Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(11):2541-2551. doi:10.7150/jca.29265 This issue Cite
Research Paper
The oncogenic potential of PRR11 gene in Tongue Squamous Cell Carcinoma cells
Chunyang Wang1,2*, Liang Yu3*, Xianyue Ren1,2*, Tong Wu1,2, Xijuan Chen1,2, Yulei Huang1,2, Bin Cheng1,2 ![]()
1. Guanghua School of Stomatology, Hospital of Stomatology, Sun Yat-sen University, Guangzhou, Guangdong, P.R. China
2. Guangdong Provincial Key Laboratory of Stomatology, Guangzhou, Guangdong 510060, P.R. China
3. Department of Thyroid and Breast Surgery, The First Affiliated Hospital of Sun Yat‑sen University, Guangzhou, Guangdong 510080, P.R. China
*Chunyang Wang, Liang Yu and Xianyue Ren contribute equally to the work.
Received 2018-8-16; Accepted 2019-4-12; Published 2019-6-2
Abstract
Background: We previously demonstrated that Proline rich 11 (PRR11) gene is associated with the development and progression of tongue squamous cell carcinoma (TSCC), but the underlying mechanism is unknown. This study aimed to investigate the molecular mechanism underlying oncogenic potential of PRR11 in TSCC cells.
Methods: Overexpression and knockdown of PRR11 were performed by plasmid transfection into SCC15 and HSC3 human TSCC cells. Expressions of mRNA and protein were assessed by qRT-PCR and Western blot, respectively. Cell proliferation and invasion were determined by CCK-8 and Transwell assay, respectively. In vivo tumor growth and cell cycle were determined by a nude mice model of subcutaneous tumorigenesis and flow cytometry, respectively.
Results: Overexpression of PRR11 significantly enhanced TSCC cells proliferation and the invasive ability of TSCC cells, whereas PRR11 knockdown in TSCC cells exhibited a reverse trend. In addition, the in vivo subcutaneous tumorigenicity assay showed that PRR11 knockdown significantly reduced tumor size and the Ki67 (a proliferation marker)expression in the tumor tissue. Flow cytometry analysis revealed that PRR11 overexpression significantly decreased the proportion of cells in S phase, whereas PRR11 knockdown in TSCC cells exhibited a reverse trend. Furthermore, PRR11 overexpression simultaneously down-regulated two cyclin-dependent kinase inhibitors (CKIs), p21 and p27 and up-regulated CDK2 and Cyclin A2 in TSCC cells. PRR11 knockdown again exhibited reverse trends of expressions of the above proteins.Conclusion: These results suggested that PRR11 promoted cell proliferation by regulating the expressions of p21, p27, CDK2 and Cyclin A to facilitate S/G phase transition in TSCC cells.
Keywords: Proline rich 11 (PRR11), tongue squamous cell carcinoma (TSCC), oncogene, S phase arrest
Introduction
Tongue squamous cell carcinoma (TSCC) is the most common type of oral and maxillofacial malignant tumors [1]. Due to the rich blood supply and frequent movement characteristics of the tongue, TSCC patients have a high risk of early cervical lymph node metastasis and a relatively poor prognosis [2]. Current treatment options for TSCC mainly include surgical resection, radiotherapy and chemotherapy. Even though the treatment outcomes for TSCC have been extensively improved in recent decades, the 5-year survival rate of TSCC remains unsatisfied [3,4]. Further investigating the molecular mechanisms of tumorigenesis of tongue cancer may help to develop new diagnostic and treatment strategies which can improve the survival rate and quality of life of TSCC patients.
Proline rich 11 (PRR11) is a tumor-associated gene located on chromosome 17q22 (5), encoding a 360-amino acid protein. In normal tissues, PRR11 gene is expressed in a very low level, but is significantly up-regulated in several tumors, such as lung [5], breast [6], gastric [7], pancreatic cancers [8], and cholangiocarcinoma [9]. In addition, PRR11 expression has been shown to be associated with the development and progression of cancers, and could be used as a prognostic indicator for these cancers [5-7,9].
As for TSCC, we have previously demonstrated that PRR11 mRNA and protein expression is markedly upregulated in surgically resected human TSCC tissues [10]. Immunohistochemical analysis in 72 paraffin-embedded TSCC specimens reveals that PRR11 expression level is significantly associated with the clinical stage, T classification, N classification of the tumor as well as the survival outcome [10]. Kaplan-Meier survival analysis suggests that patients with high-PRR11 expression in TSCC have shorter survival times as compared with those with low-PRR11 expression. Univariate and multivariate analyses indicated that PRR11 upregulation is an independent risk factor for the overall survival of TSCC patients [10]. These observations suggest that PRR11 is involved in the development and progression of TSCC. However, the molecular mechanism of the effect of PRR11 in TSCC remains to be investigated. Therefore, the purpose of the present study was to elucidate the molecular mechanism underlying oncogenic potential of PRR11 in TSCC.
Materials and Methods
Cell culture
Human TSCC cell line SCC15 was purchased from ATCC cell bank (USA). Human TSCC cell line CAL-27 was a gift from Professor Musheng Zeng (Cancer Center of Sun Yat-sen University, China). Human TSCC cell lines HSC3 and HSC4 were kindly provided by Professor Qianming Chen (Sichuan University, China). Human TSCC cell lines UM1, UM2 were gifts from Professor Hongzhang Huang (Oral and Maxillofacial Surgery department, Sun Yat-sen University, China). Human immortalized normal oral epithelial cells (NOK) and human TSCC cell line HSC-6 were donated by Professor J. Silvio Gutkind (National Institute of Dental and Craniofacial Research, USA).
The NOK cells were cultured in serum-free KSFM medium (Invitrogen, USA). CAL27, HSC-3, HSC-4, and HSC-6 cells were cultured in DMEM medium (Hyclone, USA) containing 10% FBS (Hyclone). The SCC15, UM1, and UM2 cells were cultured in DMEM/F12 (1:1) medium (Hyclone) containing 10% FBS. Cells in logarithmic growth phase were used for the experiments.
Quantitative Real-time PCR (qRT-PCR)
Total RNA was isolated using the TRIzol reagent according to the manufacturer's protocol (Invitrogen, USA). The reverse transcription was performed on 1 µg of total RNA in a final volume of 13 µl using Transcriptor First Strand cDNA Synthesis Kit (Roche, USA) following the manufacturer's instructions. Quantitative real-time PCR was carried out in triplicate by using the SYBR Green I Master (Roche) on LightCycler® 480 System (Roche). The primers set used for PRR11 were forward: 5'-GACTTCCAAAGCTGTGCTTCC-3' and reverse: 5'-CTGCATGGGTCCATCCTTTTT-3'; for 18S rRNA, forward: 5'- CCTGGATACCGCAGCTAGGA-3', reverse: 5'- GCGGCGCAATACGAATGCCCC-3'. The mRNA level was normalized to the 18S rRNA transcript level. The expression fold change of PRR11 was calculated for each sample using the 2-ΔΔC method.
Vectors construction for overexpression and knockdown of PRR11
For overexpression, PRR11 cDNA was ligated into pcGFP plasmid at BamHⅠ and EcoRⅠ sites to construct pcGFP-PRR11 plasmid. For PRR11 knockdown, two siRNAs against PRR11 were designed, and an unrelated (scrambled) sequence was used as a negative control (Scr) (Table 1). The shRNA template was generated by PCR at the following condition: 95 °C 5 min, 95 °C 30 sec, 70 °C 30 sec, 50 °C 2 min, 4 °C preservation. The shRNA was ligated into the shRNA plasmid expression vector pGPU6/GFP/Neo (GenePharma, Shanghai, China) at the BamH I and Bbs sites. The plasmid was transfected into HSC3 or SCC15 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
siRNA Primer sequences
| PRR11 RNAi#1 Primer F | CACCGTCTCGCTAAAGCACTTCAGGTTCAAGAGACCTGAAGTGCTTTAGCGAGACTTTTTTG |
|---|---|
| PRR11 RNAi#1 Primer R | GATCCAAAAAAGTCTCGCTAAAGCACTTCAGGTCTCTTGAACCTGAAGTGCTTTAGCGAGAC |
| PRR11 RNAi#2 Primer F | CACCGGATCTGCGGAAACTGCTTAGTTCAAGAGACTAAGCAGTTTCCGCAGATCCTTTTTTG |
| PRR11 RNAi#2 Primer R | GATCCAAAAAAGGATCTGCGGAAACTGCTTAGTCTCTTGAACTAAGCAGTTTCCGCAGATCC |
| Scr F | CACCGTTCTCCGAACGTGTCACGTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTG |
| Scr R | GATCCAAAAAATTCTCCGAACGTGTCACGTAATCTCTTGACGTGACACGTTCGGAGAAC |
Western Blot
The TSCC cells transfected with pcGFP-PRR11, shRNA plasmid or control plasmid for 48 h were collected in RIPA buffer (Shanghai Biocolor Bioscience & Technology Co., Ltd, China) with 10% phosphatase inhibitor (Roche, Switzerland) on ice for lysis. After centrifugation at 12,000 ×g, 4 °C for 20 min, the resulting supernatant was collected. The protein concentration was determined by BCA Protein Assay Kit (Beijing Kangwei Century Biotechnology Co., Ltd., China). Equal amount of total protein was separated on an SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, USA). The membrane was blocked with 5% non-fat milk in Tris-buffered saline/Tween 20 (TBST) buffer for 1 h at room temperature, followed by incubated with primary antibodies at 4 °C overnight. Primary antibodies included rabbit anti-human PRR11 polyclonal antibody (1:250, NOVUS, USA), rabbit anti-human Cyclin A2 polyclonal antibody (1:1000, Santa Cruz, USA), rabbit anti-human Cyclin B1 monoclonal antibody, (1:1000, Cell Signaling, USA), rabbit anti-human Cyclin D1 monoclonal antibody (1:1000, Cell Signaling), rabbit anti-human monoclonal antibody p21 (1:1000, Cell Signaling), rabbit anti-human monoclonal antibody p27 (1:1000, Cell Signaling), rabbit anti-human monoclonal antibody β-actin antibody (1:1000, Abcam, USA) were used in this study. Secondary antibody was peroxidase-conjugated goat anti-rabbit IgG (1:5000, Cell Signaling). Immunoreactive bands were visualized using Chemiluminescence Reagent Kit (Abcam, USA) in a CCD-camera imaging system (GE, USA). The bands were quantified by using ImageJ software (NIH, USA).
CCK-8 assay
Cell proliferation was assessed using Cell Counting Kit-8 (CCK-8; Guangzhou Weijia Bio Co., Ltd., China) according to the manufacturer's instruction. At 24 h prior to transfection, HSC3 or SCC15 cells (2×104 cells/100 µl/well) were seeded in a 96-well plate. Plasmid (0.1 μg/well) was transfected into cells using Lipofectamine 2000. At 0, 24, 48, or 72 h after transfection, 10 μl of CCK-8 solution was added to each well. The plates were incubated for another 4 h at 37 °C, and the optical density was measured using a microplate reader (Thermo Electron, USA) at 450 nm.
Transwell invasion assay
The invasion ability of SCC15 and HSC3 cells was determined by transwell invasion assay. After 48 h transfection, cells (3×104 cells, in 100 µl serum-free medium) were added to the upper chamber pre-coated with Matrigel (Gibco, USA) in a 24-well plate, in which the lower chamber contained 600 µl of culture medium with 15% FBS. After 24 h, the invaded cells were fixed with 4% paraformaldehyde for 20 min, followed by stained with 0.4% crystal violet solution. The cells were photographed under an inverted microscope (50× magnification, Olympus, Japan). Five fields were randomly selected for counting the invaded cells. The experiment was performed in triplicate 3 times to obtain the averaged value for statistical analysis.
Flow cytometry
Cell cycle of the TSCC cells was analyzed by flow cytometry. At 24 h before transfection, 2×105 cells in 2 mL culture medium were seeded onto a well of 6-well plate, and cells were cultured with serum-free medium. A total of 2 μg of plasmid (per well) was transfected into cells. After incubation for 4 h, the medium was changed to complete medium containing 10% FBS, and cultured for an additional 72 h. The cells were collected and treated with 70% ethanol for 1 h, followed by stained with 500 μl of PI solution (containing 0.2 mg/mL RNase A, 0.1% Triton X-100) for 30 min. Cell cycle was analyzed by using a flow cytometer (Cytomics FC500 MCL, Beckman coulter, USA), and the data were analyzed by Modfit software (Becton Dickinson, USA).
Colony forming assay
Cells (5 × 102 cells/well) were plated in 6-well plates and cultured for 10 days. After fixation with 4% formaldehyde for 5 minutes, the colonies were stained with 1% crystal violet for 30 seconds. Colonies were counted and the results were shown as the fold change compared to vector control cells.
Anchorage-independent growth ability assay
Five hundred cells were suspended in 2 mL complete medium with 0.3% agar and plated on the top of 1% complete medium agar mixture. After 10 days, viable colonies that contained more than 50 cells or were larger than 0.1 mm were counted. All experiments were performed in triplicate.
Bromodeoxyuridine labeling and immunofluorescence
Cells grown on coverslips were incubated with 5-bromodeoxyuridine (BrdU) for 1 h and stained with anti-BrdU antibody (Roche, USA) according to the manufacturer's protocol. The cells were then observed under a laser scanning microscope (Axioskop 2 plus; Carl Zeiss, Jena, Germany). BrdU-positive cells were quantitatively determined by manual counting.
Animal study
The female BALB/c nude mice (4-5 weeks old) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China) and housed in specific pathogen-free units. To evaluate the in vivo tumorigenesis effect of PRR11, the PRR11 knockdown and control HSC3 cells (2×106) in 80 μL PBS were injected subcutaneously into the flank of nude mice (5 mice/group). All animal experiments were performed in accordance with a protocol approved by our Institutional Animal Care and Use Committee.
Tumor size was measured every four days using a caliper, and tumor volume was calculated using the formula volume = (length × width 2)/2. Mice were euthanized when they met the institutional euthanasia criteria for tumor size and overall health condition. The tumors were removed, photographed and weighed.
The tumor tissue was fixed in formalin, embedded in paraffin, cut into 6-μm sections. After deparaffinization, sections were analyzed by either immunohistochemical (IHC) using anti-Ki67 (proliferation marker) or H&E staining with Mayer's hematoxylin solution according to standard procedures.
RNA extraction and real-time RT-PCR
Total RNA was extracted from cultured cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Two μg of RNA was used for first-strand cDNA synthesis in ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Real-time RT-PCR primers and probes were designed using the Primer Express version 2.0 software (Applied BioSystems). Sequences of the primers were: p21, 5'- TGTCCGTCAGAACCCATGC-3' (forward); p21, 5'- AAAGTCGAAGTTCCATCGCTC-3' (reverse); p27, 5'- ATGAGCCGCAAACTGGGTC-3' (forward); p27, 5'- AGAGCCGAACTCCACAATCTC-3' (reverse);; GAPDH, 5'-GACTCATGACCACAGTC CATGC-3' (forward); GAPDH, 5'-AGAGGCAGGGATGATGTTCTG-3' (reverse); GAPDH probe, 5'-CATCAC TGCCACCCAGAAGACTGT G(TAMRA)-3'. Expression data was normalized to internal control GAPDH. The relative expression level was determined by the 2-△△Ct method.
Statistical Analysis
The data was presented as the mean± standard deviation (SD). The t-test was used to compare the means between the two groups. For comparison among groups, data with the homogeneity of variance were analyzed using one-way ANOVA with a Fisher's Least Significant Difference (LSD) post hoc test. A Welch method was used if the variance showed heterogeneity, and a DunnettT3 method was used for multiple comparisons. The value of P<0.05 was considered statistically significant. All analyses were performed using IBM SPSS Version 17 (IBM Corporation, USA).
Results
PRR11 was up-regulated in TSCC cells
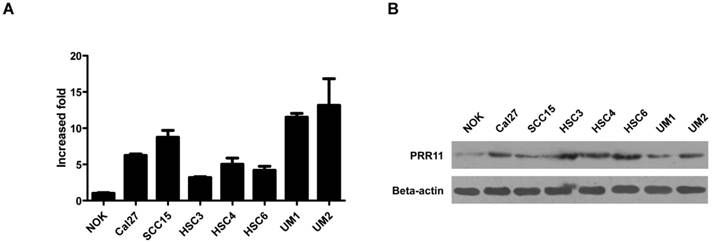
To evaluate the role of PRR11 in TSCC, we first determined the endogenous expression level of PRR11 in TSCC cell lines. The expression levels of PRR11 mRNA and protein were dramatically higher in several TSCC cell lines than in the immortalized normal oral keratinocytes (NOK) (Fig. 1), suggesting that PRR11 was up-regulated in TSCC cells. Among the assayed cell lines, both SCC15 and HSC3 cells had moderate expression level of PRR11, and these two cell lines have been reported to have the tumorigenic capacity both in vivo and in vitro. Therefore, these two cell lines were selected for the following experiments.
The mRNA (A) and protein (B) expressions of PRR11 in immortalized normal oral epithelial cells (NOK) and TSCC cell lines was detected by qRT-PCR and western blot, respectively.
PRR11 overexpression promoted proliferation and invasive ability of TSCC cells
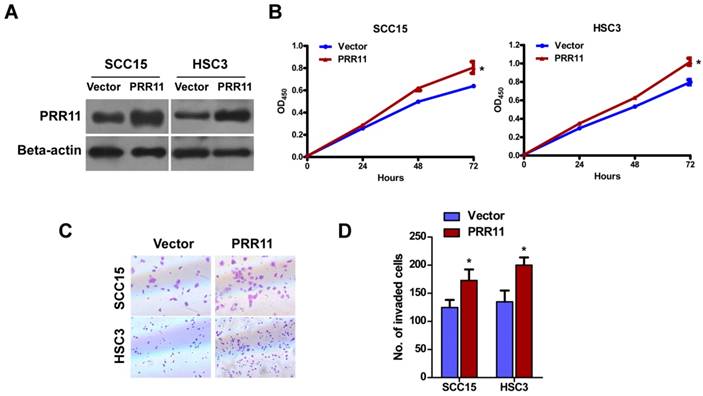
To determine the function of PRR11 in TSCC cells, pcDNA3.1-PRR11 was transfected into SCC15 and HSC3 cells for overexpression (Fig. 2A). CCK-8 assay showed that overexpression of PRR11 significantly promoted TSCC cells proliferation at 72 h (both P<0.05, Fig. 2B). Transwell assay showed that compared to the vector control group, PRR11 overexpression significantly increased the number of cells passing through the transwell chamber (both P<0.05, Fig. 2C and 2D). These results suggested that ectopic PRR11 expression enhanced proliferation and the invasive ability of TSCC cells.
PRR11 overexpression promoted proliferation and invasive ability of TSCC cells. (A) Overexpression of PRR11 in SCC15 and HSC3 cells was confirmed by Western blot. (B) Proliferation of SCC15 and HSC3 cells was determined by CCK-8 assay from 0 h to 72 h after transfection. (C) The invasive ability of SCC15 and HSC3 cells was assessed by Transwell assay at 48 h after transfection. (D) Quantitative data of the Transwell assay. *P<0.05, compared with the vector control group.
PRR11 knockdown inhibited proliferation and invasive ability of TSCC cells
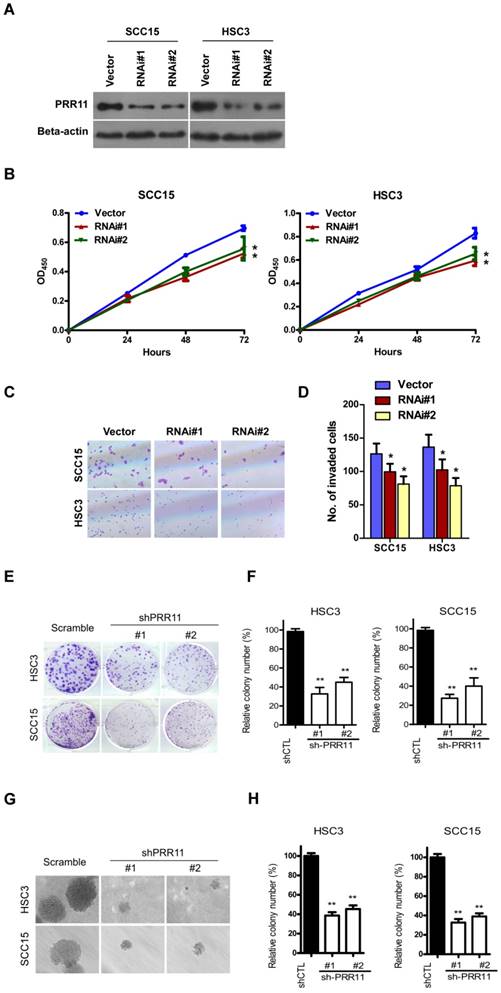
Next, we attempted to investigate the effect of PRR11 knockdown. Transfection of two shRNA-PRR11 plasmid markedly reduced the protein level of PRR11 in SCC15 and HSC3 cells (Fig. 3A). In contrast to overexpression, PRR11 knockdown significantly inhibited proliferation of TSCC cells at 72 h (both P<0.05, Fig. 3B). In addition, PRR11 knockdown significantly decreased the number of invasive SCC15 and HSC3 cells as compared to the control group (all P<0.05, Fig. 3C and 3D). Moreover, colony forming assay consistently showed that PRR11 knockdown significantly inhibited the colony forming ability of SCC15 and HSC3 cells (all P<0.01, Fig. 3E and 3F). Anchorage-independent growth assay showed that knockdown of PRR11 significantly reduced the anchorage-independent growth ability in TSCC cells as compared with the scramble control (all P<0.01, Fig. 3G and 3H). These results suggested that PRR11 knockdown reduced proliferation and the invasive ability of TSCC cells.
PRR11 knockdown inhibited proliferation and invasive ability of TSCC cells. (A) Knockdown of PRR11 in SCC15 and HSC3 cells was confirmed by Western blot. (B) Proliferation of SCC15 and HSC3 cells was determined by CCK-8 assay from 0 h to 72 h after transfection. (C) The invasive ability of SCC15 and HSC3 cells was assessed by Transwell assay at 48 h after transfection. (D) Quantitative data of the Transwell assay. (E) The colony forming ability of SCC15 and HSC3 cells was evaluated by colony forming assay. (F) Quantitative data of the colony forming assay. (G) Anchorage-independent growth assay in PRR11-knockdown or control cells. (H) Quantitative data of the anchorage-independent growth assay. **P<0.01, compared with the control group.
PRR11 knockdown inhibited subcutaneous tumorigenicity of HSC3 cells in nude mice
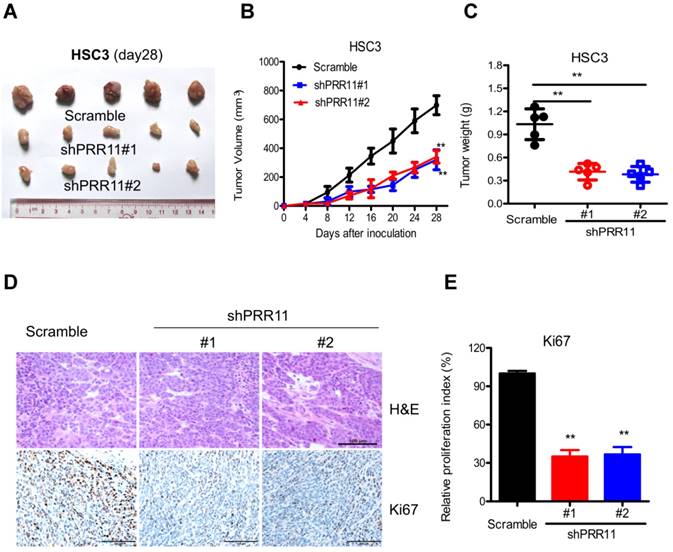
The effect of PRR11 knockdown was further evaluated in an in vivo subcutaneous tumorigenesis model. Compared to the control group, PRR11 knockdown significantly reduced tumor size (both P<0.01 at 28 days, Fig. 4A and 4B) and tumor weight (both P<0.01 at 28 days, Fig. 4C). Moreover, IHC analysis demonstrated that PRR11 knockdown significantly decreased the expression of Ki67 (a proliferation marker) in the tumor tissue (Fig. 4E). These data suggested that PRR11 knockdown inhibited subcutaneous tumorigenicity of HSC3 cells in nude mice.
PRR11 knockdown inhibited in vivo tumorigenicity of HSC3 cells in nude mice. The tumorigenicity of HSC3 cells was determined by subcutaneous tumorigenicity test (A, at day 28) in nude mice. The tumor size (B, from day 0 to day 28) and tumor weight (C, at day 28) were compared among groups. (D) The sections of tumor tissue were subjected to H&E staining (D) and IHC staining for Ki67. **P<0.01, compared with the control group.
The effect of PRR11 on the cell cycle of TSCC cells
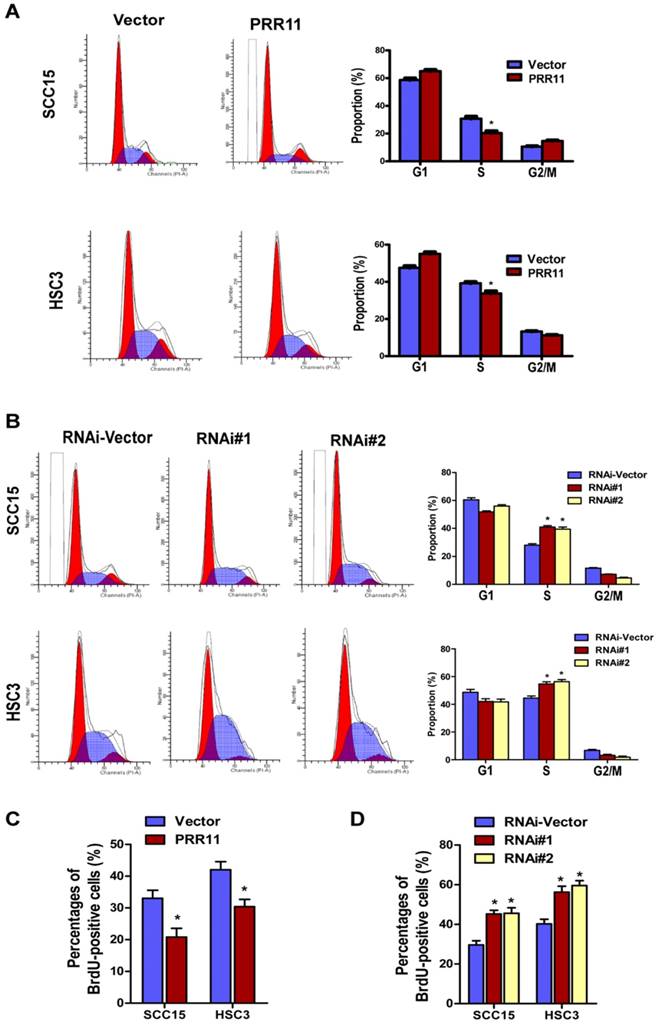
Since the oncogenic potential of PRR11 in TSCC cells was observed, we further investigated the underlying molecular mechanism. Flow cytometry analysis revealed that compared to the control group, PRR11 overexpression significantly decreased the proportion of cells in S phase (both P< 0.05, Fig. 5A), whereas PRR11 knockdown significantly increased the proportion of cells in S phase (both P< 0.05, Fig. 5B). BrdU incorporation assay showed that overexpression of PRR11 significantly increased the BrdU-positive cells as compared with the vector control (Fig. 5C, both P<0.05), whereas PRR11 knockdown significantly reduced the proportion of BrdU-positive cells in both TSCC cell lines (Fig. 5D, all P<0.05). These observations, together with the proliferation results, indicated that PRR11 knockdown induced cell cycle arrest in S phase in TSCC cells, while overexpression of PRR11 facilitated the TSCC cells to pass through the S phase.
PRR11 knockdown induced S phase arrest in TSCC cells. The cell cycle distribution of PRR11-overexpressing (A) and PRR11-silencing (B) TSCC cells was determined by flow cytometry analysis. The bar charts showed quantitative results of the distribution of cell cycle. (C) BrdU incorporation assay was performed in PRR11-overexpressing and PRR11-knockdown cells. (D) Quantitative data of the BrdU incorporation assay. *P<0.05, compared with the vector control group.
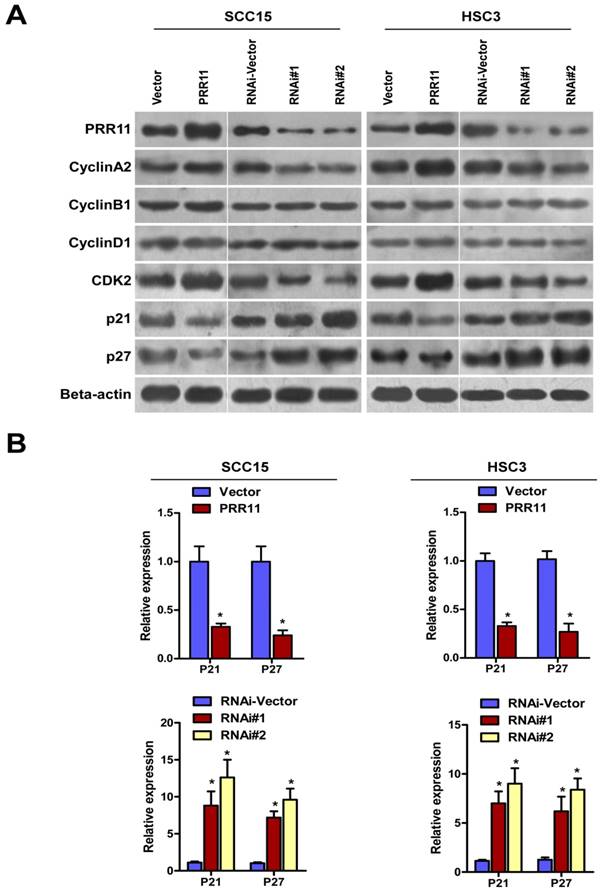
PRR11 regulated the expressions of cell cycle related proteins
To further study the effect of PRR11 in cell cycle regulation, Western blot was performed to determine cell cycle-related proteins. As shown in Fig. 6, PRR11 overexpression down-regulated two cyclin-dependent kinase inhibitors (CKIs), p21 and p27. In addition, PRR11 overexpression up-regulated CDK2 and Cyclin A2, which are responsible for S phase. However, the expressions of Cyclin B1 and Cyclin D1 were not affected. By contrast, PRR11 knockdown induced reverse trends of the expressions of these proteins. As shown in Fig. 6B, overexpression of PRR11 significantly reduced the mRNA levels of p21 and p27, whereas PRR11 knockdown significantly showed a reverse trend as compared with the control cells (Fig. 6B, all P<0.05). These results indicated that PRR11 regulated the expressions of p21, p27, CDK2 and Cyclin A to facilitate S/G phase transition and promoted cell proliferation.
PRR11 regulated the expressions of cell cycle-related proteins. (A) The protein levels of cell cycle-related proteins of PRR11-overexpressing and PRR11-silencing TSCC cells at 72 h after transfection were determined by Western blot. (B) The expression of P21 and P27 mRNA was determined using real-time PCR.
Discussion
Abnormally high expression of PRR11 has been found in several cancers. Our previous study also shows that PRR11 is highly expressed in TSCC and is associated with the development of TSCC. In this study, we investigated the molecular mechanism underlying oncogenic potential of PRR11 in TSCC cells. The results showed that overexpression of PRR11 significantly enhanced TSCC cells proliferation and the invasive ability of TSCC cells, whereas PRR11 knockdown in TSCC cells exhibited a reverse trend. In addition, the in vivo subcutaneous tumorigenicity assay showed that PRR11 knockdown significantly reduced tumor size and the expression of Ki67 in the tumor tissue. Flow cytometry analysis revealed that PRR11 overexpression significantly decreased the proportion of cells in S phase, whereas PRR11 knockdown in TSCC cells exhibited a reverse trend. Furthermore, PRR11 overexpression simultaneously down-regulated two cyclin-dependent kinase inhibitors (CKIs), p21 and p27 and up-regulated CDK2 and Cyclin A2 in TSCC cells. PRR11 knockdown again exhibited reverse trends of the above protein expressions. Taken together, these results suggested that PRR11 promoted cell proliferation by regulating the expressions of p21, p27, CDK2 and Cyclin A to facilitate S phase progression in TSCC cells. To our best knowledge, this is the first study reporting the molecular mechanism underlying oncogenic potential of PRR11 in TSCC.
In this study, our overexpression and knockdown of PRR11 experiments demonstrated that PRR11 is involved in the proliferation and the invasive ability of TSCC cells in both cellular model and in vivo nude mice tumorigenicity assay. Consistent with our findings, it has been reported that RNAi-mediated silencing of PRR11 suppressed cellular proliferation, colony formation ability, cell migration and invasion ability in lung cancer cells and inhibited tumorigenic potential in nude mice [5,11]. Chen et al. have shown that PRR11 silencing resulted in decreased cellular proliferation, cell migration, tumor growth in hilar cholangiocarcinoma cells [9]. A similar effect of PRR11 knockdown on cell growth has also been demonstrated in gastric cancer cells [7] and HeLa cervical cancer cells [5]. However, Zhang et al. [12] have reported that overexpression of PRR11 induces premature chromatin condensation, and massively attenuates proliferation of H1299 lung cancer cells, which is inconsistent with our finding. This phenomenon indicates that the mechanism of PRR11 function is complex and may differ in different types of cancer cells. Interestingly, Wang et al. have reported that PRR11 is oriented in a head-to-head configuration with SKA2 gene, and the transcription of both genes is driven by a core bidirectional promoter [11]. Expression of both PRR11 and SKA2 was significantly up-regulated in lung cancer, and high expression of both PRR11 and SKA2 is associated with poorer prognosis in lung cancer patients [11]. Knockdown of PRR11 and/or SKA2 remarkably reduced cell proliferation, migration, and invasion in lung cancer cells. The synergistic effect of the gene pair may contribute to the phenomenon that overexpression of PRR11 in lung cancer also inhibits cell proliferation. It is worth to investigate if the association between PRR11 and SKA2 genes could be identified in TSCC cells. Invasion and metastasis of tumors is an important indicator of disease progression and is also a major cause of cancer mortality. Previous studies have reported that PRR11 silencing suppresses the invasive ability of tumor cells in lung cancer cells [5] and breast cancer [6]. Nevertheless, the effect of PRR11 overexpression on the invasive ability of tumor cells has not been reported. Our data showed that overexpression of PRR11 significantly promoted the invasive ability of TSCC cells, while PRR11 knockdown significantly decreased the invasive ability. These findings further confirm that PRR11 can promote the invasive ability of TSCC cells.
Accumulating evidence has suggested that PRR11 is involved in the regulation of cell cycle in cancer cells [5,12]. The microarray analysis by Ji et al. has revealed that PRR11 knockdown induces the dysregulation of several important genes involved in cell cycle, tumorigenesis and metastasis (e.g. CCNA1, RRM1, MAP4K4 and EPB41L3) [5]. Their study also showed that PRR11 knockdown caused significant S phase arrest in HeLa cells and lung cancer cells [5]. Zhang et al. have revealed that the amount of PRR11 started to increase in the late S phase, and was retained until just before mitotic telophase in H1299 human lung cancer cells [12]. siRNA-mediated knockdown of PRR11 results in late S phase arrest and a retardation of G2/M progression, which are accompanied by mitotic defects (multipolar spindles and multiple nuclei) [12]. These results suggest that PRR11 plays a crucial role in the regulation of cell cycle progression. In agreement with the results in lung cancer cells, our results showed that PRR11 knockdown increased the proportion of cells in S phase and inhibited cell growth, suggesting that TSCC knockdown induced S phase arrest in TSCC cells. Moreover, our data further suggested that PRR11 overexpression facilitated TSCC cells to pass through S phase, indicating PRR11 promotes TSCC cell proliferation by enhancing TSCC cells to pass through S phase.
The cell cycle is stringently controlled by numerous regulatory proteins, such as the cyclins, CDKs and the CKIs [13]. Cyclin A/Cdk2 complex plays an important role in S phase progression by phosphorylating its substrates, such as Cdh1, Rb, p21 and p27 [14]. Cyclin D/Cdk4 and Cdk6 are essential for progression from G1 to S-phase [15], while cyclin E is essential for the G1-to-S phase transition [16]. On the other hand, p21 and p27 are CKIs which can broadly interfere with the activities of cyclin D-, E-, A- and B-dependent kinase complexes to inhibit the progression of cell cycle [17]. Ji et al. have found that PRR11 silencing decreases the expression of Cyclin A1 in Hela cells, and decreases the expression of Cyclin A1 and Cyclin A2 in lung cancer cells [5]. In this study, our data showed that PRR11 overexpression up-regulated protein levels of cyclin A2 and CDK2 as well as down-regulated p21 and p27 in TSCC cells. In addition, PRR11 knockdown reversed the trends of these protein expressions. Neither overexpression nor knockdown of PRR11 had an effect on the expression levels of cyclin B1 and cyclin D1. These data suggested that PRR11 may inhibit the expression of p21 and p27, which in turn enhanced the activity of cyclin A2/CDK2, thereby promoting the S phase progression and proliferation in TSCC cells. However, the detailed molecular mechanism remains to be further elucidated.
There are still some limitations in this study. Although we found ectopic PRR11 down-regulated p21 and p27 in TSCC cells, the key molecules and signaling pathways responsible for this regulation are still unknown. In addition, the in vitro findings of this study should be further validated in an in vivo animal model. All these limitations should be addressed in the following study.
Conclusion
In summary, our results showed that PRR11 promoted proliferation and invasion of TSCC cells, at least partially, by regulating the expressions of p21, p27, CDK2 and Cyclin A to facilitate S phase progression. Our findings help better understanding the molecular mechanism of the oncogenic potential of PRR11 in TSCC cells.
Abbreviations
TSCC: Tongue squamous cell carcinoma; PRR11: Proline rich 11; qRT-PCR: Quantitative Real-time PCR; PVDF: polyvinylidene difluoride; TBST: Tris-buffered saline/Tween 20; IHC: immunohistochemical; SD: standard deviation; LSD: Least Significant Difference; NOK: normal oral keratinocytes; CKIs: cyclin-dependent kinase inhibitors.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (Grant Numbers: 81500865 and 81630025) and Science and Technology Program of Guangzhou (Grant Number: 201607010354).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rivera C. Essentials of oral cancer. Int J Clin Exp Pathol. 2015;8:11884-94
2. Guo XH, Wang JY, Gao Y. et al. Decreased adiponectin level is associated with aggressive phenotype of tongue squamous cell carcinoma. Cancer Sci. 2013;104:206-13
3. Riemann M, Knipfer C, Rohde M. et al. Oral squamous cell carcinoma of the tongue: Prospective and objective speech evaluation of patients undergoing surgical therapy. Head Neck. 2016;38:993-1001
4. Mishra A, Meherotra R. Head and neck cancer: global burden and regional trends in India. Asian Pac J Cancer Prev. 2014;15:537-50
5. Ji Y, Xie M, Lan H. et al. PRR11 is a novel gene implicated in cell cycle progression and lung cancer. Int J Biochem Cell Biol. 2013;45:645-56
6. Zhou F, Liu H, Zhang X. et al. Proline-rich protein 11 regulates epithelial-to-mesenchymal transition to promote breast cancer cell invasion. Int J Clin Exp Pathol. 2014;7:8692-9
7. Song Z, Liu W, Xiao Y. et al. PRR11 Is a Prognostic Marker and Potential Oncogene in Patients with Gastric Cancer. PLoS One. 2015;10:e0128943
8. Tan S, Jiang Z, Hou A. et al. Expression of PRR11 protein and its correlation with pancreatic cancer and effect on survival. Oncol Lett. 2017;13:4117-122
9. Chen Y, Cha Z, Fang W. et al. The prognostic potential and oncogenic effects of PRR11 expression in hilar cholangiocarcinoma. Oncotarget. 2015;6:20419-33
10. Wang C, Yu L, Hu F. et al. Upregulation of proline rich 11 is an independent unfavorable prognostic factor for survival of tongue squamous cell carcinoma patients. Oncol Lett. 2017;14:4527-34
11. Wang Y, Zhang Y, Zhang C. et al. The gene pair PRR11 and SKA2 shares a NF-Y-regulated bidirectional promoter and contributes to lung cancer development. Biochim Biophys Acta - Gene Regul Mech. 2015;1849:1133-44
12. Zhang C, Zhang Y, Li Y. et al. PRR11 regulates late-S to G2/M phase progression and induces premature chromatin condensation (PCC). Biochem Biophys Res Commun. 2015;458:501-8
13. Casimiro MC, Crosariol M, Loro E. et al. Cyclins and cell cycle control in cancer and disease. Genes Cancer. 2012;3:649-57
14. Oakes V, Wang W, Harrington B. et al. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle. 2014;13:3302-11
15. Bertoli C, Skotheim JM, De Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518-28
16. Ohtsubo M, Theodoras AM, Schumacher J. et al. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612-24
17. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-93
Author contact
![]() Corresponding author: Professor Bin Cheng, Guanghua School of Stomatology, Hospital of Stomatology, Guangdong Provincial Key Laboratory of Stomatology, Sun Yat-sen University, 56 Lingyuanxi Road, Guangzhou, Guangdong 510060, P.R. China. E-mail: chengbinsysu.edu.cn; Tel: 86 138 2974 1695; Fax: 86 020-83822807
Corresponding author: Professor Bin Cheng, Guanghua School of Stomatology, Hospital of Stomatology, Guangdong Provincial Key Laboratory of Stomatology, Sun Yat-sen University, 56 Lingyuanxi Road, Guangzhou, Guangdong 510060, P.R. China. E-mail: chengbinsysu.edu.cn; Tel: 86 138 2974 1695; Fax: 86 020-83822807