Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(15):3571-3581. doi:10.7150/jca.28428 This issue Cite
Research Paper
LncRNA SNHG16 Promotes Hepatocellular Carcinoma Proliferation, Migration and Invasion by Regulating miR-186 Expression
Hang Chen, Molin Li, Ping Huang ![]()
National Key Clinical Department, Department of Hepatobiliary Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing Medical University, Chongqing 400000, P.R. China.
Received 2018-7-10; Accepted 2019-4-30; Published 2019-6-9
Abstract
Long non-coding RNA (lncRNA) and microRNA (miRNA) play an important role in genesis and progression of tumors. The aim of this study was to explore the expression, biological function and molecular mechanism of small nucleolar RNA host gene 16 (SNHG16) in HCC. RT-qPCR was conducted to evaluate the expression level of SNHG16 in HCC tissues and cell lines. Our findings showed for the first time that SNHG16 was up-regulated in HCC tissues and cell lines. The expression of SNHG16 in cancer tissues was highly correlated with tumor size, TNM stage, ALT expression level and HBV DNA level. Moreover, cell proliferation, migration and invasion were detected by CCK-8 assay, transwell migration assay and transwell invasion assay, respectively. Xenograft tumor experiment was used to determine the biological function of SNHG16 in vivo. As revealed by our data, SNHG16 accelerated the proliferation, migration and invasion of HCC cell. SNHG16 facilitated tumor formation in vivo. Next, the relationship between SNHG16, miR-186 and ROCK1 were analyzed using bioinformatics analysis, qRT-PCR, luciferase reporter assay and western blot. Further molecular mechanism studies reported that the expression of SNHG16 was negatively correlated with the level of miR-186 and SNHG16 directly bound to miR-186. SNHG16 and miR-186 repressed each other. Notably, rescue experiments were conducted and showed that miR-186 reversed the effect of SNHG16 on cell. Taken together, SNHG16 promoted HCC cell proliferation, migration and invasion by functioning as a competitive endogenous RNA (ceRNA) to negatively regulate miR-186 expression. Our data suggested that SNHG16 might be a potential biomarker and a new therapeutic target for HCC.
Keywords: SNHG16, miR-186, HCC, Proliferation, Migration, Invasion
Introduction
Hepatocellular carcinoma (HCC) is the second most common cause of cancer-related deaths worldwide [1]. The characteristics of high metastasis and invasiveness lead to a high recurrence rate [2] thereby resulting in a poor prognosis [3]. The pathogenesis of HCC involved in many signaling pathways, and the molecular mechanism underlying malignant progression of HCC is quite ambiguous. Therefore, it is imperative to study the molecular mechanism of HCC, and explore possible tumor markers and therapeutic targets.
MicroRNA (miRNA) and long non-coding RNA (lncRNA) have been considered as a new biomarkers and therapeutic target [4,5]. MiRNAs, non-coding RNAs with a size of 20-25 nucleotides, are highly conserved and derived from hairpins or double-stranded RNA precursors [6]. The incomplete base complementation between the miRNA and the target mRNA can suppress the expression of targeted mRNA [7], and a full complementation leads to the degradation of targeted mRNA [8]. MiRNAs serve an important role in growth, apoptosis, differentiation, metabolism and immunity of cells and molecules, and is considered as potential new targets for cancer diagnosis and treatment [9]. Multiple studies have indicated that miR-186 expression was markedly decreased in ovarian cancer [10], non-small cell lung cancer [11], esophageal cancer [12] and prostate cancer [13]. MiR-186 exerts tumor inhibitory functions by inhibiting proliferation, migration, invasion and inducing apoptosis in HCC [14].
Thousands of non-coding RNA (ncRNA) genes have been discovered by the development of high-throughput RNA sequencing technology [15]. LncRNAs are defined as transcripts longer than 200 nucleotides (nt) that are 5ˊcapped and 3ˊpolyadenylated, yet this class of transcripts has limited coding potential [16]. LncRNA expressions in a variety of tumors, such as breast cancer [17], liver cancer [18], colorectal cancer [19], gastric cancer [20] and other tumor tissues have significant differences. These abnormally expressed lncRNAs may play a role in processes such as chromatin modification, X chromosome silencing, transcription, translation, gene imprinting, dose compensation effects, protein activity regulation and RNA variably splicing regulation [21]. LncRNAs can function biologically through multiple molecular mechanisms. The most common is acting as competing endogenous RNA (ceRNA) to regulate gene expression by sequestering miRNAs, thereby affecting cell function [22]. We predicted by bioinformatics that SNHG16 can interact with miR-186 as ceRNA in HCC. SNHG16 is located on human chromosome 17q25.1. It's reported that SNHG16 was involved in multifarious kinds of cancer, including, breast cancer [17], glioma [23] and colorectal cancer [19]. However, the upstream event underlying SNHG16 deregulation in each cancer type may also be different and requires further research. Especially, the role of SNHG16 in HCC is still unclear.
Our study showed for the first time that SNHG16 was significantly up-regulated in HCC tissues and cells. In addition, the expression of SNHG16 was correlated with tumor size, TNM stage, ALT expression level and HBV DNA level. SNHG16 promoted cell proliferation, migration and invasion. SNHG16 facilitated tumor formation in vivo. Mechanistically, SNHG16 and miR-186 directly interacted with and repressed each other. SNHG16 acted as a ceRNA for miR-186 to facilitate proliferation, migration and invasion of HCC cell. These results contribute to a better understanding of the pathogenesis of HCC and facilitate the development of diagnosis and therapy for HCC.
Materials and Methods
Tissue specimens
50 cases of HCC tissues and matched adjacent nontumorous liver tissues (>3 cm away from the cancerous tissue) were obtained from the First Affiliated Hospital of Chongqing Medical University during August 2015 to December 2017. Patients did not received chemotherapy or radiotherapy before surgery and HCC was confirmed by pathological examinations. Tissues were placed in the RNA preserving solution, overnight at 4°C, and preserved at -80 °C until RNA extraction. The protocol of this experiment was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University and complied with the ethical guidelines of the 1975 Declaration of Helsinki.
Cell culture and transfection
Human HCC cell lines (Hep-3B, Huh7, Sk-hep-1, SMMC-7721 and PLC) and normal liver cell line (HL-77O2) were obtained from the China Center for Type Culture Collection (Wuhan, China). Hep-3B, Huh7 and Sk-hep-1 cells were cultured in DMEM medium (Gibco, Carlsbad, CA,USA) containing 10% fetal bovine serum (PAN, Bavaria, Germany) and SMMC-7721, PLC and HL-77O2 cells were cultured in RPMI1640 medium containing 10% fetal bovine serum. Cells were cultured at 5% CO2 and 37 °C.
PLVX-ZsGreen-hsa-miR-186 (miR-186 mimic), pLVX-ZsGreen (miR-186 mimic control), pLVX-tdTomato-hsa-miR-186 inhibitor (miR-186 inhibitor) and pLVX-tdTomato (miR-186 inhibitor control) were purchased from GeneCopeia (Guangzhou, China). Cells were cultured in a six-well plate. Plasmid was transferred into cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol, when cells reached 50-70% confluence. Cells were collected for later experiments after 48h transfection.
HBLV-h-SNHG16-GFP-PURO (SNHG16), HBLV-GFP-PURO (NC), HBLV-h-SNHG16-shRNA1-GFP-PURO (sh-SNHG16-1), HBLV-h-SNHG16-shRNA2-GFP-PURO (sh-SNHG16-2), HBLV-h-SNHG16-shRNA3-GFP-PURO (sh-SNHG16-3), HBLV-GFP-PURO (sh-NC), polybrene and puromycin were purchased from Hanbio Biotechnology Co., Ltd (Shanghai, China). Cells were seeded at 1.25*106/well in a 6-well plate. Lentivirus-mediated vector was infected into cells using polybrene, when cells reached 50-70% density. Puromycin (2ug/ml) was added to remove uninfected cells.
Relative quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissues or cell using the TRIzol reagent (Invitrogen, USA). The cDNA was synthesized using the All-in-One Fist-Strand cDNA Synthesis Kit (GeneCopeia, Guangzhou, China) or the miRNA First Strand cDNA Synthesis Kit (GeneCopeia). Real-time PCR (RT-PCR) was performed using the All-in-One qPCR Mix Kit (GeneCopeia) or the MicroRNAs qPCR Kit (GeneCopeia). All qRT-PCRs were performed with CFX96TM Real-Time PCR System (Bio-Rad, USA). The RNA primers were: SNHG16, 5′-GTTCCTCTAAGTAATCGCCATGCGTTCT-3′ (forward), 5′-CATTTCAGTTTACAATCCTTGCAGTCCC-3′ (reverse); ROCK1, 5′-AAAGCCTTACTGTCGATTGCC-3′ (forward), 5′-AGGGTAATGCAACTTCCACTG-3′ (reverse); GAPDH, 5′-CAGCTAGCCGCATCTTCTTTT-3′ (forward), 5′-GTGACCAGGCGCCCAATAC-3′ (reverse); miR-186, 5′-CGACGCGTCGGGTTTACAGAACACCCATCA-3′ (forward), 5′-CCATCGATGGGGCACAGCAACAAAAGACT-3′ (reverse); U6, 5′-CTCGCTTCGGCAGCACA-3′ (forward), 5′-AACGCTTCACGAATTTGCGT-3′ (reverse). GAPDH or U6 was the reference.
CCK-8 assay
Cell proliferation was assessed using Cell Counting Kit-8 assay (CCK-8, Hanbio Biotechnology Co., Ltd., Shanghai, China). Briefly, cells were seeded in a 96-well plate at a density of 2500 cells/well, and cell viability was determined at series time points.
Transwell migration and invasion assay
Cell migration and invasion were assessed using transwell chambers (8.0 μm pore size; EMD Millipore, Billerica, MA, USA) and Matrigel (diluted 1:9) (Corning Inc., USA), respectively. For migration assay, the infected Hep-3B cells (4×105) or Sk-hep-1 cells (2×105) were resuspended in 200 ul serum-free MEM medium and seeded into the upper chamber, and 500ul DMEM medium containing 10% serum was added to the bottom chamber. For invasion assay, cells-containing medium were seeded into the upper chamber of Matrigel-coated (Corning Inc., USA), and 500ul DMEM medium containing 10% serum was added to the bottom chamber. After 24 h, the upper chamber cells were removed with cotton swabs, the lower membrane cells were fixed in 4% paraformaldehyde for 30 minutes, and then stained with 0.1% crystal violet (Beyotime, Jiangsu, China) for 30 minutes. Cells were counted under an upright microscope (Nikon, Japan, 200× magnification).
Western-blot analysis
Total protein was extracted from cells. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Beyotime, Jiangsu, China) and then transferred onto PVDF membrane (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skimmed at room temperature for 2 h. Anti-ROCK1 (1:500, Ruiyingbio, Suzhou, China), anti-GAPDH (1:500, Ruiyingbio) were added overnight at 4°C. The membranes were subsequently incubated with goat anti-rabbit IgG secondary antibody conjugated to horseradish peroxidase (1:5,000, Ruiyingbio) at room temperature for 2h. Finally, proteins were visualized using a WestrenBright ECL Kit (Advansta, USA).
Luciferase reporter assay
The reporter vector pmiRGLO-SNHG16-wild-type (SNHG16-wt) or miRGLO-SNHG16-mutant (SNHG16-mut) containing the predicted miR-186 binding sites were purchased from GenePharma (Shanghai, China). SNHG16-wt or SNHG16-mut was co-transfected with miR-186 mimic or miR-186 mimic control into cells using Lipofectamine 2000 (invitrogen, USA). After 48 h, firefly and renilla luciferase activities were measured with the Dual Luciferase Reporter assay system (Promega, USA). The luciferase activities were normalized with the renilla luciferase activity.
Xenograft tumor model
4-week female BALB/c nude mice were obtained from the Laboratory Animal Center Chongqing Medical University (Chongqing, China). 5×106 cells infected with SNHG16, NC or sh-SNHG16-2, sh-NC were injected subcutaneously into the hip back of mice (4 mice in each group). Tumor volumes were measured every 4 days, and the size of the tumors was calculated: length × width2 /2(mm3). All mice were sacrificed on the 28th day, and tumors were removed, weighed and photographed.
Statistical analysis
All the statistical analyses were performed using the GraphPad software 6.0 (GraphPad Inc., San Diego, CA, USA) and SPSS program (version 24.0 SPSS, Chicago ,IL,USA). Each experiment was repeated at least three times. Data were presented as Mean±SD (standard deviation). Differences were considered as statistically significant at P<0.05.
Results
SNHG16 was up-regulated in HCC tissues and cell lines
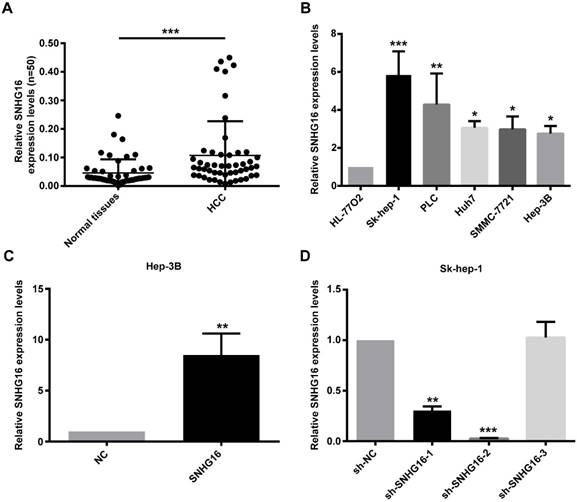
RT-qPCR was performed to detect the expression of SNHG16 in 50 pairs of HCC tissues and paired adjacent normal liver tissues. The results of qRT-PCR revealed that the expression level of SNHG16 was up-regulated in HCC tissues in comparison with adjacent non-tumor liver tissues (Figure 1A). In order to investigate the correlation between SNHG16 and clinicopathological variables, 50 HCC patients were divided into high and low expression groups according to the average expression level of SNHG16 in HCC tissues. We performed Chi-square test and found that the SNHG16 level was correlated with the tumor size, TNM stage; ALT level and HBV DNA level (Table 1). These indicated that SNHG16 may be a biomarker for HCC.
Expression of SNHG16 in HCC tissues and cell lines. (A). Expression levels of SNHG16 in 50 pairs of HCC tissues and paired adjacent normal liver tissues were evaluated by qRT-PCR. (B). The relative expression of SNHG16 in five HCC cell lines (Hep-3B, Huh7, Sk-hep-1, SMMC-7721 and PLC) and immortal liver cell line (HL-77O2) were determined by qRT-PCR. (C). The expression of SNHG16 in Hep-3B cells infected with NC or SNHG16. (D). qRT-PCR was conducted to confirm the knockdown efficiency of sh-SNHG16-1, sh-SNHG16-2 and sh-SNHG16-3 on SNHG16. *p<0.05, **p<0.01 and ***p<0.001.
Association between SNHG16 expression and clinicopathological characteristics in HCC (n=50)
| clinicopathological factors | Number (n=50) | SNHG16 expression | p | ||
|---|---|---|---|---|---|
| High | Low | ||||
| Gender | Male | 41 | 20 | 21 | 0.636 |
| Female | 9 | 3 | 6 | ||
| Age | ≥50 | 27 | 10 | 17 | 0.168 |
| <50 | 23 | 13 | 10 | ||
| Tumor size (cm) | ≥5 | 32 | 19 | 13 | 0.011 |
| <5 | 18 | 4 | 14 | ||
| TNM stage | III-IV | 30 | 20 | 10 | 0.001 |
| I-II | 20 | 3 | 17 | ||
| HBsAg | + | 41 | 20 | 21 | 0.636 |
| - | 9 | 3 | 6 | ||
| HBV DNA | + | 31 | 19 | 12 | 0.006 |
| - | 19 | 4 | 15 | ||
| ALT(U/L) | ≥40 | 25 | 16 | 9 | 0.011 |
| <40 | 25 | 7 | 18 | ||
| AST(U/L) | ≥40 | 30 | 17 | 13 | 0.064 |
| <40 | 20 | 6 | 14 | ||
| AFP(ug/L) | ≥400 | 21 | 9 | 12 | 0.704 |
| <400 | 29 | 14 | 15 | ||
TNM, tumor, node, metastasis; HBsAg, Australia antigen; HBV, hepatitis B virus;
ALT, alanine aminotransferase; AST, aspartate aminotransferase; AFP, alphafetoprotein.
Furthermore, the expression of SNHG16 in five HCC cell lines (Hep-3B, Huh7, Sk-hep-1, SMMC-7721 and PLC) and normal liver cell line (HL-77O2) was validated. The results indicated that HCC cell lines had a higher SNHG16 expression level compared with normal liver cell line. The lowest level was detected in Hep-3B, and the highest level was noted in Sk-hep-1 (Figure 1B). Therefore, NC and SNHG16 were infected into Hep-3B cells, and shRNA-NC(sh-NC) and three shRNAs for SNHG16 (sh-SNHG16-1, sh-SNHG16-2 and sh-SNHG16-3) were infected into Sk-hep-1 cells. The results of qRT-PCR demonstrated that expression of SNHG16 was significantly increased in Hep-3B cells after infected with SNHG16 relative to Hep-3B cells infected with NC (Figure 1C). Compared with the SK-hep-1 cells infected with sh-NC, SK-hep-1 cells infected with sh-SNHG16-2 markedly downregulated SNHG16 expression (Figure 1D). Therefore, Hep-3B cells stably overexpressing SNHG16 or SK-hep-1 cells stably knocking down SNHG16 and their control cells were selected by puromycin screen test for subsequent experiments.
SNHG16 promoted HCC cell proliferation, migration and invasion in vitro
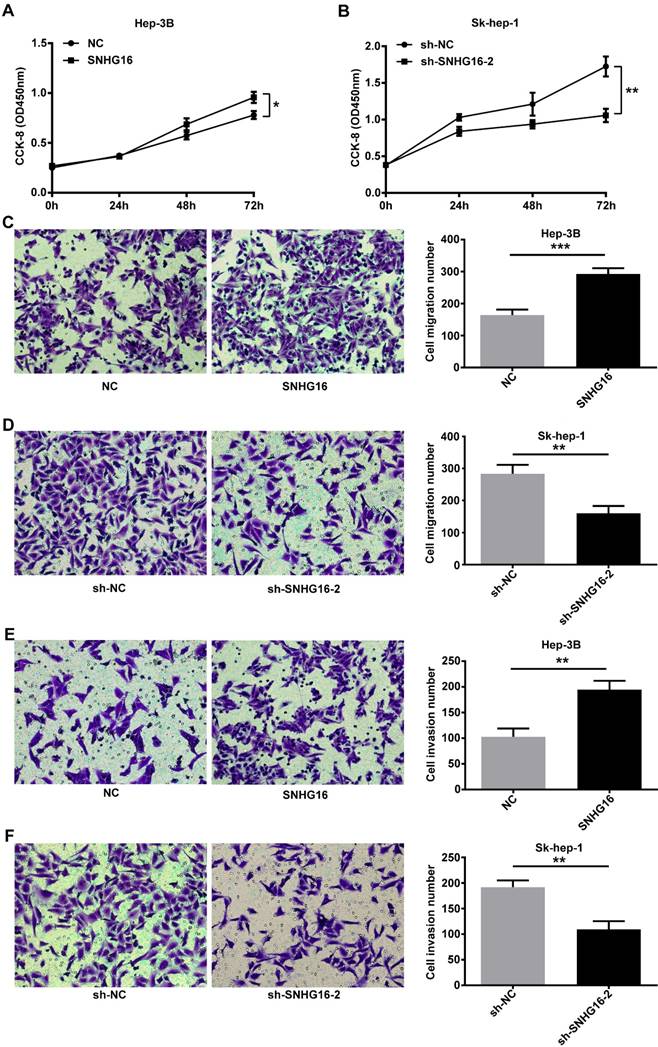
CCK-8 assay indicated that the proliferation activity of Hep-3B cells infected with SNHG16 was improved (Figure 2A). In contrast, the proliferation activity of SK-hep-1 cells infected with sh-SNHG16-2 was lower than that of the sh-NC group (Figure 2B). Transwell migration assay and transwell invasion assay indicated that the migration and invasion of Hep-3B cells infected with SNHG16 significantly increased compared with the control group (Figure 2C and 2E), while the migration and invasion of Sk-hep-1 cells infected with sh-SNHG16-2 obviously decreased compared with the sh-NC group (Figure 2D and 2F). Taken together, these results demonstrated that SNHG16 promoted cell proliferation, migration and invasion in vitro.
SNHG16 infulenced HCC cell proliferation, migration and invasion in vitro. (A). CCK-8 was performed to determine cell proliferation when SNHG16 over-expressed in Hep-3B cells. (B). CCK-8 was performed to determine cell proliferation when SNHG16 knockdown in Sk-hep-1 cells. (C and E). Cell migration and invasion were confirmed by using transwell migration assay and transwell invasion assay after infected with SNHG16 or NC into Hep-3B cells. (D and F). Transwell migration assay and transwell invasion assay were carried out to verify cell migration and invasion when down-expression of SNHG16 in Sk-hep-1 cells. *p<0.05, **p<0.01 and ***p<0.001.
SNHG16 promoted tumor proliferation in vivo
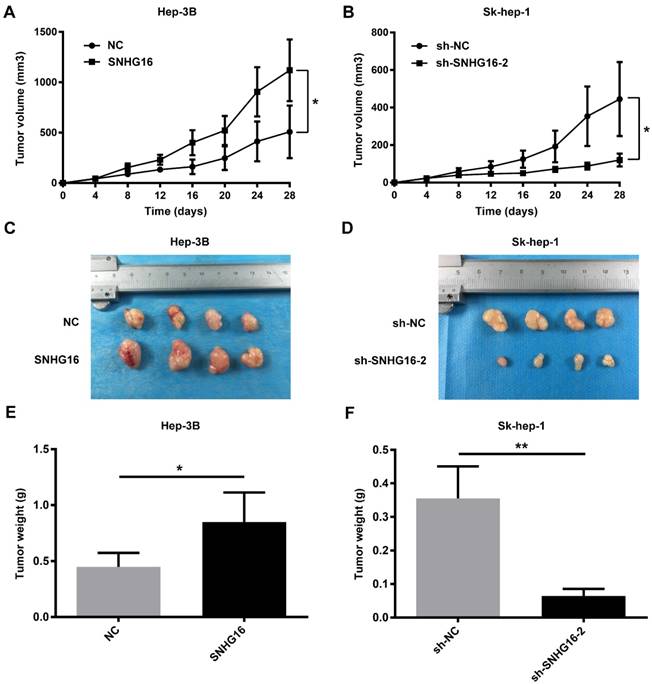
Nude mouse tumorigenesis test was carried out to confirm whether SNHG16 could enhance tumor growth in vivo. As demonstrated in Figure 3, compared with the NC group, the rate of tumor growth and tumor volumes of the SNHG16 group were significantly increased (Figure 3A and 3C). In contrast, compared with the sh-NC group, the rate of tumor growth and tumor volumes of the sh-SNHG16-2 group were significantly decreased (Figure 3B and 3D). Moreover, the tumor weight of xenograft tumor of the SNHG16 group was dramatically heavier than that of the NC group (Figure 3E). On contrary, the tumor weight of xenograft tumor of the sh-SNHG16-2 group was significantly lower than that of the sh-NC group (Figure 3F). Thus, these results indicated that SNHG16 could enhance tumor development in vivo.
SNHG16 infulenced HCC cell proliferation in vivo. Hep-3B cells (5×106cells) infected with SNHG16 or NC and Sk-hep-1 cells (5×106cells) infected with sh-SNHG16-2 or sh-NC were subcutaneously injected into the hip back of the nude mice (n = 4 per group). (A and B). The rate of tumor growth was calculated every 4 days for 28 days. (C-F). The size of tumor and tumor weight were recored and weighted on the 28th day. *p<0.05, **p<0.01 and ***p<0.001.
SNHG16 interacted with miR-186 in HCC
LncRNAs interact with miRNAs and contribute to disease. Our previous data manifested that miR-186 was lowly expressed in HCC cells and tissues and played the role of a tumor suppressor [14]. Therefore, online software program RNA22-HSA (https://cm.jefferson.edu/rna22/Interactive/) and NPInter (http://www.bioinfo.org/NPInter) were performed to predict the potential lncRNAs that interact with miR-186. Date suggested that SNHG16 may interact with miR-186 in HCC.
In order to explore the relationship between SNHG16 and miR-186, the expression of miR-186 and SNHG16 in 28 pairs of HCC and corresponding normal tissues were measured by qRT-PCR. The level of miR-186 was negatively correlated with that of SNHG16 (Figure 4A) (r=-0.5691, p=0.0046). Moreover, the expression of miR-186 in five HCC cell lines (Hep-3B, Huh7, Sk-hep-1, SMMC-7721 and PLC) and normal liver cell line (HL-77O2) was detected by qRT-PCR. The results indicated that HCC cell lines had a lower miR-186 expression level compared with normal liver cell line(Figure 4B).
The relationship between SNHG16 and miR-186 in HCC. (A).The negative correlation between SNHG16 and miR-186 expression in HCC tissues. (B). The relative expression of miR-186 in five HCC cell lines (Hep-3B, Huh7, Sk-hep-1, SMMC-7721 and PLC) and normal liver cell line (HL-77O2) were verified by qRT-PCR. (C). The relative expression of miR-186 in Hep-3B cells injected with NC or SNHG16 was detected by qRT-PCR. (D). The relative expression of miR-186 in Sk-hep-1 cells injected with sh-NC or sh-SNHG16-2 was detected by qRT-PCR. (E). qRT-PCR disclosed that ectopic expression of miR-186 decreased SNHG16 expression in Hep-3B cells. (F). qRT-PCR disclosed that knockdown of miR-186 increased SNHG16 expression in Sk-hep-1 cells. (G). Schematic of forecasted wild type or mutated miR-186 binding sequences in SNHG16. (H and I). Luciferase activity in HCC cell co-transfected with SNHG16-wt or SNHG16-mut and miR-186 mimic or miR-186 mimic control. (J-M). Effect of SNHG16 and miR-186 interactions on ROCK1 expression was quantified by Western blot and qRT-PCR. *p<0.05, **p<0.01 and ***p<0.001
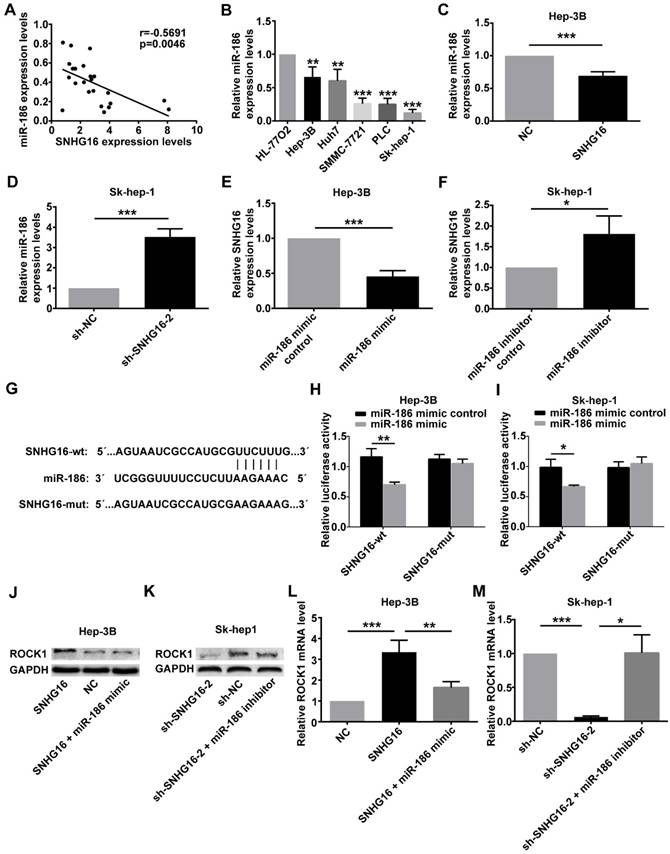
At the same time, qRT-PCR was carried out to determine the expression of miR-186 in Hep-3B cells infected with the SNHG16 or NC and in Sk-hep-1 cells infected with sh-SNHG16-2 or sh-NC. The results of qRT-PCR revealed that overexpression of SNHG16 inhibited miR-186 expression in Hep-3B cells (Figure 4C), whereas downregulation of SNHG16 induced miR-186 expression in Sk-hep-1 cells (Figure 4D). Simultaneously, the expression of SNHG16 was detected following transfection of miR-186 mimic or miR-186 mimic control into Hep3B cells, transfecting miR-186 inhibitor or miR-186 inhibitor control into Sk-hep-1cells. Interestingly, these data suggested that compared with the corresponding control group, miR-186 mimic significantly inhibited the expression of SNHG16 (Figure 4E), whereas miR-186 inhibitor significantly promoted SNHG16 expression (Figure 4F).
Dual luciferase reporter assay was performed to further explore the direct binding between SNHG16 and miR-186. We constructed luciferase reporter vectors containing the predicted wild-type or mutated miR-186 binding sites in SNHG16 (SNHG16-wt or SNHG16-mut) (Figure 4G). SNHG16-wt or SNHG16-mut was co-transfected with miR-186 mimic or miR-186 mimic control into cells. The results demonstrated that the luciferase activity of SNHG16-wt was significantly inhibited by miR-186 mimic, but miR-186 mimic control not affected SNHG16-wt luciferase activity (Figure 4H and 4I). Previous researches have shown that miR-186 inhibits the expression of ROCK1 in HCC [14]. The western blot and qRT-PCR results suggested that miR-186 mimic reversed the promotion effect of SNHG16 on ROCK1 (Figure 4J and 4L), and the inhibitory effect of sh-SNHG16-2 on ROCK1 was attenuated by miR-186 inhibitor (Figure 4K and 4M).
These results together indicated that the expression of SNHG16 and miR-186 was negatively correlated with each other; SNHG16 and miR-186 directly interacted with and repressed each other. SNHG16 functioned as a ceRNA of miR-186 in HCC.
SNHG16 affected cell proliferation, migration and invasion by regulating the expression of miR-186
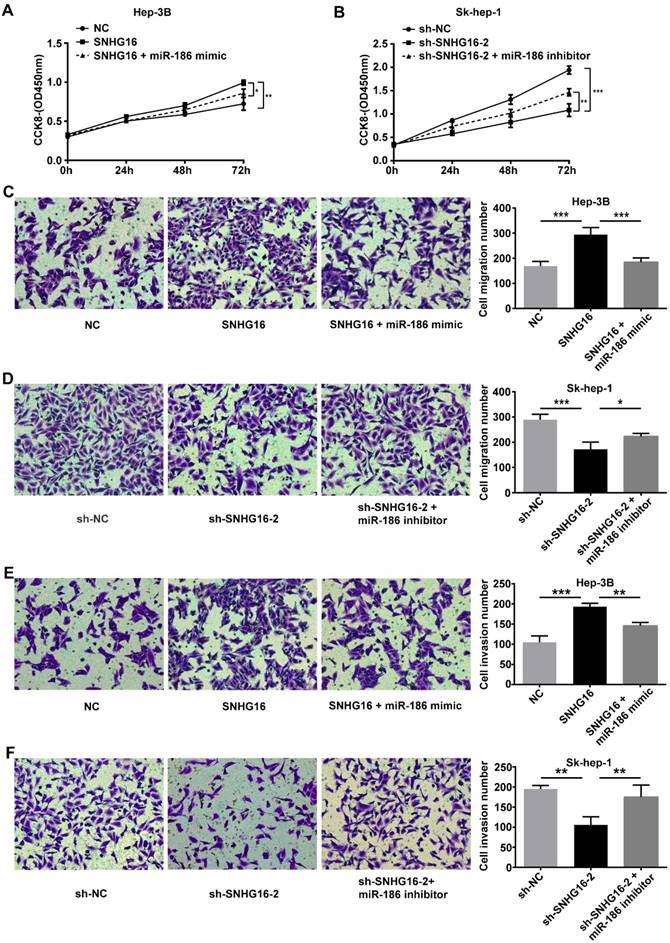
To further investigate whether SNHG16 exerted its biological functions via negatively regulating miR-186 expression, we performed rescue experiments. NC, SNHG16 and SNHG16+miR-186 mimic were transfected into Hep-3B cells, respectively. sh-NC, sh-SNHG16-2 and sh-SNHG16-2+miR-186 inhibitor were transfected into Sk-hep-1 cells, respectively. As shown by CCK-8 assay, transwell migration assay and transwell invasion assay, ectopic expression of miR-186 reversed the promotion of SNHG16 on Hep3B cell proliferation, migration and invasion (Figure 5 A, 5C and 5E). Meanwhile, the inhibitory effect of sh-SNHG16-2 on the proliferation, migration and invasion of Sk-hep-1 cells was abloished by miR-186 inhibitor (Figure 5 B, 5D and 5F). These data together revealed that SNHG16 promoted cell proliferation, migration and invasion by negatively regulating miR-186 expression.
The effect of SNHG16 on proliferation, migration and invasion of HCC cell by regulating the expression of miR-186. Hep-3B cells of SNHG16 over-expression were infected with miR-186 mimic, and Sk-hep-1 cells of SNHG16 knockdown were infected with miR-186 inhibitor. (A and B) CCK-8, (C and D) transwell migration assay and (E and F) transwell invasion assay were conducted to assess cell proliferation, migration and invasion, respectively. *p<0.05, **p<0.01 and ***p<0.001.
Discussion
Accumlating evidence has indicated that lncRNAs play an important role in the development of many diseases, including tumors. LncRNAs have many biological functions such as transcription, chromosome remodeling, translation and shearing [24]. LncRNA has gained considerable attention by functioning as a ceRNA [25]. Our previous data mainfested that miR-186 was lowly expressed in HCC cells and tissues and played the role of a tumor suppressor [14]. Therefore, online software program RNA22-HAS (https://cm.jefferson.edu/rna22/Interactive/) and NPInter (http://www.bioinfo.org/NPInter) were performed to predict the potential lncRNAs that interact with miR-186. Date suggested that SNHG16 might associate with miR-186 in HCC.
Previous studies have reported that SNHG16 expression is up-regulated in colorectal cancer and regulated by the Wnt signaling pathways, which may affect genes involved in lipid metabolism through ceRNA-related mechanisms [19]. SNHG16 is highly expressed in gastric cancer tissues. Inhibition of SNHG16 expression could inhibit the proliferation, migration and invasion of gastric cancer cells, and increase apoptosis [20]. Competitively binding miR-98 with E2F5, SNHG16 expression is up-regulated in breast cancer tissues and induces breast cancer cell migration [17]. In a recent study, SNHG16 was used as a competitive endogenous RNA to regulate PRMT5 expression through direct sponge miR-4518, which acts as an oncogenic lncRNA to promote glioma tumorigenesis [23]. However, there is little knowledge about the expression, function and molecular mechanism of SNHG16 in HCC.
Therefore, we conducted a series of experiments to investigate the expression, function and molecular mechanism of SNHG16 in HCC. In this study, SNHG16 was up-regulated in HCC tissues and cell lines. SNHG16 expression was correlated with tumor size, TNM stage, ALT expression level and HBV DNA level. Up-regulation of SNHG16 expression increased proliferation, migration and invasion of HCC cell. Down-regulation of SNHG16 expression reduced proliferation, migration and invasion of HCC cell. Furthermore, in vivo experiments confirmed that SNHG16 overexpression significantly promoted tumor growth in nude mice, and SNHG16 knockdown markedly inhibited tumor growth in nude mice. These results indicated that SNHG16 exerted its oncogenic role in HCC tumorigenesis and progression.
More and more evidences have indicated that miRNAs are involved in various physiological and pathological processes. MiRNAs could directly bind to the target mRNA to induce mRNA deregulation or translational repression [26]. Studies have found that miR-186 has a suppressive role in ESCC progression via SKP2-mediated pathway [12]. MiR-186 functions as tumor suppressor in NSCLC by targeting ROCK1 [27]. It also affects the proliferation, invasion and migration of human gastric cancer by inhibition of Twist1 [28]. MiR-186 expression is down-regulated in HCC and inhibits proliferation, migration, and invasion of HCC cell [14]. However, there is little knowledge about the role of the interaction of miR-186 with SNHG16 in the progression of HCC. Therefore, we confirmed the interaction of miR-186 and SNHG16 in HCC cell by qRT-PCR and dual luciferase assays. As revealed by our data, the expression of SNHG16 was negatively correlated with the expression of miR-186 (r= -0.5691, p=0.0046), SNHG16 and miR-186 directly interacted with and repressed each other. In addition, the results of qRT-PCR and western blotting demonstrated that SNHG16 might function as a ceRNA for miR-186 to regulate ROCK1 expression. ROCK1 is a target of miR-186 that plays important roles in regulating cell polarity and migration [14, 29]. Subsequently, rescue experiments were performed and showed that miR-186 could reverse the effect of SNHG16 on HCC cell. Therefore, it might be explained that SNHG16 could act as a ceRNA via regulating miR-186 to facilitate proliferation, migration and invasion of HCC cell.
Our present study contributes to a better understanding of the pathogenesis of HCC and facilitates the development of diagnosis and therapy for HCC. However, the limitations of this study should not be ignored. Above all, we only detected the expression of SNHG16 in 50 pairs of HCC tissues and matched non-tumor tissues. Whether SNHG16 can be used as a biomarker for the diagnosis of HCC requires more clinical sample data, also the expression levels of SNHG16 in the patient's blood should be evaluated. Next, our work was not perfect for the study of the regulatory relationship between SNHG16, miR-186 and ROCK1, so the exact mechanism of SNHG16-miR-186-ROCK1 axis in HCC requires further study. Lastly, SNHG16 may target multiple miRNAs for biological function in CRC [19]. Whether SNHG16 can exert its function by acting as a ceRNA to regulate multiple miRNAs in HCC also requires further study.
In summary, our study is the first to show that SNHG16 was highly expressed in HCC tissues and cells and that promoted HCC cell proliferation, migration and invasion by functioning as a ceRNA to regulate miR-186. Our findings indicated that SNHG16 may be a potential biomarker for clinical diagnosis of HCC and a new therapeutic target for the treatment of HCC.
Abbreviations
AFP: alphafetoprotein; ALT: alanine aminotransferase; AST: aspartate aminotransferase; CCK-8: Cell Counting Kit-8 assay; ceRNA: competitive endogenous RNA; DMEM: Dulbecco's modified Eagle's medium; HBsAg: Australia antigen; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; LncRNA: long non-coding RNA; miR-186: microRNA-186; PBS: phosphate-buffered saline; PMSF: Phenylmethanesulfonyl fluoride; PVDF: polyvinylidene difluoride; qRT-PCR: Relative quantitative real-time PCR; RIPA: Radio Immunoprecipitation Assay; ROCK1: Rho associated coiled-coil containing protein kinase 1; SNHG16: small nucleolar RNA host gene 16; TNM: tumor, node, metastasis.
Acknowledgements
We are grateful to Dr. Tinghe Yu (Director of Key Medical labortory of Obstetrics and Gynecology, The Second Affiliated Hospital, Chongqing Medical University, Chongqing, China) for the generous support of the experimental facilities.
Ethics Committee Approval and Patient Consent
Investigations involving humans will have been performed in accordance with the principles of Declaration of Helsinki. And the use of animals in experiments will have observed the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, Ad Hoc Animal Research Committee.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-386
2. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-1255
3. Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;6:1
4. Cho WC. MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol. 2010;42:1273-1281
5. Khoury S, Tran N. Circulating microRNAs: potential biomarkers for common malignancies. Biomarkers Med. 2015;9:131-151
6. Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S. et al. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750-6
7. Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U. et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344-357
8. Leucci E, Patella F, Waage J, Holmstrøm K, Lindow M, Porse B. et al. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci Rep. 2013;3:2535
9. Wang D, Qiu C, Zhang H, Wang J, Cui Q, Yin Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One. 2010:5 (9)
10. Sun KX, Jiao JW, Chen S, Liu BL, Zhao Y. MicroRNA-186 induces sensitivity of ovarian cancer cells to paclitaxel and cisplatin by targeting ABCB1. J Ovarian Res. 2015;8:80
11. Cai J, Wu J, Zhang H, Fang L, Huang Y, Yang Y. et al. miR-186 downregulation correlates with poor survival in lung adenocarcinoma where it interferes with cell cycle regulation. Cancer es. 2013;73:756-66
12. He W, Feng J, Zhang Y, Wang Y, Zang W, Zhao G. microRNA-186 inhibits cell proliferation and induces apoptosis in human esophageal squamous cell carcinoma by targeting SKP2. Lab Invest. 2016;96:317-24
13. Hua X, Xiao Y, Pan W, Li M, Huang X, Liao Z. et al. miR-186 inhibits cell proliferation of prostate cancer by targeting GOLPH. Am J Cancer Res. 2016;6:1650-60
14. Dan S, Ziyao W, Ping H. Expression and Role of MicroRNA-186 in Human Hepatocellular Carcinoma. Tumor. 2017;07:750-761
15. Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma:Mechanisms, biomarkers and therapeutic targets. Journal of Hepatology. 2017;67:603-618
16. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J. et al. The Long Noncoding RNA lncTCF7 Promotes Self-Renewal of Human Liver Cancer Stem Cells through Activation of Wnt signaling. Cell Stem Cell. 2015;16:413-25
17. Cai C, Huo Q, Wang X, Chen B, Yang Q. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485:272-278
18. Ma J, Li T, Han X, Yuan H. Knockdown of LncRNA ANRIL suppresses cell proliferation, metastasis, and invasion via regulating miR-122-5p expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144:205-214
19. Christensen LL, True K, Hamilton MP, Nielsen MM, Damas ND, Damgaard CK. et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10:1266-82
20. Lian D, Amin B, Du D, Yan W. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21:151-160
21. Yuan J, Yue H, Zhang M, Luo J, Liu L, Wu W. et al. Transcriptional profiling analysis and functional prediction of long noncoding RNAs in cancer. Oncotarget. 2016;7:8131-8142
22. Guo G, Kang Q, Zhu X, Chen Q, Wang X, Chen Y. et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene. 2015;34:1768-1779
23. Lu YF, Cai XL, Li ZZ, Lv J, Xiang YA, Chen JJ. et al. LncRNA SNHG16 Functions as an Oncogene by Sponging MiR-4518 and Up- Regulating PRMT5 Expression in Glioma. Cell Physiol Biochem. 2018;45:1975-1985
24. Wang N, Cao P, Xia W. Identification and characterization of long non-coding RNAs in response to early infection by Melampsora larici - populina using genome - wide high - throughput RNA sequencing. Tree Genetics & Genomes. 2017;13:34
25. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272-83
26. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597-610
27. Cui G, Cui M, Li Y, Liang Y, Li W, Guo H. et al. MiR-186 targets ROCK1 to suppress the growth and metastasis of NSCLC cells. Tumour Biol. 2014;35:8933-7
28. Cao C, Sun D, Zhang L, Song L. miR-186 affects the proliferation, invasion and migration of human gastric cancer by inhibition of Twist1. Oncotarget. 2016;7:79956-79963
29. Morgan-Fisher M, Wewer UM, Yoneda A. Regulation of ROCK activity in cancer. J Histochem Cytochem. 2013;61:185-98
Author contact
![]() Corresponding author: Professor Ping Huang, National Key Clinical Department, Department of Hepatobiliary Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing Medical University, 1 Youyi Road, Yuzhong, Chongqing 400000, P.R. China. E-mail: huangpingcqmu.edu.cn Telephone Number: 18623317078
Corresponding author: Professor Ping Huang, National Key Clinical Department, Department of Hepatobiliary Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing Medical University, 1 Youyi Road, Yuzhong, Chongqing 400000, P.R. China. E-mail: huangpingcqmu.edu.cn Telephone Number: 18623317078