Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(15):3501-3516. doi:10.7150/jca.29490 This issue Cite
Research Paper
KIAA0101 is a novel transcriptional target of FoxM1 and is involved in the regulation of hepatocellular carcinoma microvascular invasion by regulating epithelial-mesenchymal transition
Tao Zhang1*, Jianrong Guo1*, Jian Gu1, Ke Chen1, Zheng Wang1,2, Huili Li1 ![]() , Guobin Wang1
, Guobin Wang1 ![]() , Jiliang Wang1
, Jiliang Wang1 ![]()
1. Department of Gastrointestinal Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
2. Research Centre for Tissue Engineering and Regenerative Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
*Contributed equally
Received 2018-8-26; Accepted 2019-5-10; Published 2019-6-9
Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths due to tumor invasiveness, frequent intrahepatic dissemination and extrahepatic metastasis. However, the genes and signaling pathways that are involved remain incompletely understood. In this study, weighted gene coexpression network analysis (WGCNA) was performed to jointly analyze clinical information and gene expression data to identify key genes associated with clinical features. Through the bioinformatic analysis, the yellow module and microvascular invasion (MVI) were found to be highly associated (r=0.41) by Pearson's correlation analysis, and 20 hub genes were identified with both high gene significance (GS) and high module membership (MM) in the yellow module. Among these genes, FoxM1 and KIAA0101 were upregulated in HCC with MVI and were significantly positively correlated in HCC samples, indicating a novel regulatory network in HCC microvascular invasion. Moreover, in vitro experiments demonstrated that KIAA0101 is a direct target of FoxM1 and that KIAA0101 is required for the FoxM1-induced promotion of HCC cell invasion and migration. In addition, the FoxM1-KIAA0101 axis promotes HCC metastasis by inducing epithelial-mesenchymal transition (EMT). In summary, KIAA0101 is a novel target of FoxM1 and contributes to HCC metastasis by activating EMT. The FoxM1-KIAA0101 axis might be applied as a potential prognostic biomarker and therapeutic target for HCC.
Keywords: weighted gene coexpression network analysis, hepatocellular carcinoma, microvascular invasion, FoxM1, KIAA0101, epithelial-mesenchymal transition.
Introduction
Liver cancer is one of the most common malignant cancers and is currently the third leading cause of cancer-related deaths worldwide. In the United States, it is estimated that there were 40,710 new cases and 28,920 new deaths associated with liver cancer in 2017[1]. In China, the world's most populous country, the estimated numbers of new cases and deaths associated with liver cancer in 2015 were 466.1 and 422.1 thousand, respectively [2]. Hepatocellular carcinoma (HCC) is a major histological subtype (accounting for approximately 80%) of primary liver cancers and is a malignant tumor associated with high morbidity and mortality [3]. Although great advancements have been made in the past decades in HCC treatment, the prognosis of HCC patients remains poor, which is mainly attributed to the invasion and metastases of cancer [4].
In recent years, with the rapid technological breakthroughs of genome-wide microarrays and high-throughput sequencing, which shed new light on the research of various cancers, the application of bioinformatics tools in biological and medical studies, including research regarding cancer early diagnosis, cancer grading and prognosis prediction, has increased remarkably [5]. Bioinformatic approaches, based on data mining of microarray and high-throughput sequencing data, make it possible for us to achieve a system-level insight into the complex biological processes that underlie cancer. Weighted gene coexpression network analysis (WGCNA) is an important bioinformatics tool that can construct gene coexpression networks to explore the correlations among different gene clusters or the relationships between gene clusters and clinical features and detect hub genes that may serve as diagnostic, prognostic or therapeutic targets [6].
In a recent study, we downloaded two original microarray datasets, GSE10143 [7] and GSE20140 [8], from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). We constructed a gene coexpression network based on WGCNA and identified 12 modules associated with clinical features of HCC. Among these modules, the yellow module was highly correlated with microvascular invasion (MVI). Thus, the yellow module was selected as an interesting module and clinical feature for further study. Through bioinformatics analysis, HIST1H2BF, HIST1H2AE, BRCA1, HMGA1, CDKN3, FOXM1, NCAPH, CCNE2, MCM4, PCNA, PKM2, TOP2A, KIAA0101, TROAP, BUB1B, E2F1, CENPF, CDK2, BIRC5, and PRC1 were identified as hub genes in HCC MVI. Among these hub genes, BIRC5, BUB1B, CENPF, FOXM1, KIAA0101, MCM4, PKM2, and TOP2A have important clinical implications because of their significant overexpression in HCC with MVI and their similar expression pattern in HCC compared with their expression in normal liver tissues. Moreover, the upregulated expression of these genes is correlated with poor prognosis of HCC patients. Additionally, our bioinformatic analysis revealed that FoxM1 and KIAA0101 were notably positively correlated in HCC, and both were significantly overexpressed in tissues of HCC with MVI, indicating a potential regulatory role and network of FoxM1 and KIAA0101 in HCC MVI.
MVI in HCC is usually associated with more advanced tumor stage and disease progression. HCC patients with MVI usually do not have the opportunity to undergo curative liver resection or transplantation, and they usually have poor long-term survival outcomes, even when they undergo these treatments [9]. MVI of HCC is a multistep pathobiological process, but the mechanisms underlying this process remain unclear. Several signaling pathways and factors, such as Wnt/β-catenin, RAS/RAF/MAPK signaling, growth factor, and lncRNA MVIH, may be involved in the process [9]. Previous studies have shown that the activation of epithelial-mesenchymal transition (EMT) contributes to the development of HCC MVI [10]. FoxM1 is a member of the forkhead box (FOX) family of transcription factors and has been suggested to be an oncogenic protein that plays important roles in angiogenesis, invasion, and metastasis [11]. KIAA0101, also known as proliferating cell nuclear antigen (PCNA)-associated factor (PAF15), is a conserved PCNA-binding protein that functions as an oncogene and is upregulated in multiple cancers, including HCC [12].
In the present study, we primarily assessed and confirmed the upregulation of FoxM1 and KIAA0101 in HCC by comparing their expression profiles in HCC and normal liver tissues. We then sought the relationship between FoxM1 and KIAA0101 in HCC cells and explored their roles in cell invasion and migration to achieve a better understanding of the mechanism underlying MVI development in HCC. Our results revealed that KIAA0101 is a novel transcriptional target of FoxM1 and that the FoxM1-KIAA0101 axis promoted HCC metastasis by regulating EMT.
Materials and methods
Microarray data
Datasets related to HCC were obtained from NCBI-GEO (http://www.ncbi.nlm.nih.gov/geo), a free microarray and high-throughput sequencing database, with an accession number of GSE10143 [7]. The dataset was submitted by Yujin Hoshida, based on the GPL5474 platform (Human 6k Transcriptionally Informative Gene Panel for DASL), and included 80 HCC samples and 307 nontumor samples.
For validation, another dataset, GSE20140 [8], was also downloaded. The dataset was submitted by Yujin Hoshida, was based on the GPL8432 platform (Illumina HumanRef-8 WG-DASL v3.0), and included 165 HCC samples.
Weighted gene coexpression network construction
Weighted gene coexpression network construction and module identification of all 6144 genes in the GSE10143 dataset were performed following the protocols of WGCNA [6]. Briefly, a gene coexpression similarity measure was first calculated and used to relate every pairwise gene-gene relationship. The adjacency matrix and topological overlap matrix (TOM) were then constructed utilizing a 'soft' power adjacency function [13]. “Gene modules” are groups of genes that have high topological overlap. Modules were identified using hierarchical clustering with a dissimilarity measure (1-TOM) [14].
Interesting module detection and functional annotation
The correlations between modules and clinical features were identified by Pearson's correlation tests to reveal clinically meaningful modules. The modules that exhibited the highest correlation were selected as interesting modules and clinical features to be further studied. To explore the potential impaction of module genes on correlative clinical features, all genes of interesting modules were subjected to gene ontology (GO) function analysis and Kyoto Encyclopedia of Gene and Genome (KEGG) pathway enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [15].
Identifying hub genes and performing a correlation analysis
Genes with both high gene significance (GS) and high module membership (MM) were defined as hub genes. The preliminary relationships between hub genes and corresponding clinical features were shown by scatter plot graphs, and the correlations between them were tested with an independent sample t-test. The results were verified by the validation of the GSE20140 cohort.
Survival analysis, expression comparison and correlation analysis of hub genes
The Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/index.html) is an online tool used to perform fast and customizable analyses of RNA sequencing expression data based on the Cancer Genome Atlas (TCGA)[16] and the Genotype-Tissue Expression (GTEx) databases, which include 9736 tumor samples and 8587 normal samples[17]. The GEPIA provides key interactive and customizable functions, including correlation analysis, differential expression analysis, profiling plotting, dimensionality reduction analysis, patient survival analysis, and similar gene detection. Therefore, we could evaluate the prognostic value of the hub genes in HCC patients, compare hub gene expression in HCC tissues with the expression in nontumor liver tissues, and analyze the correlations among the hub genes.
Ethical considerations and patient tissue samples
All of the experimental protocols involving humans were approved by the ethics committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). A total of 4 pairs of HCC tissue samples, including cancer tissues and corresponding normal adjacent liver tissues, were collected from the Gastrointestinal Surgery Unit of Union Hospital. The excised tissue was embedded in paraffin or snap-frozen and stored at -80°C. Written informed consent was obtained from all patients prior to surgery for the use of their tumor tissue. All samples were from patients with HCC who had not received any chemoradiotherapy before surgery. The clinical parameters of the patients are shown in Supplementary Table 2.
Cell culture and reagents
HEK293T cells and the human liver cancer cell lines HepG2 and Huh7, obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, China), were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum (FBS) (ScienCell Research Laboratories, Inc., San Diego, CA, USA) and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.). Cells were cultured at 37°C in an incubator with 5% CO2.
SiRNA and lentiviral transfection
Small interfering RNA (siRNA) targeting KIAA0101 (si-KIAA0101) and nonsilencing control siRNA (NC) were purchased from RiboBio (Guangzhou, China). In vitro transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.). Lentivirus vectors containing FoxM1-siRNA sequence (si-FoxM1), negative control (NC), and KIAA0101 cDNA (KIAA0101) were purchased from GeneChem (Shanghai, China). To establish a stable cell line with FoxM1 downregulation, a lentiviral vector with FoxM1 siRNA was transfected into HepG2 and Huh7 cells. Then, a lentiviral vector with KIAA0101 cDNA (KIAA0101) was transfected into FoxM1-downregulated cell lines to establish KIAA0101-upregulated cell lines. All the transfection processes were conducted according to the manufacturer's protocols.
Western blot analysis
Tissues or cell proteins were extracted using cold RIPA buffer with PMSF (BOSTER Biological Technology Co. Ltd., Wuhan, China). The protein concentration was determined with a Bradford Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Total proteins (40 µg) were loaded onto SDS-PAGE gels, separated by electrophoresis and transferred onto PVDF membranes (BOSTER Biological Technology Co. Ltd.). After blocking in TBS solution containing 5% nonfat milk and 0.1% Tween 20 for 1 h, membranes were incubated with primary antibodies overnight at 4°C. Antibodies against FoxM1 were purchased from Genetex (1:1000; cat. no. GTX102170; Irvine, CA, USA); KIAA0101 was purchased from NOVUS (1:500; cat. no. NBP1-80555; Littleton, CO, USA); E-cadherin (1:500; cat. no. sc-71007), N-cadherin (1:1000; cat. no. sc-59987), and vimentin (1:500; cat. no. sc-80975) were purchased from Santa Cruz (Santa Cruz, CA, USA); SNAIL (1:1000; cat. no. # 3879T) was purchased from CST; and TWIST1 (1:1000; cat. no. 25465-1-AP), ZEB1 (1:1000; cat. no. 21544-1-AP) and GAPDH (1:1000; cat. no. 60004-1-Ig) were purchased from Proteintech. After washing with TBS solution containing 0.1% Tween 20 three times, the membranes were incubated at 37°C with HRP-linked secondary antibodies (1:3000; cat. no. #7074 or #7076; CST) for 1 h. Enhanced chemiluminescent substrate was used to visualize the immune bands.
RNA isolation, reverse transcription, and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA from tissue samples or liver cancer cells was extracted by using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Reverse transcription was conducted with HiScript® II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech Co. Ltd., Nanjing, China) according to the manufacturer's protocol. Real-time PCR was performed via the Applied Biosystems StepOne Real-Time PCR system using ChamQTM SYBR® qPCR Master Mix (High ROX Premixed) (Vazyme Biotech Co. Ltd.), containing 5 ng cDNA and 10 pM of each primer. The cycling conditions consisted of one cycle at 95°C for 30 sec and 40 cycles at 95°C for 10 sec and 60°C for 30 sec. Melting curve analysis was conducted for each PCR to confirm the specificity of the amplification. The expression levels of the target genes were normalized to GAPDH and calculated using the 2-∆∆CT method. The primers are shown in Table 2.
Immunohistochemistry (IHC)
Sections (5 µm) were cut from formalin-fixed, paraffin-embedded HCC and corresponding normal adjacent liver tissue blocks, collected on 3-ethoxy-aminoethyl-silane-treated slides, and allowed to dry overnight at 37°C. After dewaxing in xylene and rehydrating in a gradient concentration (100%, 95%, and 75%) of ethanol to distilled water, the samples were immersed in 10 mmol/L citrate buffer (pH 6.0) and processed in a thermostatic water bath for 2 h at 98°C for antigen retrieval. Then, the slides were incubated in 0.3% hydrogen peroxide to block endogenous peroxidase activity and were then incubated with anti-KIAA0101 antibody (1:200; cat. no. #81533; CST; Danvers, MA, USA) or anti-FoxM1 antibody (1:200; cat. no. GTX102170; Genetex) overnight at 4°C. Subsequently, the slides were incubated with biotinylated goat anti-rabbit IgG (antGene, Wuhan, China). After washing, diaminobenzidine and hematoxylin were added to detect immunoreactive proteins.
Wound-healing and transwell assays
For the wound-healing assay, HepG2 or Huh7 cells were seeded in 12-well plates until confluence and were then scratched with a micropipette tip. Cell migration distances were determined using an inverted microscope (Olympus, Tokyo, Japan) at 0 h and 48 h after scratching.
A transwell assay was performed to determine the migration and invasion ability using 24-well transwell plates with 8 µm pores (Corning Incorporated, Corning, NY, USA). HepG2 (10×104 cells) or Huh7 (10×104 cells) cell suspensions in FBS-free medium were seeded into a transwell chamber, which was coated with (invasion) or without (migration) Matrigel (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and the bottom chamber was filled with 30% FBS as a chemotaxin. After 24 h of incubation, the cells that failed to invade from the top of the membranes were removed, and then the invaded cells were stained with 0.05% crystal violet for visualization and counted in six random fields.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a SimpleChIP® Plus Enzymatic Chromatin IP Kit (Agarose Beads) (cat. no. #9004; CST) according to the manufacturer's instructions. Briefly, the HepG2 and Huh7 cells used for each immunoprecipitation reaction were collected and cross-linked with 1% formaldehyde, and then micrococcal nuclease was used to digest DNA to a length of approximately 150-900 bp. Immunoprecipitation was performed after incubation with anti-FoxM1 (cat. no. GTX102170; Genetex) or IgG antibody (cat. no. #2729; CST) overnight at 4°C and a subsequent incubation with ChIP-Grade Protein G Agarose Beads (cat. no. #9007; CST) for 2 h. After DNA purification, PCR was used to amplify the target sequences from the inputted and the immunoprecipitated DNA samples. The primers specific to the KIAA0101 gene promoter were as follows: forward: GCTCTGACACCATTCGGTTCT; and reverse: GGAATGGGGAAAAGGAAGCTA.
Luciferase reporter assay
The promoter region (1.0 kb sequence upstream of the transcription initiation site) containing the putative binding area (wild type, WT) or mutant area (mutant type, MUT) was cloned into pGL3-based vectors (referred to as P-KIAA-WT or P-KIAA-MUT) and transfected into HEK293T cells. The transfected cells were cotransfected with pcDNA-FoxM1, pcDNA-NC (referred to as TF-FoxM1 or TF-NC) (0.2 μg), or the corresponding negative control in a 96-well plate. Firefly luciferase activity was measured by the Dual-Luciferase Reporter Assay system (Promega, Madison, WI, USA) and normalized to Renilla luciferase activity. All experiments were repeated three times independently, and five samples were analyzed per group.
Statistical analysis
The experimental data were analyzed using GraphPad 6.0 statistical software (GraphPad Software, Inc., La Jolla, CA, USA) and are presented as the mean values ± standard deviations (SDs). Unpaired t-tests were used to compare the groups. Spearman's correlation analysis was used to analyze the correlation between FoxM1 and KIAA0101. A receiver operating characteristic (ROC) curve analysis was used to determine the diagnostic value of the hub genes for differentiating MVI-present or MVI-absent HCC. The area under the curve (AUC), sensitivity, specificity, and accuracy of the differential diagnosis abilities of these genes were calculated. All in vitro experiments and assays were repeated at least three times. When p<0.05, the values were considered statistically significant.
Results
Coexpression network construction and interesting module detection
WGCNA was performed on all 6144 genes in the 80 GSE10143 HCC samples. After removing 7 outlier samples, the connectivity between the genes in the gene network formed a scale-free network distribution when the soft-threshold power β was set to 3 (Figure 1A). Then, 12 coexpressed modules were identified and represented by different colors. The “gray” module was reserved for genes identified as not coexpressed (Figure 1B).
WGCNA network and module detection. (A) Selection of the soft-thresholding powers. The left panel shows the scale-free fit index versus the soft-thresholding power. The right panel displays the mean connectivity versus the soft-thresholding power. Power 3 was chosen because the fit index curve flattened out upon reaching a high value (>0.9). (B) Cluster dendrogram and module assignment for modules from WGCNA. Genes cluster dendrogram drawn using a dissimilarity measure (1-TOM). The colored horizontal bar below the dendrogram represent the modules. A total of 6144 genes were assigned to one of 12 modules including the gray module. (C) Each column corresponds to a clinical feature and each row to an eigengene module. Each cell contains the corresponding correlation on the first line and the p-value on the second line. The table is color-coded by correlation according to the color legend. The gray module included all the genes that could not be clustered.
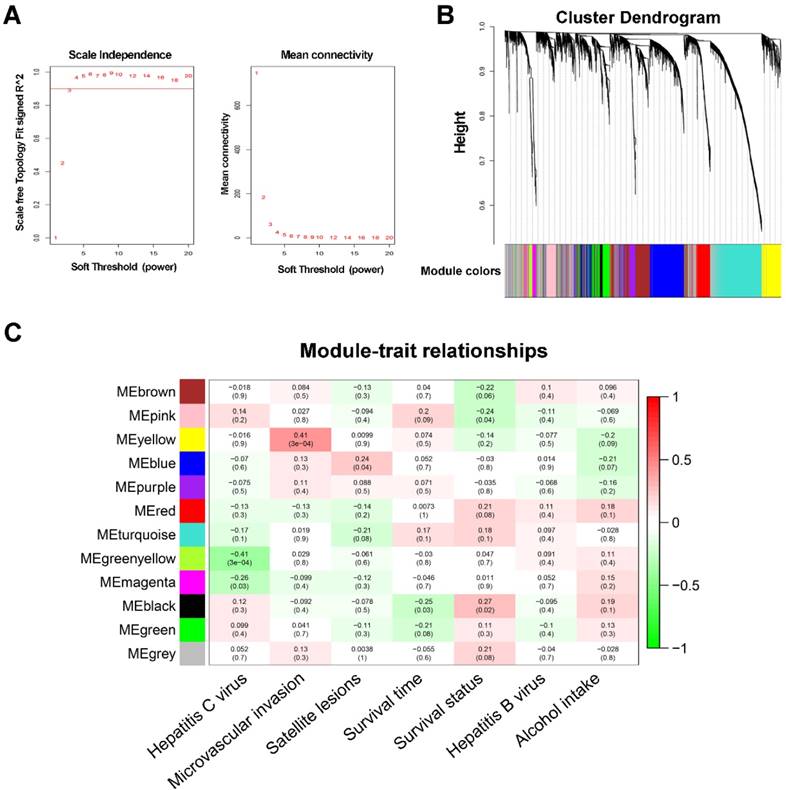
The correlations between modules and clinical features, such as hepatitis C virus, microvascular invasion, satellite lesions, survival time, survival status, hepatitis B virus, and alcohol intake, were calculated. The highest association in the module-trait relationship was found between the yellow module and MVI (r=0.41, P=3×10-4; Figure 1C) and was selected as an interesting module and clinical feature to be studied in subsequent analyses and experiments.
GO function and KEGG pathway enrichment analyses
There were 362 genes in the yellow module (Supplementary Table 1). To obtain a more in-depth understanding of the genes in the selected module, GO function and KEGG pathway enrichment analyses were conducted using DAVID. The biological processes of the yellow module were found to focus on small molecule catabolic processes (P=5.47×10-19), single-organism catabolic processes (P=7.33×10-18), cellular catabolic processes (P=1.46×10-12), sister chromatid segregation (P=2.81×10-12), and mitotic cell cycle processes (P=6.54×10-12). Additionally, metabolic pathways (P=3.26×10-12), complement and coagulation cascades (P=1.08×10-9), chemical carcinogenesis (P=9.42×10-9), metabolism of xenobiotics by cytochrome P450 (P=2.65×10-8), and the cell cycle (P=1.24×10-7) were most enriched in the KEGG pathway analysis (Supplementary Figure 1).
Identifying hub genes and correlation analysis
Twenty hub genes, which exhibited both high module membership (MM) in the interesting module and high gene significance (GS) for microvascular invasion, were identified in the yellow module (Table 1).
Top 20 hub genes in module yellow
| Genes | GS | MM |
|---|---|---|
| HIST1H2BF | 0.420235149 | 0.762627111 |
| HIST1H2AE | 0.455812738 | 0.707747124 |
| BRCA1 | 0.448998734 | 0.697984627 |
| HMGA1 | 0.482641604 | 0.677504949 |
| CDKN3 | 0.365262761 | 0.79722955 |
| FOXM1 | 0.357649431 | 0.760024888 |
| NCAPH | 0.351571886 | 0.787606906 |
| CCNE2 | 0.466445457 | 0.651511475 |
| MCM4 | 0.377719672 | 0.686865286 |
| PCNA | 0.403360726 | 0.665227696 |
| PKM2 | 0.401783039 | 0.666578589 |
| TOP2A | 0.367977988 | 0.688547317 |
| KIAA0101 | 0.453852012 | 0.644065803 |
| TROAP | 0.342888061 | 0.741025847 |
| BUB1B | 0.496349915 | 0.605109597 |
| E2F1 | 0.463173675 | 0.62003613 |
| CENPF | 0.337218685 | 0.739137297 |
| CDK2 | 0.51702651 | 0.596380963 |
| BIRC5 | 0.32077786 | 0.755315157 |
| PRC1 | 0.392212544 | 0.636585838 |
GS: gene significance; MM: module membership
Premier set used for qRT-PCR
| Primer set | Primers | Sequence(5'-3') |
|---|---|---|
| GAPDH | Forward | ACAACTTTGGTATCGTGGAAGG |
| Reverse | GCCATCACGCCACAGTTTC | |
| FoxM1 | Forward | TTAAGCAGCAGAAACGACCG |
| Reverse | TCACCGGGAACTGGATAGGT | |
| KIAA0101 | Forward | GGTGCGGACTAAAGCAGACA |
| Reverse | TTTTTGCCACTTGGGAGTTGG | |
| E-cadherin | Forward | GCCCTGCCAATCCCGATGAAA |
| Reverse | GGGGTCAGTATCAGCCGCT | |
| N-cadherin | Forward | CAGAATCGTGTCTCAGGCTCCAAG |
| Reverse | CTGCGTTCCAGGCTGGTGTATG | |
| Vimentin | Forward | GAGAACTTTGCCGTTGAAGC |
| Reverse | GCTTCCTGTAGGTGGCAA | |
| TWIST1 | Forward | CATTCTCAAGAGGTCGTGCCA |
| Reverse | CAGGCCAGTTTGATCCCAGTA | |
| ZEB1 | Forward | TAGCGAGTGGTTCTTCTGCG |
| Reverse | AGGGCTGCTGGAAGGTAAAC | |
| SNAIL | Forward | TTACACCTTTGCATACAGAACCC |
| Reverse | TTTACGATTACACCCAGACTGC |
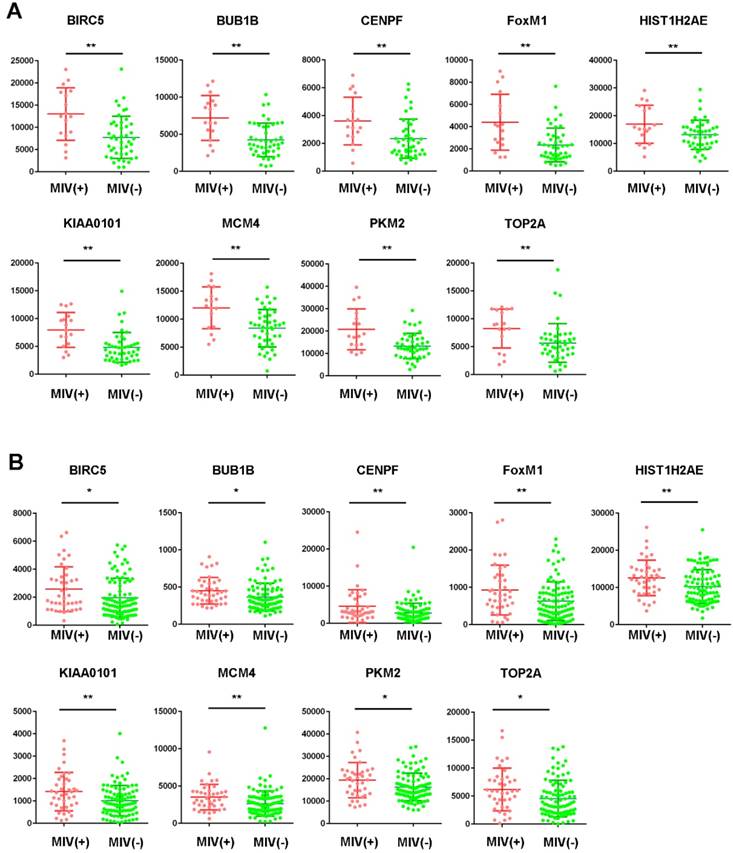
Among the 20 hub genes, a significant difference (P<0.05) between the different statuses of HCC MVI and gene expression levels were found for BIRC5, BUB1B, CENPF, FOXM1, HIST1H2AE, KIAA0101, MCM4, PKM2, and TOP2A (Figure 2A). Additionally, the correlation between the expression of these hub genes and MVI was verified by the validation of the GSE20148 dataset (Figure 2B).
Expression profile of hub genes in HCC tissues. Expression of hub genes in HCC samples with or without microvascular invasion (MVI) in the GSE10143 dataset (A) and the GSE20140 validation cohort (B). *P<0.05, **P<0.01.
Survival analysis, expression comparison and correlation analysis of hub genes
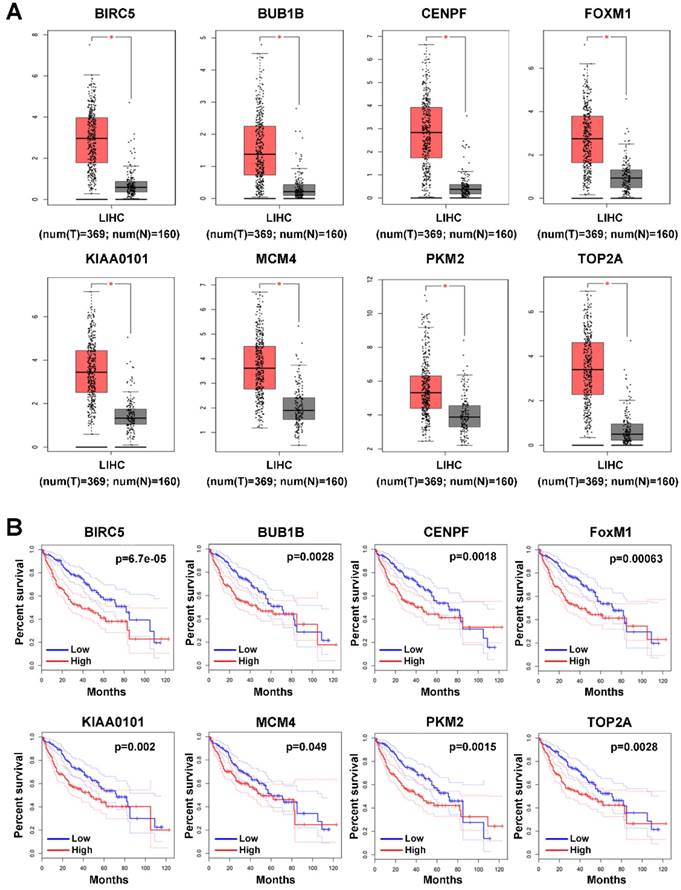
Since the 9 hub genes mentioned above were correlated with MVI, we subsequently conducted a comparative analysis of the expression of these genes between HCC and normal liver tissues to further elucidate the role of these genes in HCC using GEPIA based on the TCGA database. The expression levels of BRCA1, FOXM1, MCM4, PKM2, TOP2A, KIAA0101, BUB1B, and CENPF were significantly upregulated in HCC (log fold change (FC) ≥1, P<0.05; Figure 3A), indicating a potential role of these genes in HCC occurrence and development. Additionally, the survival analysis showed that high expression of these genes was associated with the poor prognosis of HCC patients (Figure 3B).
Expression profile and prognostic value of hub genes in HCC tumor tissues. (A) Expression profile of hub genes in HCC tumor tissues (T) and nontumor tissues (N) in GEPIA based on the TCGA and GTEx databases. Log(FC)≥1, and *P<0.05 were set as the criteria. (B) The prognostic value of the hub genes in HCC patients based on the TCGA database analyzed by GEPIA. The median expression level was set as the cut-off value.
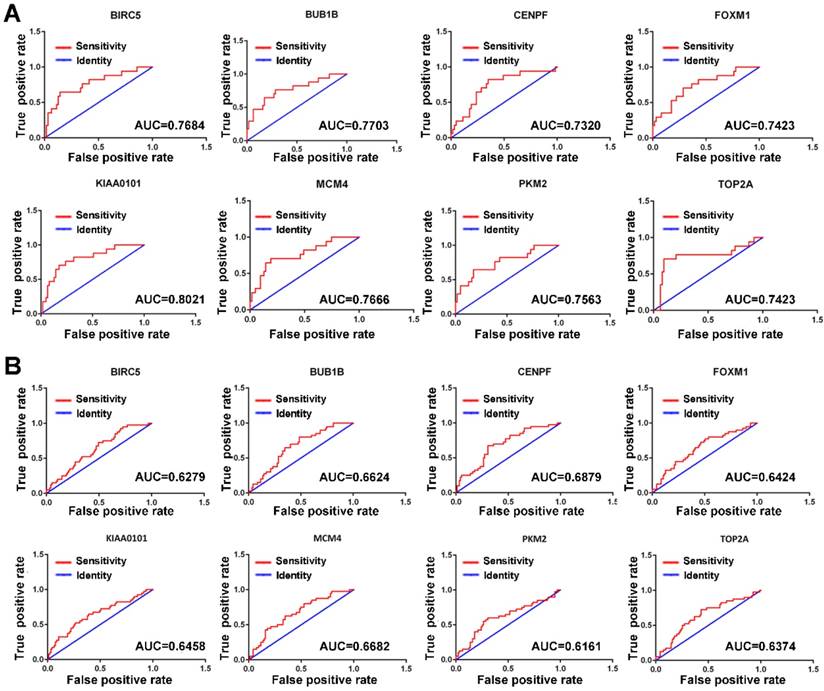
Since MVI is a major risk factor for tumor recurrence and mortality in HCC, simple and practicable means for the early assessment of the probability of developing MVI are thus of great need. The ROC curve indicated that the 8 hub genes exhibited moderate diagnostic efficiency for MVI in HCC in the GSE10143 dataset (AUC>0.7) (Figure 4A). However, the diagnostic value of these hub genes was not observed in the GSE20140 dataset (AUC<0.7) (Figure 4B). Therefore, further investigations are needed to validate the diagnostic value of these hub genes.
ROC curves of the hub genes determined by the AUC in HCC based on the GSE10143 (A) and GSE20140 (B) datasets. Red represents the sensitivity curve, and green indicates the identification line.
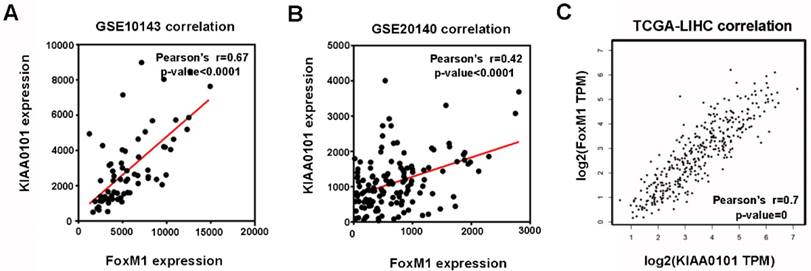
Among the 8 genes with clinical significance, FoxM1 was obviously positively correlated with KIAA0101 both in the GSE10143 and GSE20140 datasets (Figure 5A and B), as well as in the TCGA database analyzed by GEPIA (Figure 5C), indicating a potential regulatory network in HCC that warrants further research.
The expression levels of FoxM1 and KIAA0101 were significantly positively correlated in HCC patients. The positive correlation between FoxM1 and KIAA0101 in HCC patients based on the GSE10143 dataset (A), the GSE20140 dataset (B), and the TCGA database analyzed by GEPIA (C).
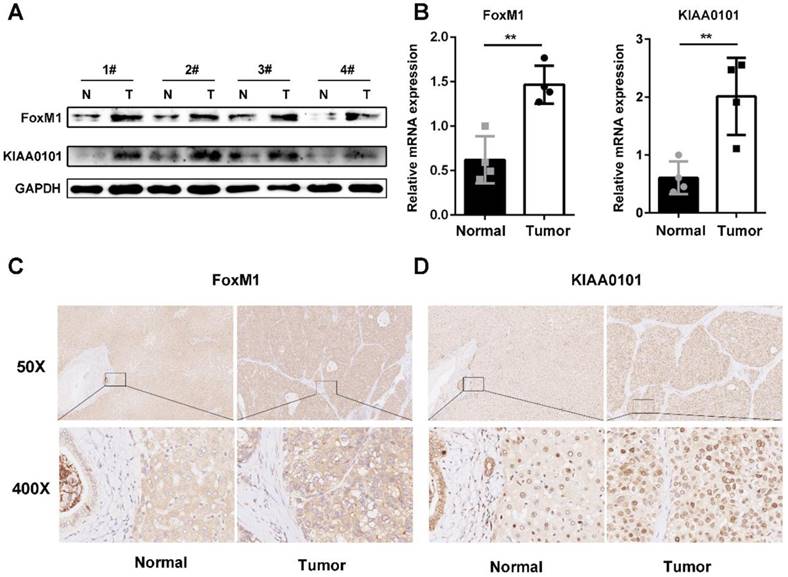
FoxM1 and KIAA0101 are overexpressed in human HCC
To further validate the microarray data, FoxM1 and KIAA0101 expression levels were detected in 4 pairs of HCC and corresponding normal adjacent liver tissue samples using western blot and qRT-PCR analysis. Both the mRNA and protein levels of FoxM1 and KIAA0101 were higher in the HCC tissues than in the normal liver tissues (Figure 6A, and B). In addition, an intense and diffuse cytoplasmic staining pattern of FoxM1 and a nuclear staining pattern of KIAA0101 was detected in the HCC tissues by IHC, whereas the corresponding normal liver tissues showed weak staining of FoxM1 and KIAA0101 (Figure 6C). Taken together, these results indicate that FoxM1 and KIAA0101 may serve as cancer-promoting proteins in HCC.
FoxM1 and KIAA0101 were both upregulated in HCC samples. Western blot (A), qRT-PCR (B), and immunohistochemical staining (C) showed the expression of FoxM1 and KIAA0101 in HCC tissues and adjacent normal tissues. N: normal tissue; T: HCC tissue. Magnification: ×50 and ×400. All data are presented as the means±SDs of at least three independent experiments. *P<0.05, **P<0.01.
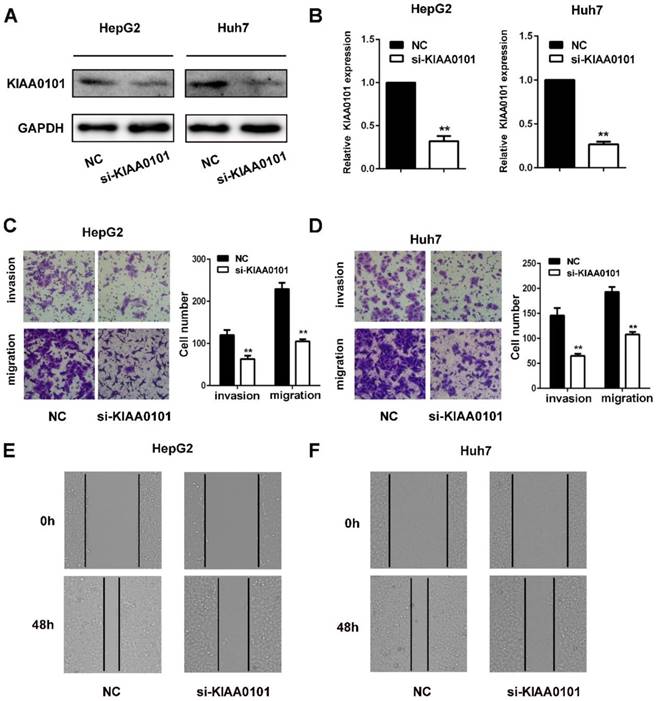
KIAA0101 promotes the migration and invasion of liver cancer cells
The invasion and migration of liver cancer cells are essential for MVI development. We explored the role of KIAA0101 in the invasion and migration of liver cancer cells. To investigate the biological roles of KIAA0101, short interference siRNAs for human KIAA0101 (si-KIAA0101) were applied to knockdown the expression of KIAA0101 in liver cancer cells. SiKIAA0101 led to significant downregulation of KIAA0101 in both HepG2 and Huh7 cells (Figure 7A, and B). In addition, both wound healing and transwell assays with or without Matrigel-coating showed that knockdown of KIAA0101 significantly repressed the migration and invasion ability of liver cancer cells (Figure 7C, D, E, and F).
KIAA0101 promotes the migration and invasion of liver cancer cells. After transfection with si-KIAA0101 as well as NC, the protein and mRNA expression of KIAA0101 in HepG2 and Huh7 cells was measured by western blot (A) and qRT-PCR (B). A transwell assay was applied to assess the invasion and migration ability of the transfected cells. Cell counts from five random microscopic fields were analyzed (C and D. Magnification: ×200). Migration ability was also assessed by a wound-healing assay (E and F. Magnification: ×40). All data are presented as the means±SDs of at least three independent experiments. *P<0.05, **P<0.01, compared with the NC group; #P<0.05, ##P<0.01, compared with the si-FoxM1 group.
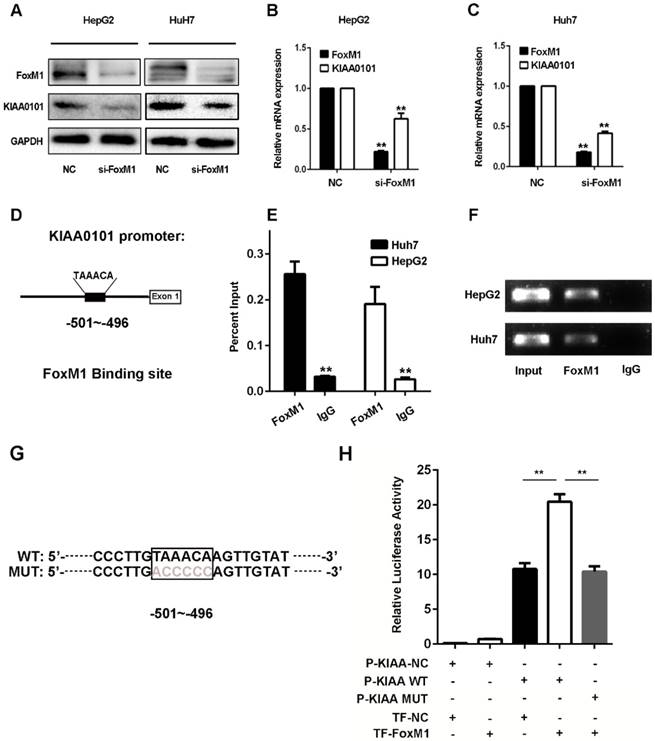
KIAA0101 is a direct target of the transcriptional factor FoxM1
Since the expression levels of FoxM1 and KIAA0101 were markedly positively correlated in HCC in the GSE10143, GSE20140, and TCGA databases and the western blot and qRT-PCR results revealed that knockdown of FoxM1 significantly repressed the expression of KIAA0101 in HepG2 and Huh7 cell lines (Figure 8A, B and C), we presumed that KIAA0101 might be the potential target of FoxM1. Additionally, sequence analysis showed a potential binding sequence, TAAACA [18], for FoxM1 in the promoter region of the KIAA0101 gene (Figure 8D). To validate the interaction between FoxM1 and KIAA0101, we performed a ChIP assay in HepG2 and Huh7 cells using FoxM1-specific antibodies (FoxM1). Compared to the sample bound to IgG, we found that the FoxM1-bound complex showed a marked enrichment of the KIAA0101 promoter (Figure 8E and F). To further validate the activation role of FoxM1 in KIAA0101 transcription, we performed a luciferase reporter assay in HEK293T cells. The result showed that FoxM1 regulated KIAA0101 mRNA expression by binding to the promoter of KIAA0101 (Figure 8G and H).
KIAA0101 is a direct target of the transcriptional factor FoxM1. After transfection with si-FoxM1 or negative control (NC), the protein and mRNA expression of FoxM1 and KIAA0101 in HepG2 and Huh7 cells were measured by western blot (A) and qRT-PCR (B and C). Schematic illustration of the KIAA0101 promoter region and the binding site of FoxM1. The KIAA0101 promoter was determined by using https://genome.ucsc.edu. The sequence is numbered from the transcription start site (exon 1). The primer set was designed to amplify the region of the promoter to amplify the potential binding site (D). ChIP assays with anti-FoxM1 antibody or IgG were performed to verify the binding between FoxM1 and the KIAA0101 promoter in HepG2 and Huh7 cells. After the reversal of cross-linking, the coimmunoprecipitated DNA was amplified by PCR using primers amplifying the FoxM1 binding-site-containing region (E). In addition, a representative gel image of the ChIP results is presented (F). The WT and MUT promoter sequences of the putative promoter regions (G). A luciferase assay revealed that FoxM1 regulated KIAA0101 mRNA expression by binding to the promoter of KIAA0101 (H). All data are presented as the means±SDs of at least three independent experiments. *P<0.05, **P<0.01.
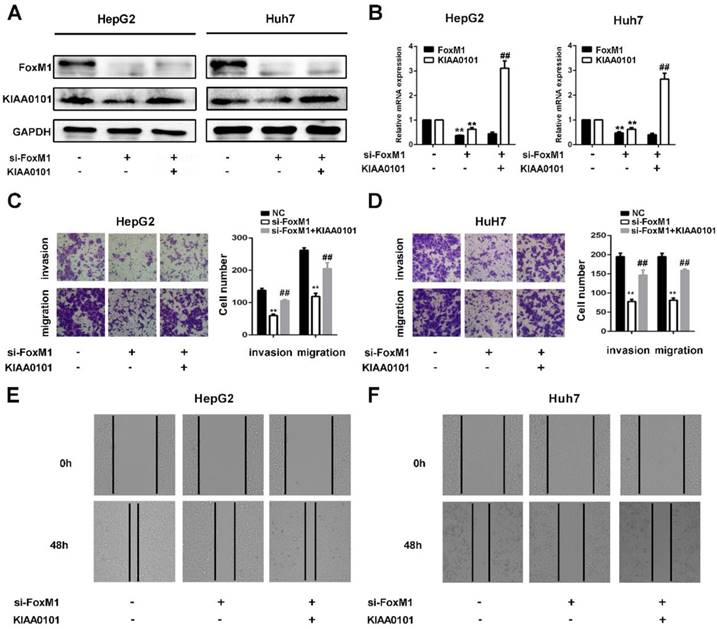
KIAA0101 is a critical target of FoxM1 in promoting migration and invasion of liver cancer cells
FoxM1 is a widely studied transcriptional factor that is crucial for metastasis, invasion, angiogenesis, and DNA repair, and its overexpression is a common feature of multiple tumors [11]. Additionally, previous studies have shown that KIAA0101 plays a vital role in cell proliferation, cell survival, DNA repair, and tumorigenesis [19]. To investigate the role of KIAA0101 in FoxM1-regulated cancer progression, LV-KIAA0101 was transfected into the si-FoxM1 stable HepG2 and Huh7 cell lines. Western blot and qRT-PCR revealed that LV-KIAA0101 restored KIAA0101 expression (Figure 9A and B). Moreover, as shown in Figure 9C, D, E, and F, the inhibitory effect of siFoxM1 on the migration and invasion capacity of HepG2 and Huh7 cells was significantly rescued by the overexpression of KIAA0101.
KIAA0101 is a critical target of FoxM1 in promoting migration and invasion of liver cancer cells. After transfection with si-FoxM1 and cotransfection with si-FoxM1 and KIAA0101 as well as NC, the protein and mRNA expression levels of FoxM1 and KIAA0101 in HepG2 and Huh7 cells were measured by western blot (A) and qRT-PCR (B). A transwell assay was applied to assess the invasion and migration ability of the transfected cells. Cell counts from five random microscopic fields were analyzed (C and D. Magnification: ×200). Migration ability was also assessed by a wound-healing assay (E and F. Magnification: ×40). All data are presented as the means±SDs of at least three independent experiments. *P<0.05, **P<0.01, compared with the NC group; #P<0.05, ##P<0.01, compared with the si-FoxM1 group.
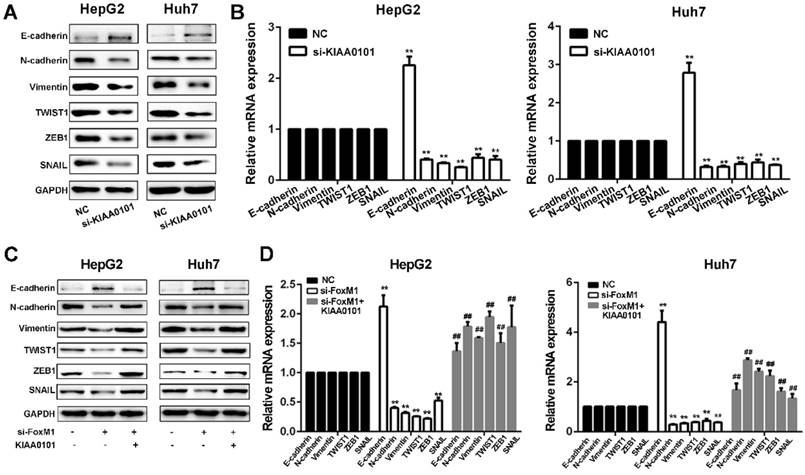
FoxM1-KIAA0101 activates the EMT profile in liver cancer cells
Since EMT is considered a central process of tumor invasion and migration [20], it may participate in the development of HCC MVI [10]. We next confirmed whether EMT is involved in the HCC MVI or metastasis regulated by the FoxM1-KIAA0101 axis. Our results showed that KIAA0101 knockdown suppressed EMT in HepG2 and Huh7 cells by upregulating E‐cadherin and downregulating N‐cadherin, vimentin, TWIST1, ZEB1, and SNAIL. Additionally, consistent with previous studies [21, 22], our results showed that increased expression of FoxM1 could induce the occurrence of EMT and that FoxM1 knockdown significantly increased E-cadherin expression and decreased N-cadherin, vimentin, SNAIL, TWIST, and ZEB1 expression, thus inhibiting EMT in HepG2 and Huh7 cells. Moreover, the overexpression of KIAA0101 could effectively reverse the changes in the expression of these proteins (Figure 10). Together, these data suggest that FoxM1 promotes EMT in liver cancer cells at least partially by regulating KIAA0101 expression.
The FoxM1-KIAA0101 axis induces the EMT profile in liver cancer cells. The expression levels of the EMT-related factors E-cadherin, N-cadherin, vimentin, TWIST1, ZEB1, and SNAIL in transfected HepG2 and Huh7 cells were assessed by western blot (A, and C) and qRT-PCR (B, and D). All data are presented as the means±SDs of at least three independent experiments. *P<0.05, **P<0.01, compared with the NC group; #P<0.05, ##P<0.01, compared with the si-FoxM1 group.
Discussion
As one of the most highly aggressive cancers in humans, HCC causes unfavorable outcomes in terms of overall survival. The poor prognosis of HCC is mainly attributed to tumor invasion, frequent intrahepatic dissemination, and extrahepatic metastasis [23]. The elucidation of the molecular mechanisms underlying HCC metastasis is of the utmost importance for the development of future strategies for treating HCC. The NCBI-GEO databases, which contain a great deal of gene expression and clinical information of multiple human malignancies, provide us with the possibility of integrative investigations and the identification of new targets with potential value in cancer treatment.
In this study, we explored HCC datasets with gene expression profiles and patient information from the NCBI-GEO database. The aim of the study was to gain molecular insights into the progression of HCC and to identify the candidate gene groups associated with the different clinical features of HCC by constructing a gene coexpression network using WGCNA. The module that most significantly correlated with the MVI of HCC was identified. The presence of MVI, such as portal venous, hepatic vein or bile duct infiltration, is considered the best indicator of an unfavorable prognosis after hepatic resection and transplantation [24]. Thus, the genes in the corresponding module were extracted to perform pathway enrichment analysis to identify signaling pathways that contributed to the pathophysiological process. A series of pathways involved in tumorigenesis and progression, including catabolic processes, mitotic cell cycle processes, and chemical carcinogenesis, were detected. In addition, we identified 8 hub genes involved in HCC tumorigenesis and MVI, including BIRC5, BUB1B, CENPF, FOXM1, KIAA0101, MCM4, PKM2, and TOP2A. The oncogenic functions of these genes in many cancers have been reported in previous studies.
Although tremendous efforts have been made to detect HCC MVI early, MVI is difficult to detect preoperatively with current conventional imaging modalities and can only be found by histopathological examination [25]. Given that MVI is a major risk factor for tumor recurrence and mortality in HCC [25], predictive molecular biomarkers for the early assessment of the probability of MVI are thus greatly needed. Previous studies have shown that abnormally high expression of the serum or tumor markers AFP[26], prothrombin induced by vitamin K absence II (PIVKA-II) [27], HSP70 [28], and α-enolase (Eno-1) [28] are closely related to the presence of MVI and could be used as diagnostic biomarkers of MVI. However, in the present study, the diagnostic value of these 8 hub genes was not significant; this finding requires further study for validation.
Among the hub genes, we found that FoxM1 and KIAA0101 were markedly positively correlated in HCC, indicating a potential novel network in the regulation of MVI in HCC. FoxM1 is a master regulator in multiple steps of tumorigenesis, including cell cycle progression, blood vessel formation, cell migration and invasion, EMT, cell differentiation and senescence, and stem cell pluripotency [11]. KIAA0101 is one of the interacting proteins of PCNA, and the role of KIAA0101 is closely related to the function of PCNA, which is involved in the regulation of DNA replication, the cell cycle, and cellular proliferation [19]. Recent studies have shown that KIAA0101 is also involved in tumor invasion and migration by activating Wnt/β-catenin signaling and stimulating EMT [29].
The overexpression of FoxM1 has been reported in a wide range of malignancies; moreover, we found that KIAA0101 was also overexpressed in multiple cancers (Supplementary Figure 2). In this study, we confirmed the overexpression of either the mRNA or protein levels of both FoxM1 and KIAA0101 in human HCC specimens from our medical center. In addition, we also found that a high level of either FoxM1 or KIAA0101 is an independent indicator of poor prognosis in HCC patients; therefore, the proper cut-off of FoxM1 or KIAA0101 expression level can be applied to determine the prognosis of HCC patients in the clinical setting. Then, we explored the relationship between FoxM1 and KIAA0101. We examined the effects of changed expression of FoxM1 on KIAA0101 at both the mRNA and protein levels in the liver cancer cell lines HepG2 and Huh7. Downregulating FoxM1 expression significantly repressed KIAA0101 expression in liver cancer cells. The results of the ChIP assay and the luciferase reporter assay further validated our finding, revealing that KIAA0101 was transcriptionally regulated by FoxM1.
Additionally, since the invasion and migration of liver cancer cells play crucial roles in MVI development, we conducted cell experiments to explore the FoxM1-KIAA0101 axis in HCC metastasis. We used LV-siRNA to stably knockdown FoxM1 in liver cancer cells. Both transwell and wound healing assays confirmed that FoxM1 is essential for HCC invasion and migration. Our rescue experiment revealed that KIAA0101 greatly contributes to the FoxM1-driven invasion and migration of liver cancer cells, indicating that the FoxM1-KIAA0101 axis plays an important role in HCC metastasis and that the metastasis of cancer cells can be potently suppressed by blocking this signaling pathway.
EMT is a process that allows polarized epithelial cells to acquire mesenchymal properties [30]. EMT is primarily regulated by three families of transcription factors: SNAIL, ZEB (ZEB1/ZEB2), and basic helix-loop-helix transcription factors (e.g., Twist1) families [30]. These transcription factors act to repress epithelial marker genes (e.g., E-cadherin) and activate genes associated with the mesenchymal phenotype (e.g., N-cadherin). EMT is regarded as a critical factor for cancer cells to initiate metastasis from the primary site to distant sites by enhancing their invasive potential by accelerating the penetration of the extracellular matrix and the vascular wall, subsequently allowing the cancer cells to enter the bloodstream [10]. Previous studies have shown that EMT-related genes, including SNAIL, FoxC1, and vimentin, are upregulated in HCC with MVI, implying that EMT is associated with the development of MVI [10]. It has been reported that FoxM1 plays a crucial role in mediating EMT by transcriptionally regulating SNAIL [31] and Twist1 [32] expression. We revealed that downregulated FoxM1 expression could contribute to the loss of the EMT phenotype by upregulating the epithelial cell marker E-cadherin and downregulating the mesenchymal cell markers N-cadherin, vimentin, ZEB1, TWIST1, and SNAIL, which was in agreement with the findings of previous studies. Our rescue experiment revealed that upregulating KIAA0101 expression notably restored the expression of these altered genes involved in EMT.
In the present study, we demonstrate the important prognostic value of the FoxM1-KIAA0101 axis in HCC patients and highlight the essential role of KIAA0101 in FoxM1-driven HCC invasion and metastasis. Thus, the FoxM1-KIAA0101 axis might be a potential prognostic biomarker and therapeutic target in HCC treatment.
Abbreviations
HCC: hepatocellular carcinoma; WGCNA: weighted gene coexpression network analysis; MVI: microvascular invasion; TCGA: The Cancer Genome Atlas; FoxM1: forkhead box M1; HIST1H2BF: histone cluster 1, H2bf; HIST1H2AE: histone cluster 1, H2ae; BRCA1: breast cancer 1; HMGA1: high mobility group AT-hook 1; CDKN3: cyclin-dependent kinase inhibitor 3; NCAPH: non-SMC condensin I complex, subunit H; CCNE2: cyclin E2; MCM4: minichromosome maintenance complex component 4; PKM2: pyruvate kinase, muscle; TOP2A: topoisomerase (DNA) II alpha; TROAP: trophinin-associated protein; BUB1B: BUB1 budding uninhibited by benzimidazoles 1 homolog beta; E2F1: E2F transcription factor 1; CENPF: centromere protein F; CDK2: cyclin-dependent kinase 2; BIRC5: baculoviral IAP repeat-containing 5 (survivin); PRC1: protein regulator of cytokinesis 1; GS: gene significance; MM: module membership; EMT: epithelial-mesenchymal transition; TOM: topological overlap matrix; GO: gene ontology; KEGG: Kyoto Encyclopedia of Gene and Genome; DAVID: Database for Annotation, Visualization and Integrated Discovery; GEPIA: The Gene Expression Profiling Interactive Analysis; GTEx: the Genotype-Tissue Expression; qRT-PCR: quantitative real-time polymerase chain reaction; IHC: immunohistochemistry; ChIP: chromatin immunoprecipitation; ROC: receiver operating characteristic curve; AUC: area under the curve; FC: fold change; PCNA: proliferating cell nuclear antigen; PIVKA-II: prothrombin induced by vitamin K absence II.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported by Natural Science Foundation of China (NO. 81570568 and NO. 81602419). Research Fund of Public welfare in Health Industry (No.201402015), Health and Family Plan Committee of China, 2014 (http://www.nhfpc.gov.cn/) support this research.
Ethics Committee Approval and Patient Consent
All procedures performed in studies involving human participants were approved by the ethical standards of the Ethics Committee of Tongji Medical College, Huazhong University of science and technology (Wuhan, China) and in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Zhu RX, Seto WK, Lai CL, Yuen MF. Epidemiology of Hepatocellular Carcinoma in the Asia-Pacific Region. Gut Liver. 2016;10:332-9
4. Kondo Y, Wada K. Intrahepatic metastasis of hepatocellular carcinoma: a histopathologic study. Human pathology. 1991;22:125-30
5. Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5:588-99
6. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
7. Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A. et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995-2004
8. Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Alsinet C. et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology. 2011;140:1501-12 e2
9. Zhang X, Li J, Shen F, Lau WY. Significance of presence of microvascular invasion in specimens obtained after surgical treatment of hepatocellular carcinoma. J Gastroenterol Hepatol. 2018;33:347-54
10. Xu ZY, Ding SM, Zhou L, Xie HY, Chen KJ, Zhang W. et al. FOXC1 contributes to microvascular invasion in primary hepatocellular carcinoma via regulating epithelial-mesenchymal transition. Int J Biol Sci. 2012;8:1130-41
11. Bella L, Zona S, Nestal de Moraes G, Lam EW. FOXM1: A key oncofoetal transcription factor in health and disease. Semin Cancer Biol. 2014;29:32-9
12. Yuan RH, Jeng YM, Pan HW, Hu FC, Lai PL, Lee PH. et al. Overexpression of KIAA0101 predicts high stage, early tumor recurrence, and poor prognosis of hepatocellular carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:5368-76
13. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17
14. Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics (Oxford, England). 2008;24:719-20
15. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57
16. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19:A68-77
17. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research. 2017;45:W98-W102
18. Littler DR, Alvarez-Fernandez M, Stein A, Hibbert RG, Heidebrecht T, Aloy P. et al. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic acids research. 2010;38:4527-38
19. Xie C, Yao M, Dong Q. Proliferating cell unclear antigen-associated factor (PAF15): a novel oncogene. The international journal of biochemistry & cell biology. 2014;50:127-31
20. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740-6
21. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X. et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. The EMBO journal. 2013;32:231-44
22. Meng FD, Wei JC, Qu K, Wang ZX, Wu QF, Tai MH. et al. FoxM1 overexpression promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma. World journal of gastroenterology. 2015;21:196-213
23. Huo TI, Lin HC, Huang YH, Wu JC, Chiang JH, Lee PC. et al. The model for end-stage liver disease-based Japan Integrated Scoring system may have a better predictive ability for patients with hepatocellular carcinoma undergoing locoregional therapy. Cancer. 2006;107:141-8
24. Yamashita Y, Shirabe K, Aishima S, Maehara Y. Predictors of Microvascular Invasion in Hepatocellular Carcinoma. Dig Dis. 2015;33:655-60
25. Du M, Chen L, Zhao J, Tian F, Zeng H, Tan Y. et al. Microvascular invasion (MVI) is a poorer prognostic predictor for small hepatocellular carcinoma. BMC cancer. 2014;14:38
26. McHugh PP, Gilbert J, Vera S, Koch A, Ranjan D, Gedaly R. Alpha-fetoprotein and tumour size are associated with microvascular invasion in explanted livers of patients undergoing transplantation with hepatocellular carcinoma. HPB (Oxford). 2010;12:56-61
27. Pote N, Cauchy F, Albuquerque M, Voitot H, Belghiti J, Castera L. et al. Performance of PIVKA-II for early hepatocellular carcinoma diagnosis and prediction of microvascular invasion. J Hepatol. 2015;62:848-54
28. Yu YQ, Wang L, Jin Y, Zhou JL, Geng YH, Jin X. et al. Identification of serologic biomarkers for predicting microvascular invasion in hepatocellular carcinoma. Oncotarget. 2016;7:16362-71
29. Wang X, Jung YS, Jun S, Lee S, Wang W, Schneider A. et al. PAF-Wnt signaling-induced cell plasticity is required for maintenance of breast cancer cell stemness. Nat Commun. 2016;7:10633
30. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
31. Wei P, Zhang N, Wang Y, Li D, Wang L, Sun X. et al. FOXM1 promotes lung adenocarcinoma invasion and metastasis by upregulating SNAIL. Int J Biol Sci. 2015;11:186-98
32. Su J, Wu S, Wu H, Li L, Guo T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/beta-catenin-FoxM1-Twist signaling. Molecular carcinogenesis. 2016;55:1962-73
Author contact
![]() Corresponding authors: Huili Li: huili_liedu.cn, Guobin Wang: wangguobinunioncom, and Jiliang Wang: jiliang_wangedu.cn
Corresponding authors: Huili Li: huili_liedu.cn, Guobin Wang: wangguobinunioncom, and Jiliang Wang: jiliang_wangedu.cn