Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(15):3427-3434. doi:10.7150/jca.30318 This issue Cite
Research Paper
Nuclear Nrf2 Activity in Laryngeal Carcinoma is Regulated by SENP3 After Cisplatin-Induced Reactive Oxygen Species Stress
Zheng Zhou*, Ji Xu*, Ximing Bao, Jiali Shi, Bin Liu, Yanqing Chen ![]() , Jiping Li
, Jiping Li ![]()
Department of Otolaryngology, Ren ji Hospital, School of Medicine, Shanghai Jiao tong University, Shanghai, China
*These authors contributed equally to this work.
Received 2018-9-30; Accepted 2019-4-25; Published 2019-6-9
Abstract
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a nuclear transcription factor that is activated by reactive oxygen species (ROS). Recent studies reported that hyperactivation of the Nrf2 pathway creates an environment that favors the survival of normal as well as malignant cells, protecting them against oxidative stress, chemotherapeutic agents, and radiotherapy. SUMO1/sentrin/SMT3 specific peptidase 3 (SENP3) reverses sumoylation of small ubiquitin-like modifier (SUMO)-conjugates. We demonstrated that Nrf2 was detected in the nuclei of laryngeal carcinoma cells, but not in cells of tissues surrounding the cancer, which correlated with the appearance of SENP3 in the nuclei. Silencing of Nrf2 in laryngeal carcinoma cell line Hep-2 significantly reduced cell viability and enhanced apoptosis rates under cisplatin, 5-fluorouracil (5-FU) and phenethyl isothiocyanate (PEITC) exposure. Cisplatin exposure induced ROS stress in Hep-2 cells in a time-dependent manner and was accompanied by increased Nrf2 and SENP3 protein accumulations, an effect reversed by the addition of the antioxidant N-acetyl-cysteine (NAC). Silencing of SENP3 led to reduced Nrf2 protein levels, whereas overexpression of SENP3 led to concomitant enhanced transcription of the Nrf2 target genes HO-1, NQO1, GCLC and GSTM1. Immunoprecipitation showed that overexpressed Nrf2 and SENP3 could be precipitated together, indicating that they were intracellular bound to each other. Our data identified intranuclear activation of Nrf2 is triggered by cisplatin-induced ROS development through the activity of SENP3. These findings provide novel insights into the Nrf2 reduced cancer cell response to the chemotherapy of laryngeal carcinoma.
Keywords: Nrf2, SENP3, Hep-2 cells, ROS, cisplatin
Introduction
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor activated by emerging reactive oxygen species (ROS). Nrf2 is a major regulator of cytoprotective responses under oxidative stress thereby preventing cancer initiation and progression. In cancer cells, activated Nrf2 resulted in chemotherapy and radiotherapy resistance and therefore promotes growth of tumor cells (1-4).
Under ROS free conditions, two Kelch ECH associating protein 1 (Keap1) molecules bind to Nrf2, which is then polyubiquitylated by the Cul3-based E3 ubiquitin ligase complex and subsequently degraded by the ubiquitin proteasome pathway. ROS development leads to inhibition of Keap1 binding by mechanisms that remain to be fully elucidated, but two mechanisms have been proposed. In one model, Keap1 thiol residue modifications leads to misalignment of lysine residues within Nrf2 which can then no longer be polyubiquitinylated. In the other model, thiol modifications cause dissociation of Cul3 from Keap1. However, it is noteworthy that in both models Nrf2 proteins escape degradation and translocate into the nucleus where they bind, in combination with small Maf proteins (sMaf), to antioxidant response elements (ARE) and direct transcription of Nrf2 target genes such as NAD(P)H quinone oxidoreductase1 (NQO1), heme oxygenase1 (HO-1), glutamate-cysteineligase (GCL) and glutathione S transferases (GSTs) (5). Other Nrf2 activation mechanisms involve p21 and p62 binding to Nrf2, which disrupts the interaction between Nrf2 and Keap1 (6, 7). p53 can reduce Nrf2 activity by an unknown mechanism and it has been suggested that strong activation of p53 inhibits Nrf2, leading to reduced antioxidant defense and cell survival responses with the promotion of cell death. In contrast, weak p53 induction of p21 activity is increased and cell survival mechanisms are dominated by the DNA damage repair functions of p53 and Nrf2-mediated cytoprotective response activation (8). However, regulation of the activity of Nrf2 is not limited to the cytosol since Nrf2 traffics to promyelocytic leukemia-nuclear bodies (PML-NBs) where the poly-SUMO-specific E3 ubiquitin ligase RING finger protein 4 (RNF4) ubiquitylates it with small ubiquitin-like modifier (SUMO)-1 and SUMO-2 polysumoylated Nrf2, which is subsequently degraded independently of Keap1 (9). SUMO1/sentrin/ SMT3 specific peptidase 3 (SENP3) is a member of the SENP family, which reverse sumoylation of SUMO-2 or SUMO-3 conjugates and to a less extent SUMO-1 modifications (10). In a previous study it has been demonstrated that SENP3 activity was enhanced by mild oxidative stress which led to accelerated cell proliferation through de-conjugation of SUMO2/3 from PML (11). Another study noted that SENP3 was overexpressed in oral squamous cell carcinoma (OSCC) and may be involved in the development of OSCC during oxidative stress (12).
In the present study, we first investigated correlations of SENP3 and Nrf2 in laryngeal carcinoma under the hypothesis that SENP3 might be a regulator of Nrf2 activity and then focused on cell chemosensitivity to cisplatin.
Materials and Methods
Human biopsies
Laryngeal specimens were taken from surgically resected tissues of laryngeal carcinoma and the corresponding surrounding tissue (n = 32). The patients had not received pre-operative chemotherapy or radiotherapy. The use of human HNC tissue specimens was evaluated and approved by the Ethical Committee of Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University.
Cell culture and treatments
The laryngeal carcinoma cell line Hep-2 was purchased from the American Type Culture Collection (Manassas, VA, US). The HEK293T derived 293FT cell line was cultured in Dulbecco's modified Eagle's medium (HyClone). All media were supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, US). When needed, the antioxidant N-acetyl-cysteine (NAC) (Sigma, St Louis, MO, US) was pre-administered for 4 h, and the proteasome inhibitor MG132 (Merck KGaA, Germany) was pre-administered for 10 h before other treatments. 5-FU and PEITC were purchased from Sigma. Cisplatin was obtained from Qilu Pharmaceutical Co., Ltd (Jinan, China). Cisplatin, 5-FU and phenethyl isothiocyanate (PEITC) were pre-administered for 24 h. PEITC was dissolved in dimethyl sulfoxide (DMSO) and was freshly diluted in culture media before use in experiments. Cell transfection was performed using Lipofectamine 2000 (Invitrogen) for plasmid DNA and siRNAs following the manufacturer's instructions. Cells were harvested between 24 h and 48 h post transfection for protein analyses.
Antibodies
The following antibodies were used in this study: mouse monoclonal antibodies against β-actin (Cat#A5441, Sigma, St Louis, MO, US), Flag (Cat#F3165, Sigma, St Louis, MO, US), HO-1 (Cat# ab13248, Abcam, Cambridge, MA, US) and GAPDH (Cat# ab8245, Abcam, Cambridge, MA, US); rabbit monoclonal antibodies against Nrf2 (Cat#ab137550, Abcam, Cambridge, MA, US); rabbit polyclonal antibodies against SENP3 (Cat#5591, Cell Signalling, Danvers, MA, US); normal rabbit IgG (Cat#5591, Cell Signalling, Danvers, MA, US).
Establishment of stable cell lines
To establish the stable SENP3-shRNA (5'UTR) Hep-2 cell line, pLVX-shRNA2 lentiviral expression vector in combination with the psPAX2 lentiviral packaging system (Takara Bio Inc., Japan) were transiently transfected into 293FT cells. After 72 h, supernatants were harvested to infect Hep-2 cells with a final concentration of 10 μg/mL polybrene. Finally, the GFP-positive cells were sorted on a FACSAria II flow cytometer (BD Biosciences, San Jose, CA, USA). The SENP3-shRNA oligonucleotides were SENP3-shRNA FW 5'-gatccgggctggaaaggttacttcaattcaagagattgaagtaacctttccagcccttttttacgcgtg-3'; SENP3-shRNA RV 5'-aattcacgcgtaaaaaagggctggaaaggttacttcaatctcttgaattgaagtaacctttccagcccg-3'. For Nrf2 knockdown, Nrf2 siRNA was transiently transfected with Lipofectamine 2000 reagent.
The siRNA oligonucleotides specific for SENP3 and Nrf2, and non-specific controls were purchased from RiboBio Co., Ltd (Guangzhou, China). Flag-SENP3 and Myc-Nrf2 were constructed with Flag and Myc tagged pCDNA-3 vectors.
Immunohistochemistry
Paraformaldehyde-fixed and paraffin-embedded sections of human laryngeal specimens were archived pathological specimens from the Ren Ji Hospitals and obtained after securing institutional approval of the experimental protocols. The immunohistochemistry for SENP3 and Nrf2 was performed using serial sections as previously describe (13).
Immunofluorescence
Paraformaldehyde-fixed and paraffin-embedded sections of human laryngeal specimens were incubated with 5% BSA containing anti-α-SENP3 (#ab5694, abcam, USA) and anti-Nrf2 primary antibody (#ab89443, abcam, USA) at 4 °C overnight. Next day, sections were stained with Alexa Fluor 488-labelled goat anti-rabbit or 555-labelled goat anti-mouse secondary antibody (Invitrogen) at 37 °C for 2 h and counterstained with DAPI (Beyotime, China) for observation. Images were taken with Zeiss 710 laser scanning confocal microscopy system on a Zeiss Axio Observer Z1 inverted microscope, equipped with a Plan-Apochromat 20 X, 0.75 NA, differential interference contrast objective and switching reconstruction method. The 8-bit original data were exported and analyzed with ImageJ software.
Immunoblotting
Immunoblotting was performed as previously described (14). Briefly, filters were incubated with rabbit antibodies against Nrf2 and SENP3 with normal anti-rabbit IgG (Jackson ImmunoResearch Inc, PA, US) as secondary antibodies and with mouse antibodies against β-actin, Flag, HO-1 and GAPDH with normal anti-mouse IgG (Jackson ImmunoResearch Inc, PA, US) as secondary antibodies.
Flag immunoprecipitation assay
Transfected cells were lysed in a lysis buffer (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA and 1% Triton X-100). Anti-FLAG M2 Affinity Gel (Cat#A2220, Sigma) was added to the cell lysates and incubated overnight at 4°C. The beads were washed 4 times in lysis buffer. After the last wash, Flag-tagged proteins were eluted in elution buffer (lysis buffer, cocktail (Roche, Diagnostics GmbH, Mannheim, Germany), 20 mM NEM (Sigma)) and then subjected to IB.
Real-time qPCR
Real-time qPCR was conducted on an ABI Prism 7500 Fast system using SYBR Green (Roche, Diagnostics GmbH) according to the manufacturer's instructions. For HO-1, the forward primer was 5'-cagtcttcgcccctgtctac-3' and the reverse was 5'-agacaggtcacccaggtagc-3'. For NQO1, the forward primer was 5'-acccttccggagtaagaagg-3' and the reverse was 5'-ttccatccttccaggatttg-3'. For GCLC, the forward primer was 5'-caaggacgttctcaagtggg-3' and the reverse was 5'-catactctggtctccaaagg-3'. For GSTM1, the forward primer was 5'-ggagacagaagaggagaagattc-3' and the reverse was 5'-gcccagctgcatatggttgt-3'.
Cell viability assessment using a cell counting kit (CCK8)
According to the manufacturer's protocol, CCK8 (Peptide Institute Inc., Osaka, Japan) solution was added to each sample and incubated for 1 h. The absorbance was recorded at 450 nm with a microplate reader (Thermo Fisher Scientific Inc., Waltham, MA, USA).
ROS detection
2', 7'-dichlorofluorescin diacetate (DCFH-DA; Sigma) was used as the ROS capturing reagent as previously described (15). DCFH-DA was deacetylated intracellularly by nonspecific esterase, which was furthered oxidized by ROS to the fluorescent compound 2, 7-dichlorofluorescein (DCF). Cells were incubated with 10 μM DCFH-DA at 37°C for 15 min and DCF fluorescence was detected by flow cytometry.
Flow cytometry assay
An Annexin V-FITC/PI cell apoptosis kit (Kaiji Inc, Nanjing, China) was used as follows: After washing with PBS, 5 × 105 cells were collected in non-EDTA pancreatin. Then 500 μL of the cells suspended in binding buffer were mixed with 5 μL of Annexin V-FITC and 5 μL propidium iodide mixed at room temperature, protected from light and reacted for 5-15 min. Cells were then mounted on a cover glass for DAPI staining and photographed with a fluorescence microscope. The excitation wavelength was Ex 488 nm and the emission wavelength Em 530 nm. Adjustments were made for the green fluorescence of Annexin V-FITC through the FITC channel (FL1); PI red fluorescence (flow Ex. 488 nm, Em ≥ 630 nm) was detected through the FL3 channel.
Statistical analysis
Statistical analyses were performed using Microsoft Excel (San Francisco, CA, US) or GraphPad Prism (La Jolla, CA, US). The relationship between the SENP3 and Nrf2 was examined using the Spearman Rank correlation coefficient. A P-value < 0.05 was considered to be statistically significant. No samples were excluded from the analysis.
Results
Accumulation of Nrf2 was enhanced in laryngeal carcinoma cells, but not in the surrounding tissue
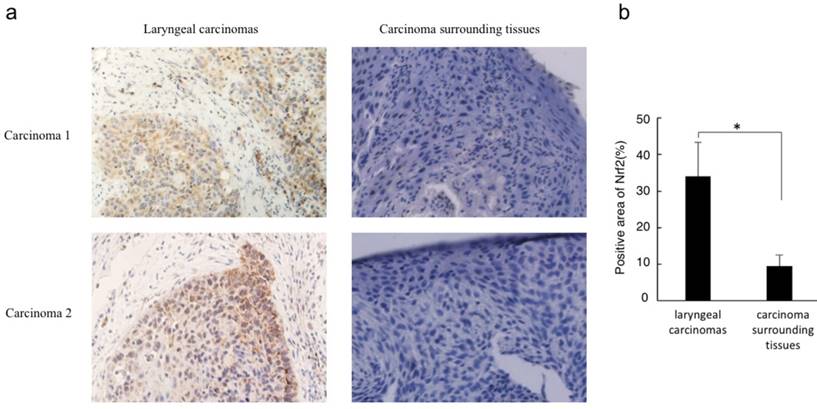
Immunohistochemistry analysis revealed that Nrf2 was highly expressed in laryngeal carcinoma cells but not in the surrounding tissues. In addition, Nrf2 was located in the cancer cell nuclei, indicating that cytosolic degradation via Keap1 had not been activated (Figure 1).
Nrf2 protein level correlates with laryngeal carcinoma. (a) Immunohistochemistry for Nrf2 was performed in sections derived from laryngeal carcinoma specimens and their corresponding carcinoma surrounding tissues. Two representative images are shown (magnification ×400). (b) The relative positive stain areas of Nrf2 in laryngeal carcinoma specimens (n=32) and their corresponding carcinoma surrounding tissues were measured and displayed in bar chart. All data were shown with the means ± s.d. of all specimen in different groups. *P<0.05.
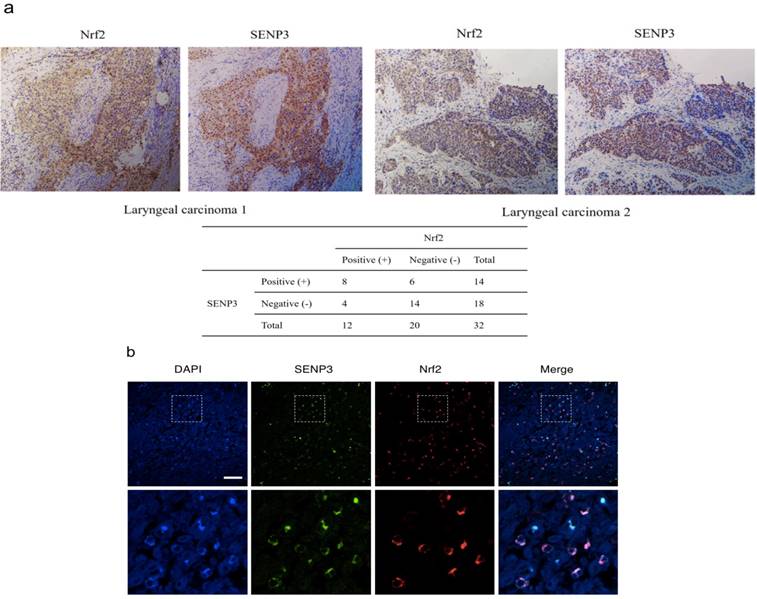
SENP3 co-localized with Nrf2 in the laryngeal carcinoma cell nuclei
The results of serial section analyses revealed that both Nrf2 and SENP3 were both accumulated in the nuclei of laryngeal cancer cells (P < 0.05, r = 0.358) (Figure 2).
SENP3 protein co-localized with Nrf2 in laryngeal carcinoma. (a) Immunohistochemistry for SENP3 and Nrf2 was performed in serial sections derived from specimens of different laryngeal carcinoma. Two representative specimens are shown (Magnification ×200). (b) Immunofluorescence for SENP3 and Nrf2 were performed in sections derived from laryngeal carcinoma specimens and representative pictures were shown. SENP3, green; Nrf2, red; 4′,6-diamidino-2-phenylindole (DAPI), blue. Scale bar, 50 μm.
Nrf2 is a critical factor for chemosensitivity and apoptosis in laryngeal cancer cells
Cell assays showed that Nrf2 silencing significantly reduced the cell viability of Hep-2 cells exposed to cisplatin, 5-FU and PEITC for 24 h in a concentration-dependent manner. The apoptotic rates of Hep-2 cells exposed to 4 μg/uL cisplatin for 18 h was 0.52% without transfection, 53.58% after Nrf2 silencing and 30.10% with transfected control siRNA (Figure 3).
Nrf2 is critical for chemosensitivity in laryngeal cancer cells. A) Hep-2 cells with Nrf2 silencing (Nrf2-siRNA) for 48 h were exposed to the indicated doses of a) cisplatin, b) 5-FU and c) PEITC for 24 h. The viable cell numbers were measured by CCK8. B) The cell apoptotic rates were determined by flow cytometry assays of a) Hep-2 cells (0.52%), b) Hep-2 cells transfected with Nrf2-siRNA (53.58%) and c) Hep-2 cells transfected with control siRNA (30.10%) and exposed to 4 μg/µL cisplatin for 18 h. The values are the means ± s.d. of three replication wells. The experiment repeated three times.
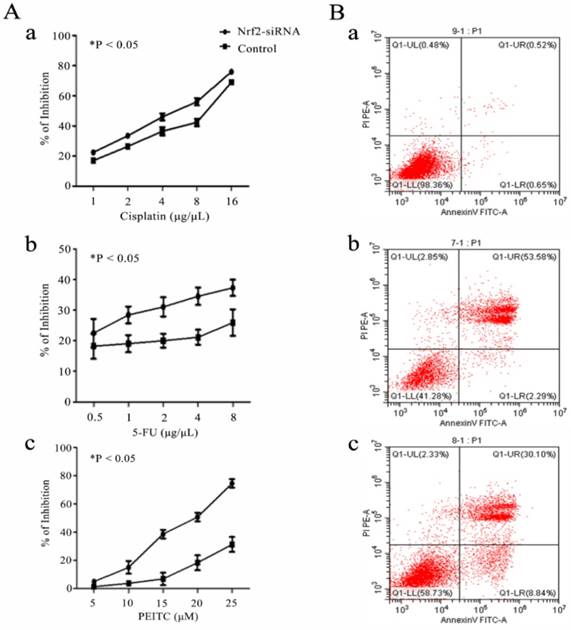
The data indicated that Nrf2 activity was not cell protecting in laryngeal carcinoma cells under chemotherapy regimens.
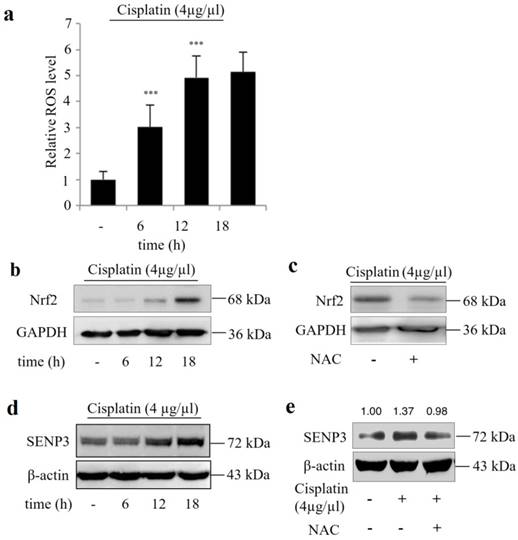
Cisplatin increases the Nrf2 and SENP3 protein levels in Hep-2 cells in a ROS-dependent manner
Figure 4a shows that cisplatin exposure induced ROS stress in Hep-2 cells in a time dependent manner and was accompanied by increased Nrf2 and SENP3 protein accumulation (Figure 4 b, d), which could be reversed by addition of the ROS inhibitor NAC (Figure 4 c, e).
Cisplatin increased the protein level of Nrf2 and SENP3 in Hep-2 cells in a ROS-dependent manner. (a) ROS level was determined by DCFH-DA staining and flow-cytometric analysis in Hep-2 cells exposed to 4 μg/µL cisplatin for the indicated time. Error bars represent ± s.d. for triplicate experiments, ***P < 0.001. (b) The level of Nrf2 was determined by IB in Hep-2 cells exposed to 4 μg/µL cisplatin for indicated times. (c) The Nrf2 level was determined by IB in Hep-2 cells exposed to 4 μg/µL cisplatin in the presence or absence of 5 mM NAC for 18 h. (d) The level of SENP3 was determined by IB in Hep-2 cells exposed to 4 μg/µL cisplatin for indicated times. (e) The SENP3 level was determined by IB in Hep-2 cells exposed to 4 μg/µL cisplatin in the presence or absence of 5 mM NAC for 18 h. The gray scale ratio of IB was analyzed with ImageJ software and displayed above the strip.
SENP3 regulates Nrf2 activation in Hep-2 cells
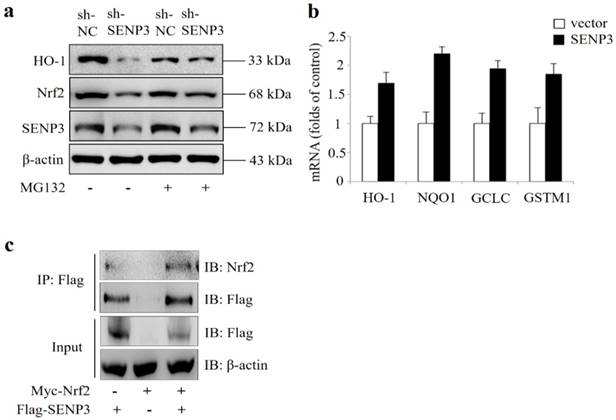
In order to evaluate whether SENP3 was a regulator of Nrf2 activity, SENP3 was stably silenced in a Hep-2 cell line (shSENP3). As shown in Figure 5a, silencing of SENP3 also led to reduced Nrf2 and reduced expression of its transcriptional target gene HO-1, which were both reversed by addition of the proteasome inhibitor MG132. Also, a qRT-PCR analysis revealed that after transient overexpression of SENP3 in Hep-2 cells, the Nrf2 target genes HO-1, NQO1, GCLC, GSTM1 were significantly upregulated (Figure 5b). In addition, immunoprecipitation experiments showed that overexpressed Nrf2 and SENP3 could be precipitated together, indicating a common intracellular binding (Figure 5c).
Critical role of SENP3 in regulating Nrf2 activation in stable SENP3 expressing Hep-2 cells. (a) The levels of Nrf2 and HO-1 were determined by IB in non-specific shRNA (sh-NC) or SENP3 shRNA (sh-SENP3) treated Hep-2 cells in the presence or absence of 10 μM MG132 for 10 h. (b) The mRNA levels of Nrf2 target genes HO-1, NQO1, GCLC and GSTM1were determined by qRT-PCR at 48 h post transfection in Hep-2 cells with overexpression of SENP3 or an empty control vector. Error bars represent ± s.d. for triplicate replication wells. (c) SENP3 and Nrf2 interaction was determined by co-IP in Hep-2 cells transfected with Flag-SENP3 and Myc-Nrf2. Co-IP was performed using Flag-M2 beads for immunoprecipitation and using anti-Flag and anti-Nrf2 antibodies for IB.
Discussion
In the present study we found that in laryngeal carcinoma cells, but not in cells of the cancer surrounding tissues, Nrf2 was localized to the nuclei, a finding in agreement with a previous study, in which nuclear Nrf2 was detected in 79% of squamous cell carcinomas of the larynx (16). In addition, another previous study found that in HepG2 cells, polysumoylated Nrf2 appeared in PML nuclear bodies (9). The reason for this cancer cell limited Nrf2 accumulation in PML nuclear bodies remains unclear but might be caused by permanent ROS stress in laryngeal carcinoma cells (17, 18), since the PML-NB formations have been reported to be related to oxidative stress (19, 20). A SUMO-dependent shuttling of the orphan nuclear receptor LRH-1 into PML bodies has been described in which the unmodified form as a separate pool of the LRH-1 was associated with transcription activity and the sumoylated fraction located in the PML-NBs, which have been suggested to be molecular reservoirs, maintaining the availability of transcription factors to active chromatin domains (21). This may well be the case also for Nrf2, since in HepG2 cells only a fraction appeared in the PML nuclear bodies (9). Another example is the desumoylation of STAT3 by SENP1 thereby de-repressing PML-dependent STAT3 transcriptional activity (22). On the other hand, polysumoylated Nrf2 was further ubiqitylated by the RING finger protein 4 (RNF4) and a nuclear fraction of unknown size subsequently degraded in HepG2 cells, an action which was inhibited by MG132, indicating that Nrf2 accumulation in PML-NBs is regulated (9).
There was a correlation of SENP3 and Nrf2 appearance in the nuclei of the laryngeal carcinoma cells (P < 0.05, r = 0.358), which is in line with reported overexpression of SENP3 in human oral squamous cell carcinoma (12). This finding underlines the probable oxidative stress environment in the cells, since SENP3 has been reported to be activated under oxidative stress conditions and leads to the disappearance of PML-NBs (11, 14).
Our in vitro experiments showed that cisplatin enhanced ROS production in Hep-2 cells in a time-dependent manner (Figure 4a), which is in agreement with the literature in which an essential component of the cisplatin effects has been attributed to the triggering of ROS production (23-27). Accordingly, silencing of Nrf2 led to a higher efficacy of the cisplatin response in Hep-2 cells, which supports a previous study in which Keap1-independent Nrf2 localization to the nuclei of esophageal squamous cell carcinoma (ESCC) cells predicted a poorer clinical response and poor progression-free survival after chemoradiotherapy (4). Concomitant with the emerging oxidative stress caused by cisplatin, the levels of Nrf2 and SENP3 were elevated (Figure 4b, d), which could be ameliorated by simultaneous application of NAC (Figure 4c, e). Furthermore, silencing of SENP3 led to reduced Nrf2 accumulation and related HO-1 transcription, which could be reversed by MG132 (Figure 5a), indicating that Nrf2 is degraded via the proteasome pathway, whereas SENP3 overexpression led to enhanced transcriptions of the Nrf2 target genes HO-1, NQO1, GCLC and GSTM1 (Figure 5b). In addition, the result of co-IP experiment showed that overexpressed Nrf2 and SENP3 could be precipitated together, indicating a common intracellular binding location (Figure 5c).
Taken together, the results suggest that in laryngeal cancer cells a fraction of Nrf2 is sumoylated and accumulates in PML-NBs, but is not transcriptionally active and only partly degraded. After the emergence of enhanced oxidative stress, SENP3 desumoylates Nrf2 leading to Nrf2 transcriptional activation in the nucleus (Figure 6).
A schema for the role of SENP3 in the regulation of Nrf2 activation.
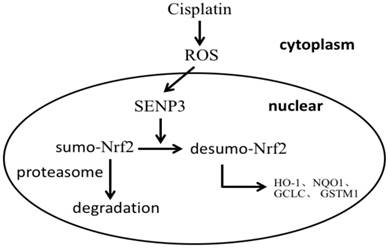
In conclusion, the results of the present study suggest that quiescent sumolyated Nrf2 activity is reinstalled by SENP3 desumoylation, which is activated by chemotherapy caused oxidative stress, leading to reduced cancer cell responses to chemotherapy.
Acknowledgements
The plasmid Flag-SENP3 was a gift from Pro. Jing Yi (Shanghai Key Laboratory of Tumor Microenvironment and Inflammation, Department of Biochemistry and Molecular Cell Biology, Institutes of Medical Sciences, Shanghai Jiao Tong University School of Medicine, Shanghai, China).
Financial Support
This work was supported by grants from Shanghai Municipal Science and Technology Commission (18ZR1423300) and Shanghai Pudong New Area Municipal Commission of Health and Family Planning (PW2017D-2).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45-49
2. Ganan-Gomez I, Wei Y, Yang H, Boyano-Adanez MC, Garcia-Manero G. Oncogenic functions of the transcription factor Nrf2. Free radical biology & medicine. 2013;65:750-64
3. Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras s ignaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158-68
4. Zhang J, Jiao Q, Kong L, Yu J, Fang A, Li M. et al. Nrf2 and Keap1 abnormalities in esophageal squamous cell carcinoma and association with the effect of chemoradiotherapy. Thorac Cancer. 2018;9:726-35
5. Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123-40
6. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D. et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663-73
7. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213-223
8. Chen W, Jiang T, Wang H, Tao S, Lau A, Fang D. et al. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal. 2012;17:1670-75
9. Malloy MT, McIntosh DJ, Walters TS, Flores A, Goodwin JS, Arinze IJ. Trafficking of the transcription factor Nrf2 to promyelocytic leukemia-nuclear bodies: implications for degradation of NRF2 in the nucleus. J Biol Chem. 2013;288:14569-83
10. Gong L, ET Yeh. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J Biol Chem. 2006;281:15869-77
11. Han Y, Huang C, Sun X, Xiang B, Wang M, ET Yeh. et al. SENP3-mediated de-conjugation of SUMO2/3 from promyelocytic leukemia is correlated with accelerated cell proliferation under mild oxidative stress. J Biol Chem. 2010;285:12906-15
12. Sun Z, Hu S, Luo Q, Ye D, Hu D, Chen F. Overexpression of SENP3 in oral squamous cell carcinoma and its association with differentiation. Oncol Rep. 2013;29:1701-6
13. Sang J, Yang K, Sun Y, Han Y, Cang H, Chen Y. et al. SUMO2 and SUMO3 transcription is differentially regulated by oxidative stress in an Sp1-dependent manner. The Biochemical journal. 2011;435:489-98
14. Yan S, Sun X, Xiang B, Cang H, Kang X, Chen Y. et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. The EMBO journal. 2010;29:3773-86
15. Yang J, Li H, Chen YY, Wang XJ, Shi GY, Hu QS. et al. Anthraquinones sensitize tumor cells to arsenic cytotoxicity in vitro and in vivo via reactive oxygen species-mediated dual regulation of apoptosis. Free radical biology & medicine. 2004;37:2027-41
16. Li C, Wu H, Wang S, Zhu J. Expression and correlation of NRF2, KEAP1, NQO-1 and HO-1 in advanced squamous cell carcinoma of the larynx and their association with clinicopathologic features. Mol Med Rep. 2016;14:5171-9
17. Baglam T, Sari M, Mine Yazici Z, Yuksel Z, Uneri C. Chemiluminescence assay of reactive oxygen species in laryngeal cancer. J Laryngol Otol. 2010;124:1091-94
18. Dwivedi R, Raturi D, Kandpal N, Dwivedi R, Singh R, Puri V. Oxidative stress in patients with laryngeal carcinoma. Indian J Cancer. 2008;45:97-99
19. Sahin U, de The H, Lallemand-Breitenbach V. PML nuclear bodies: assembly and oxidative stress-sensitive sumoylation. Nucleus. 2014;5:499-507
20. Sahin U, Lallemand-Breitenbach V, de The H. PML nuclear bodies: regulation, function and therapeutic perspectives. J Pathol. 2014;234:289-91
21. Chalkiadaki A, Talianidis I. SUMO-dependent compartmentalization in promyelocytic leukemia protein nuclear bodies prevents the access of LRH-1 to chromatin. Mol Cell Biol. 2005;25:5095-5105
22. Ohbayashi N, Kawakami S, Muromoto R, Togi S, Ikeda O, Kamitani S. et al. The IL-6 family of cytokines modulates STAT3 activation by desumoylation of PML through SENP1 induction. Biochem Biophys Res Commun. 2008;371:823-8
23. Casares C, Ramirez-Camacho R, Trinidad A, Roldan A, Jorge E, Garcia-Berrocal JR. Reactive oxygen species in apoptosis induced by cisplatin: review of physiopathological mechanisms in animal models. European archives of oto-rhino-laryngology. 2012;269:2455-59
24. Choi YM, Kim HK, Shim W, Anwar MA, Kwon JW, Kwon HK. et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PloS one. 2015;10:e0135083
25. Itoh T, Terazawa R, Kojima K, Nakane K, Deguchi T, Ando M. et al. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free radical research. 2011;45:1033-9
26. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS one. 2013;8:e81162
27. Zhang MM, Long YT, Ding Z. Cisplatin effects on evolution of reactive oxygen species from single human bladder cancer cells investigated by scanning electrochemical microscopy. Journal of inorganic biochemistry. 2012;108:115-22
Author contact
![]() Corresponding authors: Jiping Li and Yanqing Chen. Department of Otolaryngology, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China. No.160 Pujian Road, Pudong District, Shanghai 200127, China. Fax: +86 21 68383769; Tel: +86 21 68383769; E-mails: lijipingcom and doc_angelacom
Corresponding authors: Jiping Li and Yanqing Chen. Department of Otolaryngology, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China. No.160 Pujian Road, Pudong District, Shanghai 200127, China. Fax: +86 21 68383769; Tel: +86 21 68383769; E-mails: lijipingcom and doc_angelacom