Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(12):2745-2753. doi:10.7150/jca.31804 This issue Cite
Research Paper
miR-1226-3p Promotes Sorafenib Sensitivity of Hepatocellular Carcinoma via Downregulation of DUSP4 Expression
Xianqing Chen1,2,*, Wenliang Tan1,2,*, Wenxin Li1,2, Wenda Li1,2, Sicong Zhu1,3, Jinyi Zhong2, Changzhen Shang1,2, ![]() , Yajin Chen1,2,
, Yajin Chen1,2, ![]()
1. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
2. Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
3. Department of SICU, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
*These authors provided equal contribution to this work.
Received 2018-11-26; Accepted 2019-4-25; Published 2019-6-2
Abstract
Background: Sorafenib appears to increase the survival rate of hepatocellular carcinoma (HCC) patients, but its response rate is seriously limited due to drug resistance. Molecular mechanisms underlying sorafenib resistance are still unknown. Herein, we explored the possible role of miR-1226-3p in sorafenib resistance of HCC.
Methods: The miR-1226-3p expression level in HCC cell lines was evaluated by qRT-PCR. Cell viabilities to sorafenib were measured by CCK-8 assay. Cell apoptosis and proliferation were detected by flow cytometry and EdU proliferation assay. A luciferase reporter of DUSP4 3'-UTR was used for validation as a target gene of miR-1226-3p. Finally, the effects of in vivo antitumor efficacy of miR-1226-3p combined with sorafenib were evaluated by HCC tumor xenografts in nude mice.
Results: Bioinformatics analysis from Gene Expression Omnibus (GEO) datasets GSE56059 suggested that miR-1226-3p expression was downregulated in HCC patients who showed progressive disease (PD) after sorafenib treatment. SK-HEP-1 cells expressed lower levels of miR-1226-3p than HepG2 cells. We confirmed that SK-HEP-1 cells were more resistant to sorafenib compared to HepG2 cells. In addition, miR-1226-3p mimic increased cell apoptosis of SK-HEP-1 cells, whereas miR-1226-3p inhibitor significantly impaired cell apoptosis of HepG2 cells after sorafenib treatment. Moreover, we validated that miR-1226-3p directly targeted dual specificity phosphatase 4 (DUSP4), and further demonstrated that knockdown of DUSP4 reduced sorafenib resistance by regulating the JNK-Bcl-2 axis.
Conclusions: miR-1226-3p promotes sorafenib sensitivity of HCC through downregulation of DUSP4 expression, and targeting miR-1226-3p may be a novel therapeutic strategy for overcoming sorafenib resistance.
Keywords: miR-1226-3p, DUSP4, sorafenib resistance, hepatocellular carcinoma
Introduction
Liver cancer is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide, and HCC accounts for roughly 85% of all primary liver cancers [1]. Sorafenib, an oral multikinase inhibitor, is the first systemic drug for patients with advanced HCC [2]. Unfortunately, the effectiveness of sorafenib is limited on account of primary and acquired resistance [2]. Therefore, it is crucial for us to explore the molecular mechanisms underlying sorafenib resistance, as well as to evolve new strategies for the treatment of HCC patients.
As a set of small endogenously expressed non-coding RNAs of 18-22 nucleotides, microRNAs (miRNAs) inhibit translation of target mRNA through binding to their 3'untranslated regions (3'UTR) [3, 4]. Accumulating evidences revealed that the dysregulation of miRNAs is involved in HCC pathogenesis and drug resistance [5-8]. It has been reported that miR-7 overexpression can overcome sorafenib resistance in HCC [9]. Additionally, a previous study demonstrated that sorafenib resistance could be reversed by upregulation of miR-137 which downregulated ANT2 expression in HCC [10]. Other studies also reported that some miRNAs, such as miR-622, miR-486 and miR-338-3p, could affect the drug sensitivity of HCC [11-13]. Recently, limited studies revealed that miR-1226-3p functioned as a tumor suppressor and might be associated with tamoxifen resistance in breast cancer [14, 15]. However, its role in HCC remained largely unknown.
Dual specificity phosphatase 4 (DUSP4), also called MAPK phosphatase 2 (MKP2), is a member of the dual specificity phosphatase family and specifically dephosphorylates the MAP kinases ERK1/2, p38 and JNK acting as negative regulators of MAPK activity [16, 17]. The alterations of DUSP4/MKP2 expression have been reported to involve in tumor progression and drug resistance [18, 19]. It was well established that DUSP4 expression level upregulated in liver carcinomas [20]. Recently, a study has discovered that DUSP4 promotes doxorubicin resistance in gastric cancer by epithelial-mesenchymal transition (EMT) [21]. Furthermore, others demonstrated that knockdown of DUSP4 could increase the doxorubicin sensitivity of breast cancer cells [22]. Nevertheless, little is known about the role of DUSP4 in chemoresistance of HCC.
In the present study, we found that HCC patients or cells with higher expression of miR-1226-3p were more sensitive to sorafenib. We confirmed that DUSP4 was a target of miR-1226-3p, and further demonstrated that downregulation of DUSP4 significantly enhanced the anti-tumor efficiency of sorafenib in HCC cells. In addition, our results also showed that inhibiting DUSP4 markedly up-regulated the phosphorylation of JNK. Taken together, these data suggested that targeting miR-1226-3p or DUSP4 may improve therapeutic benefit to HCC patients receiving sorafenib treatment.
Materials and Methods
The Gene Expression Omnibus (GEO) data
The gene expression profile dataset GSE56059, which contained 20 micro-dissected hepatocellular carcinoma samples, was downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/). miRNA expression was correlated to clinical outcome and sorafenib-therapy. Differentially expressed miRNAs were analyzed between the sorafenib resistant HCC patients (response: progressive disease; PD) and sorafenib sensitive HCC patients (response: partial response; PR or stable disease; SD) by the GEO2R (fold changes > 2.0, p < 0.05).
Cell lines and cultures
HepG2 and SK-HEP-1 cells, obtained from the American Type Culture Collection (Manassas, VA, USA), were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Sijiqing, Zhejiang, China). All cells were fostered at 37°C in an atmosphere containing 5% CO2.
Cell viability assay
HepG2 and SK-HEP-1 cells were respectively seeded into a 96-well plate at a density of 1×104 and 0.5×104 cells/well, and treated with medium containing 10% FBS for 24 h for synchronization. Then different concentrations of sorafenib (Nexavar®) were added and the cells were cultured for 48 h. The viable cells were quantified using the Cell Counting Kit-8 (CCK-8; Beyotime, China) according to the manufacturer's protocol. Absorbance was measured at 450 nm using a microplate reader (Dynex, Chantilly, VA, USA). The absorbance value obtained from cell lines without sorafenib treatment (control) was normalized as 100%.
Cell transfection
HepG2 cells and SK-HEP-1 cells were transfected with miRNA-1226-3p inhibitor/negative control and miRNA-1226-3p mimic/negative control (RiboBio, Shanghai, China), respectively. Transfections were performed using the Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. DUSP4 siRNA and negative control siRNA (RiboBio, Shanghai, China) were transfected into SK-HEP-1 cells with Lipofectamine 2000.
Quantitative reverse transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from the cells using TRIzol reagent (Takara, Kusatsu, Japan) according to the manufacturer's instruction. Reverse transcription was performed with PrimeScript RT reagent Kit (Takara, Japan) based on the manufacturer's protocol. Next, qRT-PCR was determined with SYBR Prime Script RT-PCR Kits (Takara, Japan) to evaluate miR-1226-3p and DUSP4 expression level in different cell lines using the following primers: miR-1226-3p (forward: 5'-GCGGCTCACCAGCCCTGTGT-3'; reverse: 5'-CAGCCACAAAAGAGCACAAT-3'); DUSP4 (forward: 5'-AGCCGAGGAGAAAACTCTGG-3'; reverse: 5'-CCGTGT ATTTTTGCCGGTCG-3'); U6 (forward: 5'-CTCGCTTCGGCAGCACA-3'; reverse: 5'-AACGCTTCACGAATTTGCGT-3'); GAPDH (forward: 5'-ACAACTTTGGTATCGTGGAAGG-3'; reverse: 5'-GCCATCACGCCACAGTTTC-3'). U6 and GAPDH were used to normalize the level of miR-1226-3p and DUSP4 mRNA expression, respectively. The miR-1226-3p and DUSP4 level were calculated with the 2-ΔΔCt method. All assays were performed in triplicate. The expression levels were relative to the foldchange of the corresponding controls, which were defined as 1.0.
Western blot analysis
Cell proteins were extracted from cells lysed with lysis buffer (CWBIO, Beijing, China). The primary antibodies for PUMA, PARP, caspase-3, Bcl-2, Bax, ERK, JNK, p38, β-tubulin were purchased from Cell Signaling Technology (Danvers, MA, USA). DUSP4 antibody were purchased from Proteintech Group (Wuhan, China). Equal amounts of protein (20 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gelelectrophoresis (Beyotime, China) and then electrotransferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). After blocking in Tris-buffered saline/Tween 20 (TBST) containing 5% nonfat dry milk for 1 h, membranes were incubated with the primary antibody overnight at 4 ℃. The secondary antibodies (1:2000; abcam) were used for the detection of primary antibodies and the bands were visualized using an enhanced chemiluminescence detection kit (Millipore).
EdU Proliferation assay
Proliferation was performed using the BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime, China) according to the manufacturer's protocol. HepG2 and SK-HEP-1 cells were respectively seeded in a 6-well plate and incubated with 10% FBS-medium containing different concentrations of sorafenib for 48 h. Then, the cells were fixed in 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.3% Triton X-100 for 15 min at room temperature. Next, the cells were washed thrice using PBS, and cultured with 0.5 mL of Click Reaction Mixture for 30 min in the dark. Finally, cell nucleus was counterstained with 1.0 mL of 1× Hoechst (1:1000) for 10 min. The fluorescence microscope (Olympus, Tokyo, Japan) was used to count the numbers of proliferative cells (EdU-positive) in three random fields of view per slide.
Flow cytometric analysis of apoptosis
The percentage of cell apoptosis was determined using flow cytometry. Briefly, the cells were seeded in a 6-well plate and cultured for 48 h. Then, the cells were trypsinized and washed with PBS twice. Next, the cells were resuspended in 200 μL of 1× binding buffer and incubated with 5 μL of fluorescein isothiocyanate Annexin V (Clontech, Beijing, China) for 15 minutes at darkness. Finally, the cells were incubated 10 μL of propidium iodide (Clontech, Beijing, China) and detected by the flow cytometer (BD Biosciences, San Jose, CA). The data was analyzed using FlowJo software (BD Biosciences).
Dual luciferase reporter assay
DUSP4 3'-UTR luciferase reporter gene plasmid was constructed by GenePharma Inc (Shanghai, China). Plasmids named pDUSP4-WT and pDUSP4-Mut were generated via subcloning downstream of the luciferase vector. The HepG2 cells were co-transfected with pDUSP4-miR-1226-3p-UTR-WT or pDUSP4-miR-1226-3p-UTR-Mut by Lipofectamine2000. After 48 h, cells were assayed using Dual Luciferase Reporter Gene Assay Kit (Beyotime, China) according to the manufacturer's instructions. Luminescent signals were quantified with a luminometer (Centro XS3 LB 960; Berthold Technologies, Germany), and each value for firefly luciferase activity was normalized by renilla luciferase activity.
Mice xenograft models and immunohistochemistry analysis
Animal experiments were approved by the Animal Research Committee of Sun Yat-Sen University, and all procedures strictly followed the NIH Guide for Care and Use of Laboratory Animals. Female nude mice at age of 5-6 weeks were purchased from the Sun Yat-Sen University Laboratory Animal Center (Guangzhou, China). Approximately 5.0×106 SK-HEP-1/miR-1226-3p or SK-HEP-1/control cells were suspended in 100 μL PBS and injected subcutaneously into the right side of the posterior flank of female nude mice. When the average tumor size reached approximately 50 mm3, sorafenib (30 mg/kg) was administered via intraperitoneal injection every other day. All mice were killed after 2 weeks. The primary tumors were excised and analysed by immunohistochemistry of DUSP4 antibody (Proteintech Group, Wuhan, China) and PCNA antibody (Proteintech Group, Wuhan, China).
Statistical analysis
Statistical analysis was performed using SPSS 20.0 software (Chicago, IL, USA), and the experimental data were presented as mean ± S.D. Two-group comparisons were performed with Student's t-test. Multiple group comparisons were analyzed with one-way analysis of variance. p <0.05 was considered statistically significant.
Results
HepG2 cells express more miR-1226-3p compared to SK-HEP-1 cells, and are more sensitive to sorafenib than SK-HEP-1 cells
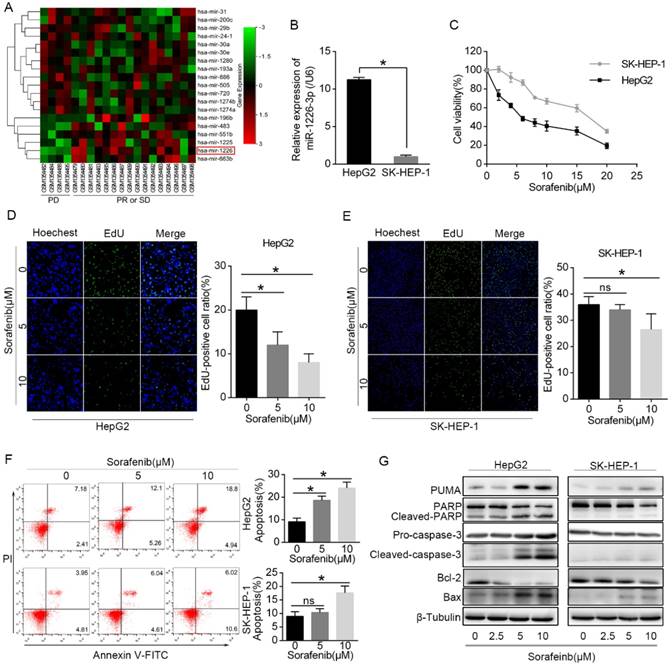
Differentially expressed miRNAs were analyzed between the sorafenib resistant HCC patients (response: PD) and sorafenib sensitive HCC patients (response: PR or SD). We found that miR-1226-3p expression was downregulated in HCC patients who showed progressive disease (PD) after sorafenib treatment (Fig 1A). Next, we examined the expression levels of miR-1226-3p in HCC cells by qRT-PCR, and discovered that HepG2 cells expressed higher levels of miR-1226-3p compared to SK-HEP-1 cells (Fig 1B). Cell viability assay was performed to detect the sorafenib sensitivity of HepG2 cells and SK-HEP-1 cells. CCK-8 assay results showed that sorafenib induced lower cell viability in HepG2 cells than in SK-HEP-1 cells (Fig 1C). Consistently, the EdU cell proliferation assay also revealed that sorafenib more significantly inhibited cell proliferation of HepG2 cells than SK-HEP-1 cells (Fig 1D, E). Meanwhile, we examined the cells apoptosis induced by sorafenib in HepG2 and SK-HEP-1 cells via flow cytometry, and found that sorafenib induced more apoptosis in HepG2 cells compared to SK-HEP-1 cells (Fig 1F). Then, western blot analysis showed that exposure to sorafenib significantly increased the expressions of PUMA, cleaved-PARP, cleaved-caspase-3 and Bax (pro-apoptotic protein), but decreased the expressions of Bcl-2 (anti-apoptotic protein) in HepG2 cells, which was scarcely observed in SK-HEP-1 cells (Fig 1G). Taken together, these data confirmed that HepG2 cells expressed higher levels of miR-1226-3p compared to SK-HEP-1 cells, and HepG2 cells were more sensitive to sorafenib than SK-HEP-1 cells.
HepG2 cells expressed higher levels of miR-1226-3p compared to SK-HEP-1 cells, and were more sensitive to sorafenib than SK-HEP-1 cells. (A) Using the GEO datasets (GSE56059), unsupervised hierarchical clustering of the miRNAs differentially expressed in sorafenib resistant HCC patients (response: PD) and sorafenib sensitive HCC patients (response: PR or SD) was constructed (fold changes > 2.0, p< 0.05). The red color scale (log2 foldchange) represents a higher expression level, and the green color scale represents a lower expression level. (B) The expression levels of miR-1226-3p in HepG2 and SK-HEP-1 cells were measured by qRT-PCR. (C) HepG2 and SK-HEP-1 cells were cultured with sorafenib (0-20 μM) for 48 h and cell viability was analyzed by CCK-8 assay. (D, E) EdU proliferation assays of HepG2 and SK-HEP-1 cells exposed to sorafenib (0, 5, 10μM) for 48 h were performed. (F) HepG2 and SK-HEP-1 cells were cultured with sorafenib (0, 5, 10μM) for 48 h, and the apoptosis rate was determined by flow cytometric analysis. (G) Western blot analysis of PUMA, PARP, caspase-3, Bcl-2, Bax and β-tubulin were performed after HepG2 cells and SK-HEP-1 cells treated with 0/2.5/5/10 µM sorafenib for 48 h. (PD: progressive disease; PR: partial response; SD: stable disease) (*p < 0.05).
miR-1226-3p promotes sorafenib sensitivity of HCC cells
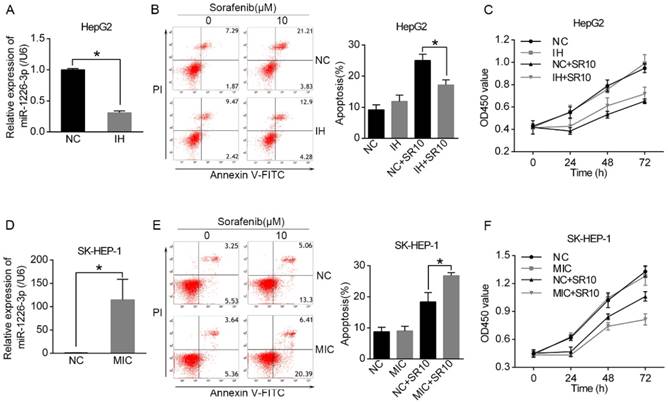
It has been reported that miR-1226-3p is associated with regulation of proliferation and drug resistance of cancer cells [14, 15]. As shown in Figure 1B, HepG2 cells expressed higher levels of miR-1226-3p compared to SK-HEP-1 cells. Then, we transfected miRNA-1226-3p inhibitor in HepG2 cells and miRNA-1226-3p mimic in SK-HEP-1 cells to investigate whether miR-1226-3p expression levels could affect sorafenib sensitivity (Fig 2A, D). Next, the rate of apoptosis was examined through flow cytometry analysis and the cell viability assay was tested by CCK-8. We found that sorafenib induced less apoptosis and higher cell viability of HepG2 cells after transfected miR-1226-3p inhibitor (Fig 2B, C). In addition, sorafenib-induced apoptosis was increased by ectopic expression of miR-1226-3p in SK-HEP-1 cells (Fig 2E). Furthermore, SK-HEP-1 cells which were co-treated with miR-1226-3p mimic and sorafenib showed lower cell viability (Fig 2F). These results suggested that the upregulation of miR-1226-3p promoted sorafenib sensitivity of HCC cells.
The miR-1226-3p expression was related to sorafenib sensitivity of HCC. (A, D) miR-1226-3p expression levels were analyzed by qRT-PCR after HepG2 cells transfected with miR-1226-3p inhibitor/negative control and SK-HEP-1 cells transfected with miR-1226-3p mimic/negative control. (B, C) The apoptosis and viability of HepG2 cells transfected with miRNA-1226-3p inhibitor/negative control were respectively measured by flow cytometric analysis and CCK-8 assay after treating with 0 or 10 µM sorafenib for 48 h. (E, F) The flow cytometric analysis and CCK-8 assay were performed to analyze the apoptosis and viability of SK-HEP-1 cells transfected with miRNA-1226-3p mimic/negative control after treating with 0 or 10 µM sorafenib for 48 h. (NC: negative control. IH: miRNA-1226-3p inhibitor. MIC: miRNA-1226-3p mimic. SR10: 10 µM sorafenib) (*p < 0.05)
miR-1226-3p downregulates DUSP4 expression
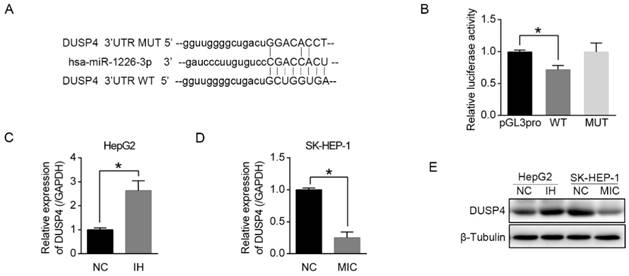
To elucidate the mechanism by which miR-1226-3p promoted sorafenib sensitivity in HCC, we performed the database analysis using two publicly available bioinformatics tools (TargetScan and OncomiR). We searched the candidates in apoptosis-related genes containing potential binding sites for miR-1226-3p in their 3'-UTRs and found DUSP4 as a target gene of miR-1226-3p. The predicted interactions between miR-1226-3p and the 3'-UTRs of DUSP4 are shown in Figure 3A. Next, we constructed a luciferase assay using reporter plasmid containing wild type (WT) or mutant (MUT) sequence of 3'UTR region of DUSP4. As shown in Figure 3B, ectopic expression of WT miR-1226-3p significantly decreased luciferase activity levels compared to control, whereas that of MUT miR-1226-3p did not. Furthermore, transfection of miR-1226-3p inhibitor in HepG2 cells increased DUSP4 mRNA and protein expression levels, while transfection of miR-1226-3p mimic in SK-HEP-1 cells decreased them (Fig 3C-E). These results demonstrated that DUSP4 was a target of miR-1226-3p and miR-1226-3p could directly downregulates DUSP4 expression.
miR-1226-3p downregulated DUSP4 expression. (A) The binding sites for miR-1226-3p in the 3'-UTRs of DUSP4 was determined. (B) HepG2 cells were co-transfected miR-1226-3p with pGL3-3'UTR DUSP4 (wild type or mutated) or pGL3-promoter vector (control), and the luciferase reporter assays were conducted to confirm the relationships between miR-1226-3p and DUSP4. (C, D) Relative expression of DUSP4 was measured respectively by qRT-PCR after HepG2 cells transfected with miRNA-1226-3p inhibitor/negative control and SK-HEP-1 cells transfected with miRNA-1226-3p mimic/negative control. (E) Western blot analysis of DUSP4 and β-tubulin were performed. (NC: negative control. IH: miRNA-1226-3p inhibitor. MIC: miRNA-1226-3p mimic) (*p < 0.05)
Knockdown of DUSP4 increases sorafenib sensitivity and activates the JNK signaling pathway
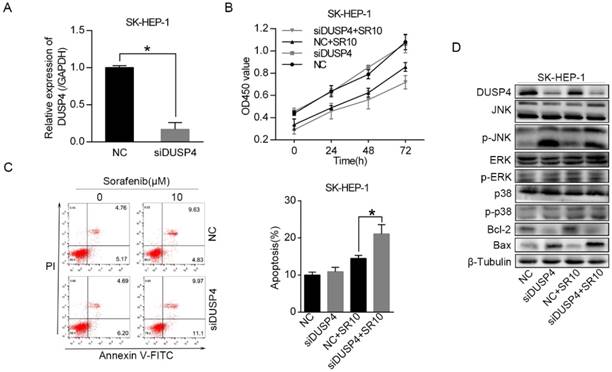
To test whether DUSP4, a target gene of miR-1226-3p, could regulate sorafenib sensitivity in HCC, we knocked down DUSP4 using DUSP4 siRNA in SK-HEP-1 cells (Fig 4A). Then, cell viability assay and the flow cytometry analysis were measured. We observed that knockdown of DUSP4 induced lower cell viability and more apoptosis in SK-HEP-1 cells after sorafenib treatment (Fig 4B, C). It has been reported that DUSP4 can dephosphorylate the MAP kinases ERK1/2, p38 and JNK [16]. Thus, we performed western blot analysis to explore the relationship between DUSP4 and the MAP kinases. After the transfection of DUSP4 siRNA, DUSP4 protein expression level was remarkably downregulated (Fig 4D). Moreover, knockdown of DUSP4 also notably increased the protein levels of phosphorylated JNK in SK-HEP-1 cells exposed to sorafenib or not, but the levels of ERK and p38 kinase phosphorylation were not distinctly altered (Fig 4D). Additionally, we discovered that Bax protein levels was increased and Bcl-2 protein levels was decreased in SK-HEP-1 cells after knockdown of DUSP4 (Fig 4D). These results revealed that knockdown of DUSP4 promoted sorafenib sensitivity and activated the JNK signaling pathway in SK-HEP-1 cells.
Knockdown of DUSP4 increased sorafenib sensitivity and activated the JNK signaling pathway. (A) DUSP4 was knocked down in SK-HEP-1 cells by siRNA and qRT-PCR was performed to analyze DUSP4 expression. (B, C) The viability and apoptosis of SK-HEP-1 cells transfected with DUSP4 siRNA or negative control siRNA were respectively measured by CCK-8 assay and flow cytometric analysis after treating with 0 or 10 µM sorafenib for 48 h. (D) Western blot analysis of DUSP4, JNK, ERK, p38, Bcl-2, Bax and β-tubulin were performed. (NC: negative control. siDUSP4: DUSP4 siRNA. SR10: 10 µM sorafenib) (*p < 0.05)
miR-1226-3p overexpression promotes the in vivo antitumor effects of sorafenib
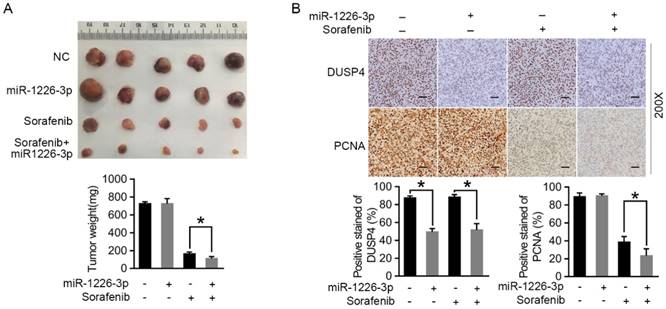
To assess the effect of miR-1226-3p on sorafenib sensitivity of HCC cells in vivo, we constructed the SK-HEP-1 cells which could stably expressed miR-1226-3p or miR-NC by miR-1226-3p plasmid (pcDNA/miR-1226-3p) or negative control plasmids (pcDNA/miR-NC), and evaluated the effect in the mouse subcutaneous injection mode. As shown in Figure 5A, tumors derived from mice inoculated with SK-HEP-1/miR-1226-3p were more sensitive to sorafenib therapy than tumors derived from mice inoculated with SK-HEP-1/NC. In addition, immunohistochemistry analysis revealed that DUSP4 expression levels were significantly reduced in SK-HEP-1/miR-1226-3p xenografts compared to SK-HEP-1/NC xenografts with or without sorafenib (Fig 5B). There were no different PCNA positivity in SK-HEP-1/miR-1226-3p xenografts and SK-HEP-1/NC xenografts without sorafenib, whereas it was lower in SK-HEP-1/miR-1226-3p xenografts than in SK-HEP-1/NC xenografts with sorafenib therapy (Fig 5B). Taken together, these results showed that miR-1226-3p overexpression could promote the in vivo anti-tumor effects of sorafenib.
miR-1226-3p overexpression promoted the in vivo antitumor effects of sorafenib. (A) Representative photos of tumors at endpoint. Graph showing the weight of tumors generated in each group. (B) DUSP4 and PCNA expression levels were analysed by immunohistochemistry in SK-HEP-1/miR-1226-3p xenografts and SK-HEP-1/control xenografts with or without sorafenib treatment. Scale bars: 50 µm (200×) (NC: negative control) (*p < 0.05).
Discussion
Sorafenib is currently regarded as the first-line treatment option for advanced HCC patients, whereas the development of drug resistance often prevents its long-term efficacy. Numerous reports demonstrated that the dysregulation of miRNAs was involved in sorafenib resistance of HCC [9, 11, 23-26]. In recent years, some studies have showed that miR-1226-3p exerts antitumor functions and is related to tamoxifen resistance in human breast cancer [14, 15]. However, the role of miR-1226-3p in HCC has not been characterized.
By bioinformatics analysis from the GEO database, we found that HCC patients with low expression of miR-1226-3p were more resistant to sorafenib. Meanwhile, our results showed that miR-1226-3p expression levels were higher in HepG2 cells than in SK-HEP-1 cells. Interestingly, it has been illustrated that HepG2 cells are more sensitive to sorafenib compared to SK-HEP-1 cells [27], and our results also confirmed this conclusion. These data indicated that miR-1226-3p might affect the sorafenib sensitivity of HCC. Consequently, our results showed that downregulation of miR-1226-3p expression decreased sorafenib sensitivity, while upregulation of miR-1226-3p expression increased sorafenib sensitivity in HCC. In vivo, miR-1226-3p overexpression also enhanced the antitumor effects of sorafenib against HCC. To the best of our knowledge, this is the first study which systematically investigates the possible relationship between miR-1226-3p and sorafenib resistance of HCC. Our findings indicate that miR-1226-3p may serve as a novel predictor of sorafenib sensitivity in HCC patients.
DUSP4 is an inducible nuclear phosphatase whose primary function is to regulate the MAPK pathway. Some reports have showed that DUSP4 plays an essential role in the regulation of cancer cells and drug resistance [28, 29]. However, the role of DUSP4 in cancer is contradictory. Some studies described that DUSP4 is a cancer suppressor gene, and that loss or mutation of DUSP4 is involved in the growth of a variety of tumors, including breast cancer, large B cell lymphoma and other primary tumors [29-31]. However, other studies reported that DUSP4 is tumorigenic and overexpression of DUSP4 promotes cancer development and progression [19, 32]. Otília Menyhart et al. found that DUSP4 was associated with increased resistance against anti-HER2 therapy in breast cancer [33]. Moreover, it has been reported that knockdown of DUSP4 increases doxorubicin sensitivity of breast cancer cells [22]. In the present study, we also discovered that knockdown of DUSP4 could enhance sorafenib sensitivity of HCC. Meanwhile, we had demonstrated that miR-1226-3p overexpression could directly downregulate DUSP4 expression, which significantly enhanced sorafenib sensitivity of HCC. Thus, our results suggested the possible mechanism that miR-1226-3p enhanced sorafenib sensitivity of HCC through downregulating DUSP4 expression.
Resistance to sorafenib is a major challenge to improve HCC patient prognosis. Many factors, including the JNK signaling pathway, play a pivotal role in sorafenib resistance of HCC [34-36]. The Jun N-terminal kinase (JNK) is often deregulated in cancers affecting proliferation, differentiation, apoptosis and migration, and can both phosphorylate and regulate the expression of several members of the Bcl-2 protein family, such as Bax and Bcl-2 [37, 38]. Recently, a study has demonstrated that JNK activation can enhance sorafenib sensitivity of HCC [39]. In addition, Ying-Ying Liang reported that JNK activation improved the anti-tumor effects of sorafenib in renal cell carcinoma [40]. Consistent with these previous reports, we had found that the protein levels of phosphorylated JNK was significantly increased after knockdown of DUSP4, and the anti-tumor effects of sorafenib in HCC was also promoted. Besides, we noted that Bcl-2 protein levels were distinctly decreased probably because of the JNK activation after transfection of DUSP4 siRNA. Several reports have confirmed that Bcl-2 family proteins can affect the HCC cells response to sorafenib [8, 24], and the repression of Bcl-2 enhance sorafenib-induced apoptosis in malignant cancer cells [41]. Therefore, based on our results, we might draw a conclusion that miR-1226-3p promoted sorafenib sensitivity of HCC due to the JNK activation and Bcl-2 suppression.
In summary, we demonstrated that miR-1226-3p promoted sorafenib sensitivity of HCC by downregulation of DUSP4 expression in vitro and in vivo, which providing us with a novel understanding of the mechanisms underlying sorafenib resistance. Further research is necessary to assess the roles of miR-1226-3p in the clinical context and determine the side effects of interfering with miR-1226-3p regulation in animals or humans.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (no. 81672419; no. 81572398), the Science and Technology Planning Project of Guangdong Province (no. 2017A010105003; no. 2016A020216010; no. 2015A050502023). Yajin Chen and Changzhen Shang designed the experiments; Xianqing Chen and Wenliang Tan performed the experiments and wrote the manuscript; Wenxin Li and Wenda Li revised the manuscript; Sicong Zhu interpreted the data and Jinyi Zhong made the pictures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol Res. 2013;43:147-54
3. Fabris L, Ceder Y, Chinnaiyan AM, Jenster GW, Sorensen KD, Tomlins S. et al. The Potential of MicroRNAs as Prostate Cancer Biomarkers. Eur Urol. 2016;70:312-22
4. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
5. Vasuri F, Visani M, Acquaviva G, Brand T, Fiorentino M, Pession A. et al. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J Gastroenterol. 2018;24:2647-60
6. Fornari F, Milazzo M, Galassi M, Callegari E, Veronese A, Miyaaki H. et al. p53/mdm2 feedback loop sustains miR-221 expression and dictates the response to anticancer treatments in hepatocellular carcinoma. Mol Cancer Res. 2014;12:203-16
7. Pollutri D, Patrizi C, Marinelli S, Giovannini C, Trombetta E, Giannone FA. et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis. 2018;9:4
8. Yang F, Li QJ, Gong ZB, Zhou L, You N, Wang S. et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol Cancer Res Treat. 2014;13:77-86
9. Kabir TD, Ganda C, Brown RM, Beveridge DJ, Richardson KL, Chaturvedi V. et al. A microRNA-7/growth arrest specific 6/TYRO3 axis regulates the growth and invasiveness of sorafenib-resistant cells in human hepatocellular carcinoma. Hepatology. 2018;67:216-31
10. Lu AQ, Lv B, Qiu F, Wang XY, Cao XH. Upregulation of miR-137 reverses sorafenib resistance and cancer-initiating cell phenotypes by degrading ANT2 in hepatocellular carcinoma. Oncol Rep. 2017;37:2071-8
11. Dietrich P, Koch A, Fritz V, Hartmann A, Bosserhoff AK, Hellerbrand C. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut. 2018;67:1328-41
12. Sun H, Cui C, Xiao F, Wang H, Xu J, Shi X. et al. miR-486 regulates metastasis and chemosensitivity in hepatocellular carcinoma by targeting CLDN10 and CITRON. Hepatol Res. 2015;45:1312-22
13. Xu H, Zhao L, Fang Q, Sun J, Zhang S, Zhan C. et al. MiR-338-3p inhibits hepatocarcinoma cells and sensitizes these cells to sorafenib by targeting hypoxia-induced factor 1alpha. PLoS One. 2014;9:e115565
14. Jin C, Rajabi H, Kufe D. miR-1226 targets expression of the mucin 1 oncoprotein and induces cell death. Int J Oncol. 2010;37:61-9
15. Zhou Q, Zeng H, Ye P, Shi Y, Guo J, Long X. Differential microRNA profiles between fulvestrant-resistant and tamoxifen-resistant human breast cancer cells. Anticancer Drugs. 2018;29:539-48
16. Low HB, Zhang Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016;16:85-98
17. Gaggianesi M, Turdo A, Chinnici A, Lipari E, Apuzzo T, Benfante A. et al. IL4 Primes the Dynamics of Breast Cancer Progression via DUSP4 Inhibition. Cancer Res. 2017;77:3268-79
18. Cushman SM, Jiang C, Hatch AJ, Shterev I, Sibley AB, Niedzwiecki D. et al. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: results from CALGB 80203 (Alliance). Clin Cancer Res. 2015;21:1078-86
19. Kim H, Jang SM, Ahn H, Sim J, Yi K, Chung Y. et al. Clinicopathological significance of dual-specificity protein phosphatase 4 expression in invasive ductal carcinoma of the breast. J Breast Cancer. 2015;18:1-7
20. Caroline Nunes-Xavier CR-MPR. Dual-Specificity MAP Kinase Phosphatases as Targets of Cancer Treatment. Anti-Cancer Agents in Medicinal Chemistry. 2011;11:109-32
21. Kang X, Li M, Zhu H, Lu X, Miao J, S" D. et al. DUSP4 promotes doxorubicin resistance in gastric cancer through epithelial-mesenchymal transition. Oncotarget. 2017;8:94028-39
22. Liu Y, Du F, Chen W, Yao M, Lv K, Fu P. Knockdown of dual specificity phosphatase 4 enhances the chemosensitivity of MCF-7 and MCF-7/ADR breast cancer cells to doxorubicin. Exp Cell Res. 2013;319:3140-9
23. Mao K, Zhang J, He C, Xu K, Liu J, Sun J. et al. Restoration of miR-193b sensitizes Hepatitis B virus-associated hepatocellular carcinoma to sorafenib. Cancer Lett. 2014;352:245-52
24. Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A. et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52:698-704
25. Xu J, Lin H, Li G, Sun Y, Chen J, Shi L. et al. The miR-367-3p Increases Sorafenib Chemotherapy Efficacy to Suppress Hepatocellular Carcinoma Metastasis through Altering the Androgen Receptor Signals. EBioMedicine. 2016;12:55-67
26. Xu Y, Huang J, Ma L, Shan J, Shen J, Yang Z. et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371:171-81
27. Tan W, Zhu S, Cao J, Zhang L, Li W, Liu K. et al. Inhibition of MMP-2 Expression Enhances the Antitumor Effect of Sorafenib in Hepatocellular Carcinoma by Suppressing the PI3K/AKT/mTOR Pathway. Oncol Res. 2017;25:1543-53
28. Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE. et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. 2012;18:1052-9
29. Balko JM, Schwarz LJ, Bhola NE, Kurupi R, Owens P, Miller TW. et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013;73:6346-58
30. Saigusa S, Inoue Y, Tanaka K, Toiyama Y, Okugawa Y, Shimura T. et al. Decreased expression of DUSP4 is associated with liver and lung metastases in colorectal cancer. Med Oncol. 2013;30:620
31. Schmid CA, Robinson MD, Scheifinger NA, Muller S, Cogliatti S, Tzankov A. et al. DUSP4 deficiency caused by promoter hypermethylation drives JNK signaling and tumor cell survival in diffuse large B cell lymphoma. J Exp Med. 2015;212:775-92
32. Groschl B, Bettstetter M, Giedl C, Woenckhaus M, Edmonston T, Hofstadter F. et al. Expression of the MAP kinase phosphatase DUSP4 is associated with microsatellite instability in colorectal cancer (CRC) and causes increased cell proliferation. Int J Cancer. 2013;132:1537-46
33. Menyhart O, Budczies J, Munkacsy G, Esteva FJ, Szabo A, Miquel TP. et al. DUSP4 is associated with increased resistance against anti-HER2 therapy in breast cancer. Oncotarget. 2017;8:77207-18
34. Niu L, Liu L, Yang S, Ren J, Lai PBS, Chen GG. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim Biophys Acta Rev Cancer. 2017;1868:564-70
35. Nguyen TV, Sleiman M, Moriarty T, Herrick WG, Peyton SR. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials. 2014;35:5749-59
36. Lee S, Lee M, Kim JB, Jo A, Cho EJ, Yu SJ. et al. 17beta-estradiol exerts anticancer effects in anoikis-resistant hepatocellular carcinoma cell lines by targeting IL-6/STAT3 signaling. Biochem Biophys Res Commun. 2016;473:1247-54
37. Bubici C, Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol. 2014;171:24-37
38. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537-49
39. Feng XQ, Rong LW, Wang RX, Zheng XL, Zhang L, Zhang L. et al. Luteolin and sorafenib combination kills human hepatocellular carcinoma cells through apoptosis potentiation and JNK activation. Oncol Lett. 2018;16:648-53
40. Liang YY, Deng XB, Zeng LS, Lin XT, Shao XF, Wang B. et al. RASSF6-mediated inhibition of Mcl-1 through JNK activation improves the anti-tumor effects of sorafenib in renal cell carcinoma. Cancer Lett. 2018;432:75-83
41. Rahmani M, Aust MM, Attkisson E, Williams DC Jr, Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089-98
Author contact
![]() Corresponding authors: Changzhen Shang and Yajin Chen. Address correspondence to Changzhen Shang, Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, No.107 Yanjian West Road, Yuexiu District, Guangzhou, Guangdong 510120, China. E-mail: shangcz_sysucom or Yajin Chen, Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, Guangzhou 510120, China. E-mail: chenyajsysu.edu.cn
Corresponding authors: Changzhen Shang and Yajin Chen. Address correspondence to Changzhen Shang, Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, No.107 Yanjian West Road, Yuexiu District, Guangzhou, Guangdong 510120, China. E-mail: shangcz_sysucom or Yajin Chen, Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, Guangzhou 510120, China. E-mail: chenyajsysu.edu.cn