenetic mechanisms [6]. The activation of CT genes in cancer samples made them oncogene candidates and their remained high immunogenicity made them perfect immunotherapeutic targets for cancer treatment [7, 8]. In addition, associations between genetic variants in CT genes and the susceptibility of cancers have been described in previous studies. For example, genetic variants in HORMAD2 and GPATCH2 were reported associated with lung cancer risk [9, 10], and variants in CTNNA2, CCDC33 and SPAG17 showed significant association with the susceptibility of breast cancer [11, 12]. All these studies suggested that genetic variants in CT genes could also contribute to the development of cancers.
Therefore, systematic analysis of the associations between genetic variants in CT genes and lung cancer risk could help identify more novel lung cancer susceptibility loci. In our previous study, we performed a systematic identification of CT genes in 19 cancer types based on multiple public-available databases [13]. As a result, 876 novel CT genes in 19 cancer types were recognized. In lung cancer, we identified 268 CT genes (including 61 known CT genes) that were activated in at least 2% of cancer samples [13]. This finding provided us an unprecedented opportunity to explore the associations between genetic variants in CT genes and the susceptibility of lung cancer.
In this study, a two-stage case control study was performed. The NJMU GWAS, which has been established in our previous study, was used to screen candidate lung cancer risk variants [9]. These promising variants were further validated in an independent Chinese population with a total of 1,056 lung cancer cases and 1,053 controls based on the Sequenom MassARRAY iPLEX platform. This study would identify novel lung cancer susceptibility loci in Chinese population and help reveal the roles of genetic variants in CT genes in the development of lung cancer.
Materials and Methods
Study subjects
Two independent datasets were used in this study. The NJMU GWAS contained 2,331 lung cancer cases and 3,077 controls, and was used as screening dataset. The detailed information about the study subjects in NJMU GWAS was described previously [9]. Genotype imputation for NJMU GWAS was performed using IMPUTE2 and Shapeit v2 with the 1000 Genomes Project (the Phase III integrated variant set release, across 2504 samples) as the reference [14]. The validation dataset consisted of 1,056 lung cancer cases and 1,053 controls. All the cases were histopathologically confirmed patients and were recruited from the First Affiliated Hospital of Nanjing Medical University and Jiangsu Cancer Hospital. Controls were obtained from a screening program for non-infectious diseases conducted in Jiangsu Province and matched to the cases for age and gender. All the participants have signed the informed consent acknowledgement and this study was approved by the ethical review board of Nanjing Medical University.
Screening for candidate risk variants in CT genes
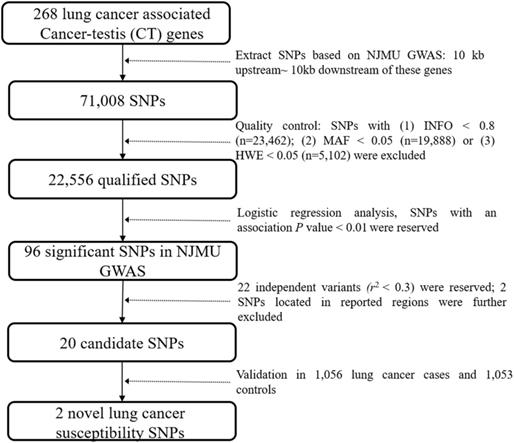
As shown in Figure 1, genetic variants in 268 lung cancer associated CT genes (Supplementary Table 1, including the 10 kb upstream and downstream of these genes) were extracted based on NJMU GWAS. Initially, 71,008 SNPs were reserved. Further, SNPs satisfied any of the following criteria were excluded: (1) imputation quality score INFO < 0.8; (2) minor allele frequency (MAF) < 0.05; (3) hardy-weinberg equilibrium (HWE) < 0.05. This resulted in 22,556 qualified SNPs reserved for association analysis. Among them, 96 SNPs showed significant association (P < 0.01) with lung cancer risk in NJMU GWAS. Furthermore, SNPs that were located in previously reported regions or showed a high correlation (r2 > 0.3) with each other (SNPs with the least P value were reserved) were excluded. Finally, 20 independent SNPs were retained as lung cancer risk variant candidates.
Flowchart of this study.
Variants validation using Sequenom platform
In this study, genotyping in the validation stage was performed using the Sequenom MassARRAY iPLEX platform (Sequenom Inc. San Diego, CA, USA) according to the protocol [15, 16]. Primers for 17 out of 20 candidate SNPs were successfully constructed. In particular, rs144031443 and rs150492976 were replaced by rs75932085 (r2 = 0.66, Chinese Han population) and rs4726004 (r2 = 1), respectively. The genotyping experiment was performed by technicians who were blinded to the status of case or control. Cases and controls were mixed in each 384-well plate with two water samples as blank controls. In addition, 5% of samples were randomly selected for repeat genotyping, which yielded a concordance rate of 100%. Primer sequences used in this study were shown in Supplementary Table 2.
Functional annotations, pathway and Gene Ontology (GO) analyses
To gain insight into the potential functions of our identified SNPs and their related variants (r2 ≥ 0.6), several public-available databases, including HaploReg v4.1 (https://pubs.broadinstitute.org/mammals/haploreg/), ENCODE, rSNPBase (http://rsnp.psych.ac.cn/index.do) [17, 18], SNP2TFBS (https://ccg.epfl.ch/snp2tfbs/) [19], PINES (http://genetics.bwh.harvard.edu/pines/index.html) and RegulomeDB (http://regulome.stanford.edu/) [20], were used for functional annotations. Pathway enrichment and GO analyses were performed based on DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/home.jsp) [21].
Statistical analysis
The chi-square test was used for the comparison of categorical variables between cases and controls, while the Student's t test was adopted for continuous variables. Associations between genetic variants and lung cancer risk were evaluated (Odd Ratios, OR and 95% confidence intervals, 95%CI) using logistical regression analysis. Age, sex, smoking status and principal components were adjusted. The differential gene expression analysis was conducted using RNA sequence data (107 paired lung cancer and adjacent normal tissues) from the Cancer Genome Atlas (TCGA) database, and was further validated in 60 pairs of lung tumor and normal tissues from GSE19804. The expression quantitative trait loci (eQTL) analysis was performed based on the GTEx v7 database (383 lung tissues). OPRM1 and NLRP8 co-expressed genes were screened based on lung cancer RNA-seq (TCGA) using Pearson correlation analysis. All the analyses were performed using R (3.5.2) or Plink 1.9.
Results
Characteristics of study subjects
The characteristics of study subjects were shown in Table 1. In brief, a total of 3,387 lung cancer cases and 4,130 controls were included in this study. Lung adenocarcinoma (LUAD) was the most common histological type and accounted for more than 50% of all the lung cancer cases. Lung squamous cell carcinoma (SCC) accounted for 35.3% and 21.8% in NJMU GWAS and validation samples, respectively. The distribution of age, sex and smoking status was comparable between lung cancer cases and controls in validation samples (P > 0.05).
Characteristics of subjects in screening and validation stages.
| Characteristics | NJMU GWAS | Validation | |||||
|---|---|---|---|---|---|---|---|
| Cases | Controls | P a | Cases | Controls | P a | ||
| Sample size | 2331 | 3077 | 1056 | 1053 | |||
| Gender | <0.001 | 0.782 | |||||
| Male | 1711(73.4%) | 2086(67.8%) | 700(66.3%) | 704(66.9%) | |||
| Female | 620(26.6%) | 991(32.2%) | 356(33.7%) | 349(33.1%) | |||
| Smoking | <0.001 | 0.432 | |||||
| Yes | 1506(64.6%) | 1309(42.5%) | 464(43.9%) | 444(42.2%) | |||
| No | 825(35.4%) | 1768(57.5%) | 592(56.1%) | 609(57.8%) | |||
| Age (years) | 0.124 | 0.317 | |||||
| Mean | 60.05±10.27 | 60.47±9.66 | 61.40±9.59 | 60.98±9.95 | |||
| < 60 | 1111(47.7%) | 1429(46.4%) | 422(40.0%) | 384(36.5%) | |||
| ≥ 60 | 1220(52.3%) | 1648(53.6%) | 634(60.0%) | 669(63.5%) | |||
| Histology | |||||||
| Squamous cell carcinoma | 822(35.3%) | / | 230(21.8%) | / | |||
| Adenocarcinoma | 1304(55.9%) | / | 622(58.9%) | / | |||
| Other | 205(8.8%) | / | 204(19.3%) | / | |||
a The chi-square test was used for gender, smoking and histology. Student's t test was adopted for age.
Two novel lung cancer susceptibility variants
As mentioned above, 17 of 20 lung cancer candidate SNPs were successfully genotyped in validation stage. Associations between 17 candidate variants and lung cancer risk were shown in Table 2. Notably, only rs6941653 and rs402969 were significant in both screening (rs6941653, OPRM1, T > C, OR = 1.24, 95%CI: 1.12-1.38, P = 2.40×10-5; rs402969, NLRP8, C > T, OR = 1.15, 95%CI: 1.04-1.26, P = 0.006) and validation datasets (rs6941653, OPRM1, T > C, OR = 1.18, 95%CI: 1.01-1.37, P = 0.039; rs402969, NLRP8, C > T, OR = 1.16, 95%CI: 1.02-1.33, P = 0.028). Therefore, rs6941653 and rs402969 were considered as novel lung cancer risk variants in Chinese population.
Associations between 17 candidate SNPs and lung cancer risk in screening and validation datasets.
| SNP | BP | CytoBand | Gene | Allele a | NJMU GWAS | Validation | Combined | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95%CI) b | P b | OR (95%CI) c | P c | OR (95%CI) | P | |||||||
| rs6941653 | chr6:154531020 | 6q25.2 | OPRM1 | C/T | 1.24(1.12,1.38) | 2.40E-05 | 1.18(1.01-1.37) | 0.039 | 1.22(1.12-1.33) | 7.84E-06 | ||
| rs6851719 | chr4:94120480 | 4q22.2 | GRID2 | C/A | 1.18(1.08,1.29) | 3.82E-04 | 1.05(0.92-1.20) | 0.462 | 1.14(1.06-1.23) | 6.15E-04 | ||
| rs8139987 | chr22:25131425 | 22q11.23 | PIWIL3 | T/G | 1.28(1.12,1.47) | 4.01E-04 | 0.92(0.76-1.12) | 0.402 | 1.09(0.79-1.51) | 0.588 | ||
| rs79727953 | chr7:102449418 | 7q22.1 | FBXL13 | A/C | 1.24(1.10,1.39) | 4.76E-04 | 1.22(0.98-1.45) | 0.073 | 1.23(1.12-1.36) | 1.53E-05 | ||
| rs77027865 | chr1:152863288 | 1q21.3 | SMCP | G/A | 1.34(1.12,1.59) | 1.13E-03 | 1.14(0.88-1.46) | 0.321 | 1.27(1.10-1.46) | 8.12E-04 | ||
| rs3123484 | chr1:182884429 | 1q25.3 | SHCBP1L | T/C | 0.75(0.63,0.89) | 1.34E-03 | 1.43(1.12-1.82) | 0.002 | 1.03(0.55-1.94) | 0.928 | ||
| rs145033304 | chr19:56514378 | 19q13.43 | NLRP5 | T/C | 0.86(0.78,0.94) | 1.37E-03 | 0.84(0.66-1.07) | 0.169 | 0.86(0.79-0.93) | 3.10E-04 | ||
| rs12645087 | chr4:178779663 | 4q34.3 | RP11-389E17.1 | T/C | 0.80(0.69,0.92) | 1.70E-03 | 1.02(0.85-1.23) | 0.827 | 0.90(0.71-1.14) | 0.364 | ||
| rs9478496 | chr6:154333183 | 6q25.2 | OPRM1 | C/T | 1.30(1.10,1.54) | 2.56E-03 | 0.96(0.75-1.22) | 0.737 | 1.13(0.84-1.52) | 0.415 | ||
| rs144031443 | chr4:94171881 | 4q22.2 | GRID2 | A/G | 0.76(0.63,0.91) | 2.70E-03 | 0.86(0.62-1.20) | 0.376 | 0.78(0.67-0.92) | 2.30E-03 | ||
| rs17135666 | chr16:1942405 | 16p13.3 | MEIOB | T/C | 0.79(0.68,0.92) | 2.96E-03 | 1.05(0.84-1.31) | 0.669 | 0.90(0.68-1.19) | 0.458 | ||
| rs7546603 | chr1:182529584 | 1q25.3 | RGSL1 | C/T | 1.20(1.06,1.35) | 4.50E-03 | 1.03(0.86-1.23) | 0.747 | 1.15(1.04-1.26) | 6.76E-03 | ||
| rs79461429 | chr4:93761350 | 4q22.2 | GRID2 | G/A | 1.18(1.05,1.32) | 5.94E-03 | 1.14(0.97-1.37) | 0.118 | 1.17(1.06-1.29) | 1.38E-03 | ||
| rs402969 | chr19:56450708 | 19q13.43 | NLRP8 | T/C | 1.15(1.04-1.26) | 6.00E-03 | 1.16(1.02-1.33) | 0.028 | 1.15(1.07-1.24) | 2.38E-04 | ||
| rs150492976 | chr7:150875087 | 7q36.1 | GBX1 | T/C | 0.86(0.78,0.96) | 7.05E-03 | 0.99(0.85-1.15) | 0.855 | 0.90(0.83-0.99) | 0.025 | ||
| rs175150 | chr22:17311027 | 22q11.1 | XKR3 | A/C | 1.14(1.04,1.26) | 7.96E-03 | 1.00(0.87-1.14) | 0.960 | 1.09(1.00-1.18) | 0.041 | ||
| rs60813831 | chr19:43930119 | 19q13.31 | TEX101 | C/G | 0.89(0.82,0.97) | 8.12E-03 | 0.93(0.83-1.05) | 0.246 | 0.90(0.84-0.97) | 4.48E-03 | ||
a: Minor/Major allele; b: Age, gender, smoking pack-years and PCA were adjusted; c: Age, gender and smoking status were adjusted. SNPs that showed consistent association results and had an association P value < 0.05 in both NJMU GWAS and validation datasets were marked in bold.
Stratification analysis and interaction analysis
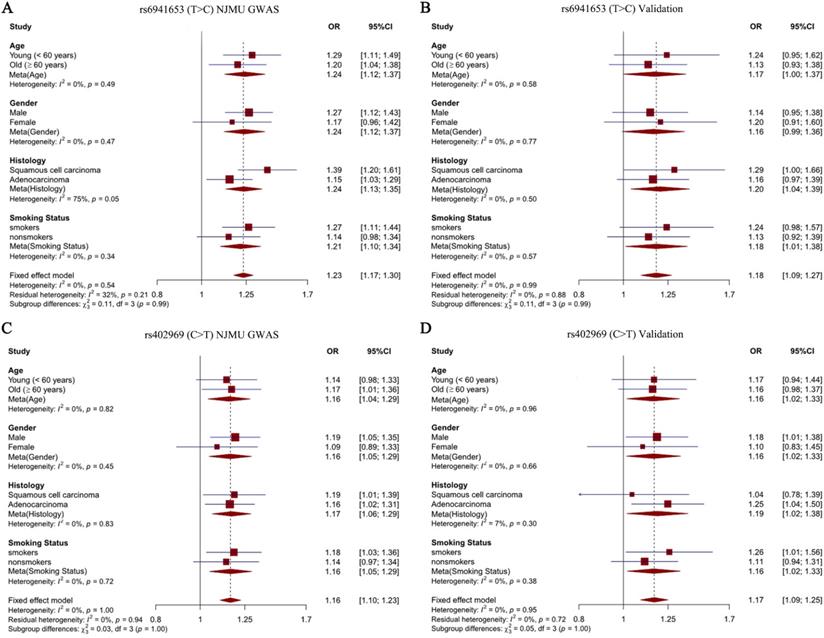
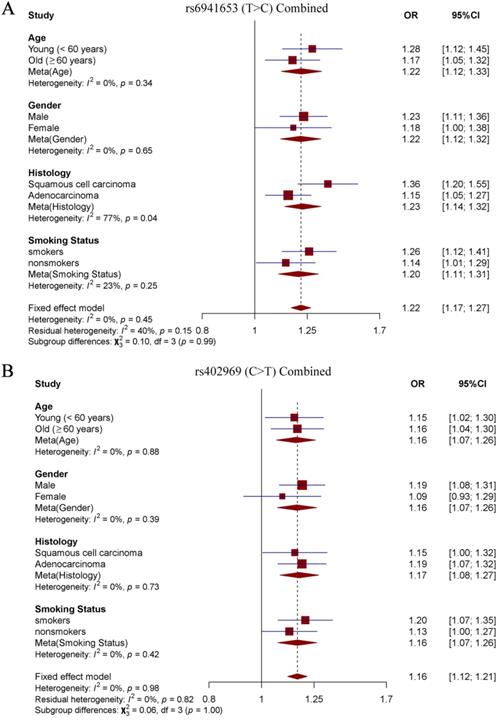
To explore the effects of our identified two SNPs in different subgroup populations, stratification analysis was further performed. Study subjects were divided into four subgroups according to age (young < 60 years old vs old ≥ 60 years old), sex (males vs females), smoking status (smokers vs nonsmokers) and histological subtypes (SCC vs LUAD). Notably, we observed a significantly stronger effect of rs6941653 in SCC (NJMU GWAS: OR = 1.39; Validation: OR = 1.29; Combined: OR = 1.36) than that in LUAD (NJMU GWAS: OR = 1.15; Validation: OR = 1.16; Combined: OR = 1.15, I2 = 77%, P = 0.04, Figure 2A-2B, Figure 3A). In contrast to rs6941653, SNP rs402969 showed a similar effect on lung cancer risk in various subgroup populations (Figure 2C-D, Figure 3B). In addition, interactions between our identified lung cancer risk variants (rs6941653 and rs402969) and smoking were evaluated. However, no significant interaction was found (Supplementary Table 3, P for interaction > 0.05). Similarly, there was no significant interaction between variant rs6941653 and rs402969 (Supplementary Table 4, P for interaction > 0.05).
Forest plot of rs6941653 and rs402969 in each subgroup. (A) Forest plot of rs6941653 in NJMU GWAS; (B) Forest plot of rs6941653 in validation dataset; (C) Forest plot of rs402969 in NJMU GWAS; (D) Forest plot of rs402969 in validation samples.
Forest plot of rs6941653 and rs402969 in combined datasets. (A) Forest plot of rs6941653 in combined data; (B) Forest plot of rs402969 in combined dataset.
Functional annotations and eQTL analysis
In order to suggest the potential functions of our identified two novel variants and their related SNPs, functional annotations were performed. As shown in Table 3, rs6941653 was not located in regulatory element regions, such as promoter, enhancer, transcription factor (TF) binding sites or DNase peak. Strikingly, rs9397692 (r2 = 0.70 with rs6941653) was an eQTL and could influence the binding of transcription factor NFATC2. What's more, rs9397692 had a Regulome DB score of 4, suggesting that it was located in TF binding site and DNase peak. All these results indicated that rs9397692 might be the functional variant, which could affect the binding of specific transcription factor NFATC2. In the second risk loci, rs805165 (r2 = 0.90 with rs402969) was predicted to be an eQTL and could modify the affinity to TF BRCA1. Consistently, the Regulome DB score of rs805165 was 5 (TF binding or DNase peak). Taken together, rs805165 might be the functional variant in the second loci. Furthermore, eQTL analysis in lung tissues was conducted based on GTEx v7 database. Unfortunately, the expression of OPRM1 and NLRP8 in lung tissues was not sufficient for eQTL calculation. Therefore, associations between our identified variants and the expression of their host genes (OPRM1 and NLRP8) were not evaluated. Notably, rs805165 was found to be significantly associated with the expression of AC010525.7 and AC024580.1 (Supplementary Figure 1). However, the functions of AC010525.7 and AC024580.1 have not been reported in previous studies.
Functional annotations for our identified SNPs and their related variants (r2 ≥ 0.6).
| r2 | SNP | Motifs | PINES a | Regulome DB b | Regulation c | eQTL d | SNP2TFBS e |
|---|---|---|---|---|---|---|---|
| 0.75 | rs10485060 | HNF4, Pax-4, RXRA | 0.433 | 6 | no | yes | HNF4G, HNF4A, SOX10 |
| 0.80 | rs2272381 | Foxp1, Hoxd8, Pou3f2 | 0.297 | 6 | yes | yes | / |
| 0.95 | rs2293537 | 7 altered motifs | 0.473 | No Data | yes | no | Crx |
| 0.65 | rs35184807 | AP-1 | 3'-UTR | 5 | yes | no | / |
| 0.77 | rs60145555 | AP-4, ELF1 | 0.178 | 5 | no | no | / |
| 0.95 | rs61307239 | EBF, ZEB1 | 0.372 | 6 | no | no | / |
| 0.70 | rs62434770 | 5 altered motifs | 0.281 | No Data | no | no | Pdx1, Prrx2 |
| 0.68 | rs62436491 | Foxd3, Foxj1 | 0.868 | 5 | no | no | / |
| 0.92 | rs6900677 | Myf | 0.379 | 6 | no | no | / |
| 0.81 | rs6921548 | 6 altered motifs | 0.048 | 5 | yes | no | / |
| 1.00 | rs6941653 | Maf, PTF1-beta | 0.485 | No Data | no | no | / |
| 0.70 | rs72574410 | 4 altered motifs | 0.232 | 6 | no | no | SPIB |
| 0.75 | rs74439078 | 5 altered motifs | 0.414 | 6 | no | no | / |
| 0.73 | rs9371779 | BDP1, E2F | 0.337 | 6 | no | no | / |
| 0.95 | rs9383694 | 9 altered motifs | 0.438 | 5 | yes | no | / |
| 0.94 | rs9383695 | BATF, Irf, SP1 | 0.367 | No Data | yes | no | / |
| 0.78 | rs9384190 | 5 altered motifs | 0.408 | 6 | no | yes | RUNX1, RUNX2 |
| 0.75 | rs9397179 | Pou2f2 | 0.369 | 6 | no | no | / |
| 0.70 | rs9397692 | NF-AT, NF-AT1 | 0.086 | 4 | no | yes | NFATC2 |
| 0.94 | rs9397696 | 6 altered motifs | 0.601 | No Data | no | no | Klf4, Klf1 |
| 0.74 | rs9478516 | 10 altered motifs | 0.498 | 6 | no | no | NFATC2 |
| 0.74 | rs9478517 | DMRT5, Foxa | 0.498 | 6 | no | no | Sox5 |
| 0.91 | rs9479791 | AP-1 | 0.424 | No Data | no | yes | / |
| 0.87 | rs451276 | 11 altered motifs | 0.621 | No Data | no | no | Klf4, Klf1 |
| 1.00 | rs402969 | Hbp1, TCF12, ZBRK1 | 0.512 | 5 | no | no | / |
| 0.90 | rs395589 | 10 altered motifs | 0.512 | 6 | no | no | / |
| 0.92 | rs381249 | SIX5 | 0.512 | 6 | no | no | / |
| 0.90 | rs448020 | Evi-1, PEBP, RXRA | 0.512 | 5 | no | no | / |
| 0.86 | rs370095 | 4 altered motifs | 0.668 | No Data | no | no | / |
| 0.90 | rs371382 | 5 altered motifs | 0.396 | No Data | no | no | Pax2 |
| 0.90 | rs805166 | 5 altered motifs | 0.408 | No Data | no | no | ZNF263 |
| 0.90 | rs809275 | Ets, TBX5, YY1 | 0.408 | 3a | yes | no | / |
| 0.90 | rs805165 | BHLHE40, BRCA1, Maf | 0.475 | 5 | no | yes | BRCA1 |
| 0.69 | rs809800 | GR, STAT | 0.475 | 6 | no | no | / |
| 0.90 | rs805164 | 6 altered motifs | 0.475 | No Data | no | no | SP1 |
| 0.68 | rs413691 | / | 0.483 | 6 | yes | no | MEF2C, MEF2A |
| 0.90 | rs429498 | 6 altered motifs | 0.483 | No Data | no | no | SP2 |
| 0.82 | rs409402 | 5 altered motifs | 0.68 | 5 | yes | no | / |
| 0.81 | rs393535 | NRSF, Zfx | 0.68 | No Data | yes | no | Zfx |
| 0.90 | rs810903 | STAT | 0.672 | No Data | no | yes | / |
| 0.85 | rs306508 | 4 altered motifs | 0.406 | No Data | no | yes | / |
| 0.85 | rs306507 | / | missense | No Data | no | yes | / |
| 0.85 | rs306506 | 8 altered motifs | missense | No Data | no | no | BRCA1 |
| 0.85 | rs7343161 | Irf, SIX5, STAT | 0.623 | 5 | no | yes | / |
| 0.85 | rs306502 | Cdx | 0.685 | No Data | no | no | / |
| 0.85 | rs306501 | 5 altered motifs | 0.763 | 6 | no | yes | / |
a: PINES (http://genetics.bwh.harvard.edu/pines/index.html) provided a powerful in silico method to prioritize functional variants. SNPs with lower P values were more likely to be functional variants. b: Scores for regulatory elements based on RegulomeDB website (http://regulome.stanford.edu/); “3a”: TF binding + any motif + DNase peak; “4”: TF binding + DNase peak; “5”: TF binding or DNase peak; “6”: other. c: Proximal regulation or Distal regulation based on rSNPBase database (http://rsnp.psych.ac.cn/index.do); d: eQTL with experimental evidence based on rSNPBase database. e: The potential transcription factors were predicted based on SNP2TFBS (https://ccg.epfl.ch/snp2tfbs/).
Differential gene expression, pathway and GO analyses
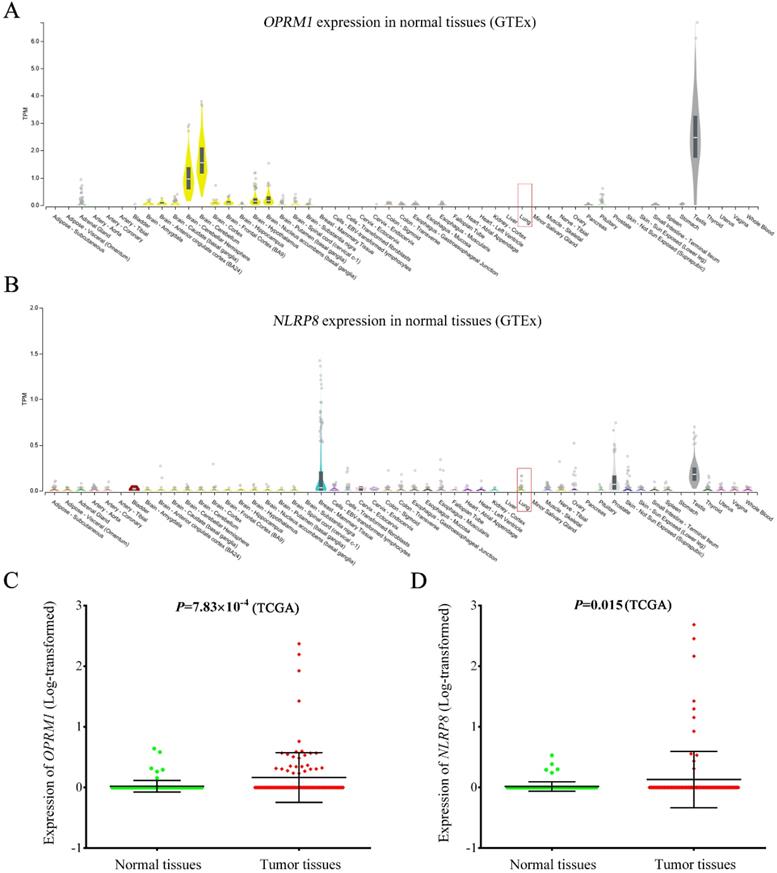
As shown in Figure 4, OPRM1 was not expressed in normal lung tissues (GTEx database), but showed abundant expression in testis and brain tissues (Figure 4A). The consistent finding was observed in lung cancer adjacent normal tissues based TCGA database. However, the expression of OPRM1 was significantly elevated in lung tumor tissues compared to that in normal tissues (P = 7.83 × 10-4, Figure 4C). Similarly, NLRP8 expression was aberrantly upregulated in tumor tissues (Figure 4D, P = 0.015). These findings were further validated in 60 paired lung tumor tissues and adjacent normal tissues from GSE19804 (Supplementary Figure 2). Pathway enrichment analysis showed that OPRM1 co-expressed genes (Fold enrichment: 14.0, P = 2.10 × 10-214) and NLRP8 co-expressed genes (Fold enrichment: 6.7, P = 3.10 × 10-110, Supplementary Table 5) were enriched in the olfactory transduction pathway. The GO analysis indicated that OPRM1 co-expressed genes might participate in various biological processes, such as immune response, signaling pathway and cell differentiation. The co-expressed genes with NLRP8 could take part in the sensory perception, keratinocyte differentiation and G-protein coupled receptor signaling pathway.
Expression of OPRM1 and NLRP8 in normal lung tissues and lung tumor tissues. OPRM1 (A) and NLRP8 (B) expression in a variety of normal tissues based on GTEx database. Lung tissue was marked in red box; The expression of OPRM1 (C) and NLRP8 (D) in 107 pairs of lung cancer tissues and adjacent normal tissues based on TCGA database.
Discussion
Although lung cancer GWASs have identified dozens of lung cancer susceptibility loci, more lung cancer associated variants remained to be identified [3]. Cancer-testis genes were recognized as promising therapeutic targets for cancers due to their high immunogenicity in tumor tissues [22, 23]. However, the roles of genetic variants in CT genes in the development of lung cancer have not been revealed in previous studies.
In the current study, a two-stage case-control study was performed to systematically evaluate the associations between genetic variants in CT genes and the risk of lung cancer in the Chinese population. A total of 22,556 qualified SNPs from 268 lung cancer associated CT genes were initially analyzed and 17 candidate SNPs were genotyped using the Sequenom platform. Finally, two variants (rs6941653 in OPRM1 and rs402969 in NLRP8) were identified as novel susceptibility loci of lung cancer in Chinese population.
SNP rs6941653 was located in the intron of OPRM1 (opioid receptor mu 1, 6q25.2), which encoded one of the opioid receptors [24]. Previous studies revealed that genetic variants in OPRM1 could modulate the dependence to multiple drugs or chemical agents, including nicotine, cocaine, alcohol [25-27]. Due to these properties, OPRM1 has been reported associated with the progression of Alzheimer's disease, Parkinson's disease, Schizophrenia and so on [28-30]. In lung cancer, Lennon FE et al found that OPRM1 (also known as MOR) expression was elevated in several human non-small cell lung cancer cell lines. What's more, overexpression of OPRM1 significantly promoted the proliferation of lung cancer in vitro and in vivo [31, 32]. Consistently, OPRM1 was recognized as a novel CT gene in lung cancer because of its aberrant expression in lung tumor tissues according to our previous study [13]. In the current study, rs6941653 in OPRM1 was further identified associated with lung cancer risk in Chinese population. Strikingly, stratification analysis indicated that rs6941653 showed a stronger effect in the SCC population. With regard to the potential mechanisms, we speculated that nicotine dependence induced by OPRM1 might partially account for this. Functional annotations for rs6941653 and their LD variants suggested that rs9397692, which was in the DNase peak and could affect the binding of TF NFATC2, might be the functional variant in these loci.
The rs402969 was located in the upstream of NLRP8, which belonged to the member of the nucleotide-binding oligomerization domain/ leucine rich repeat/ pyrin domain containing (NLRP) subfamily [33]. This gene family was involved in innate immunity, inflammasome formation and mammalian reproduction [34-36]. However, the functions and roles of NLRP8 in cancers have not been described in the previous study. In this study, NLRP8 expression was found significantly upregulated in lung cancer tissues. What's more, rs805165 (LD with rs402969) was predicted in the binding sites of TF BRCA1 or DNase peak, suggesting a potential role as a regulator of gene expression. As expected, rs805165 showed a significant association with the expression of AC010525.7 and AC024580.1. Nevertheless, the functions of them have not been elucidated. Unfortunately, the expression of NLRP8 in lung tissues was not sufficient for eQTL evaluation. Function studies were needed to reveal the role of NLRP8 in lung cancer and identify the potential functional variants in these loci.
In conclusion, we performed a systematic evaluation of the associations between genetic variants in CT genes and the risk of lung cancer. Two novel lung cancer susceptibility loci were successfully identified in Chinese population. This study could improve our understanding of the roles of CT genes in the tumorigenesis of lung cancer. However, functional studies were wanted to verify the functional variants and reveal the potential mechanisms.
Abbreviations
CT: cancer-testis; GWAS: Genome-wide association studies; SNP: single nucleotide polymorphisms; MAF: minor allele frequency; HWE: hardy-weinberg equilibrium; GO: Gene Ontology; OR: Odd Ratios; CI: confidence intervals; eQTL: expression quantitative trait loci; TCGA: The Cancer Genome Atlas; SCC: squamous cell carcinoma; LUAD: lung adenocarcinoma; TF: transcription factor.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors would like to thank all the patients, research staff and students who participated in this study. This work was supported by National Natural Science of China (81972175), Major Program of Science and Technology Foundation of Jiangsu Province (No. BE2016790 and BE2018746), the Program of Jiangsu Medical Innovation Team (No. CXTDA2017006), Natural Science Foundation of Jiangsu Province (No. BK20181496), Jiangsu Top Expert Program in Six Professions (WSW-007), Jiangsu Medical Young Talent Project (No. QNRC2016566) and Jiangsu Province 333 Talents Project (No. BRA2017545).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Dai J, Shen W, Wen W, Chang J, Wang T, Chen H. et al. Estimation of heritability for nine common cancers using data from genome-wide association studies in Chinese population. Int J Cancer. 2017;140:329-36
3. Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:363-79
4. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12-27
5. Brien GL, Valerio DG, Armstrong SA. Exploiting the Epigenome to Control Cancer-Promoting Gene-Expression Programs. Cancer Cell. 2016;29:464-76
6. Salmaninejad A, Zamani MR, Pourvahedi M, Golchehre Z, Hosseini Bereshneh A, Rezaei N. Cancer/Testis Antigens: Expression, Regulation, Tumor Invasion, and Use in Immunotherapy of Cancers. Immunol Invest. 2016;45:619-40
7. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615-25
8. Gordeeva O. Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin Cancer Biol. 2018;53:75-89
9. Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z. et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat Genet. 2011;43:792-6
10. Byun J, Schwartz AG, Lusk C, Wenzlaff AS, de Andrade M, Mandal D. et al. Genome-wide association study of familial lung cancer. Carcinogenesis. 2018;39:1135-40
11. Haryono SJ, Datasena IG, Santosa WB, Mulyarahardja R, Sari K. A pilot genome-wide association study of breast cancer susceptibility loci in Indonesia. Asian Pac J Cancer Prev. 2015;16:2231-5
12. Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S. et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92-4
13. Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N. et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun. 2016;7:10499
14. Li Z, Pu Z, Fan J, Li N, Zhu M, Zhang J. et al. Fine mapping in TERT-CLPTM1L region identified three independent lung cancer susceptibility signals: A large-scale multi-ethnic population study. Mol Carcinog. 2018;57:1289-99
15. Trembizki E, Smith H, Lahra MM, Chen M, Donovan B, Fairley CK. et al. High-throughput informative single nucleotide polymorphism-based typing of Neisseria gonorrhoeae using the Sequenom MassARRAY iPLEX platform. J Antimicrob Chemother. 2014;69:1526-32
16. Hang D, Zhou W, Jia M, Wang L, Zhou J, Yin Y. et al. Genetic variants within microRNA-binding site of RAD51B are associated with risk of cervical cancer in Chinese women. Cancer Med. 2016;5:2596-601
17. Guo L, Du Y, Chang S, Zhang K, Wang J. rSNPBase: a database for curated regulatory SNPs. Nucleic Acids Res. 2014;42:D1033-9
18. Tian J, Chang J, Gong J, Lou J, Fu M, Li J. et al. Systematic Functional Interrogation of Genes in GWAS Loci Identified ATF1 as a Key Driver in Colorectal Cancer Modulated by a Promoter-Enhancer Interaction. Am J Hum Genet. 2019;105:29-47
19. Kumar S, Ambrosini G, Bucher P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017;45:D139-D44
20. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790-7
21. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57
22. Grizzi F, Mirandola L, Qehajaj D, Cobos E, Figueroa JA, Chiriva-Internati M. Cancer-testis antigens and immunotherapy in the light of cancer complexity. Int Rev Immunol. 2015;34:143-53
23. Whitehurst AW. Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol. 2014;54:251-72
24. Crist RC, Berrettini WH. Pharmacogenetics of OPRM1. Pharmacol Biochem Behav. 2014;123:25-33
25. Chen YT, Tsou HH, Kuo HW, Fang CP, Wang SC, Ho IK. et al. OPRM1 genetic polymorphisms are associated with the plasma nicotine metabolite cotinine concentration in methadone maintenance patients: a cross sectional study. J Hum Genet. 2013;58:84-90
26. Clarke TK, Crist RC, Kampman KM, Dackis CA, Pettinati HM, O'Brien CP. et al. Low frequency genetic variants in the mu-opioid receptor (OPRM1) affect risk for addiction to heroin and cocaine. Neurosci Lett. 2013;542:71-5
27. Otto JM, Gizer IR, Deak JD, Fleming KA, Bartholow BD. A cis-eQTL in OPRM1 is Associated with Subjective Response to Alcohol and Alcohol Use. Alcohol Clin Exp Res. 2017;41:929-38
28. Xu C, Liu G, Ji H, Chen W, Dai D, Chen Z. et al. Elevated methylation of OPRM1 and OPRL1 genes in Alzheimer's disease. Mol Med Rep. 2018;18:4297-302
29. Cormier-Dequaire F, Bekadar S, Anheim M, Lebbah S, Pelissolo A, Krack P. et al. Suggestive association between OPRM1 and impulse control disorders in Parkinson's disease. Mov Disord. 2018;33:1878-86
30. Ding S, Chen B, Zheng Y, Lu Q, Liu L, Zhuge QC. Association study of OPRM1 polymorphisms with Schizophrenia in Han Chinese population. BMC Psychiatry. 2013;13:107
31. Lennon FE, Mirzapoiazova T, Mambetsariev B, Salgia R, Moss J, Singleton PA. Overexpression of the mu-opioid receptor in human non-small cell lung cancer promotes Akt and mTOR activation, tumor growth, and metastasis. Anesthesiology. 2012;116:857-67
32. Lennon FE, Mirzapoiazova T, Mambetsariev B, Poroyko VA, Salgia R, Moss J. et al. The Mu opioid receptor promotes opioid and growth factor-induced proliferation, migration and Epithelial Mesenchymal Transition (EMT) in human lung cancer. PLoS One. 2014;9:e91577
33. Duenez-Guzman EA, Haig D. The evolution of reproduction-related NLRP genes. J Mol Evol. 2014;78:194-201
34. Martinon F, Gaide O, Petrilli V, Mayor A, Tschopp J. NALP inflammasomes: a central role in innate immunity. Semin Immunopathol. 2007;29:213-29
35. MacDonald JA, Wijekoon CP, Liao KC, Muruve DA. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life. 2013;65:851-62
36. Tian X, Pascal G, Monget P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol. 2009;9:202
Author contact
![]() Corresponding authors: Liang Chen, Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, No. 300, Guangzhou Road, Nanjing 210029, China. Tel: +86-13913872727. Fax: +86-25-83718836. E-mail: clbright0909edu.cn or Quan Zhu, Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, No. 300, Guangzhou Road, Nanjing 210029, China. Tel: +86-13505152928. Fax: +86-25-83718836. Email: chest2006com.
Corresponding authors: Liang Chen, Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, No. 300, Guangzhou Road, Nanjing 210029, China. Tel: +86-13913872727. Fax: +86-25-83718836. E-mail: clbright0909edu.cn or Quan Zhu, Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, No. 300, Guangzhou Road, Nanjing 210029, China. Tel: +86-13505152928. Fax: +86-25-83718836. Email: chest2006com.