Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(11):2488-2500. doi:10.7150/jca.29528 This issue Cite
Review
Regulatory Molecules and Corresponding Processes of BCR-ABL Protein Degradation
Han-Qing Zhu, Feng-Hou Gao ![]()
Department of Oncology, Shanghai 9th People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200011, China
Received 2018-8-27; Accepted 2019-5-2; Published 2019-6-2
Abstract
The BCR-ABL fusion protein with strong tyrosine kinase activity is one of the molecular biological bases of leukemia. Imatinib (Gleevec), a specific targeted drug for the treatment of chronic myeloid leukemia (CML), was developed for inhibiting the kinase activity of the BCR-ABL fusion protein. Despite the positive clinical efficacy of imatinib, the proportion of imatinib resistance has gradually increased. The main reason for the resistance is a decrease in sensitivity to imatinib caused by mutation or amplification of the BCR-ABL gene. In response to this phenomenon, the new generation of tyrosine kinase inhibitors (TKIs) targeting the BCR-ABL fusion protein was developed to solve the problem. However this strategy only selectively inhibits the tyrosine kinase activity of the BCR-ABL protein without eliminating the BCR-ABL protein, it does not fundamentally cure the BCR-ABL-positive leukemia patients. With the accumulation of the knowledge of cellular molecular biology, it has become possible to specifically eliminate certain proteins by cellular proteases in a specific way. Therefore, the therapeutic strategy to induce the degradation of the BCR-ABL fusion protein is superior to the strategy of inhibiting its activity. The protein degradation strategy is also a solution to the TKI resistance caused by different BCR-ABL gene point mutations. In order to provide possible exploration directions and clues for eliminating the BCR-ABL fusion protein in tumor cells, we summarize the significant molecules involved in the degradation pathway of the BCR-ABL protein, as well as the reported potent compounds that can target the BCR-ABL protein for degradation.
Keywords: BCR-ABL fusion protein, Tyrosine kinase activity, Inhibitor, Protein degradation
Introduction
The c-ABL gene of the long arm of chromosome 9 can be translocated to the breakpoint concentration region (BCR) on the long arm of chromosome 22 to produce the oncogenic BCR-ABL fusion gene. The BCR-ABL fusion gene can be seen in more than 95% CML patients. Meanwhile, it is also found in other types of leukemia patients, including ~25% adult B-cell acute lymphoblastic leukemia (B-ALL) patients, ~30% mixed phenotype acute leukemia (MPAL) patients, <1% acute myeloid leukemia (AML) patients, as well as in occasional lymphoma and myeloma patients [1, 2]. The BCR-ABL fusion protein encoded by the fusion gene has strong tyrosine kinase activity and is the significant molecular biological basis for the pathogenesis of these diseases. Therefore, it has also become an important molecular target for such leukemia.
Imatinib (Gleevec) specifically inhibits the tyrosine kinase activity of the fusion protein by blocking the binding of ATP to BCR-ABL protein, thereby inhibiting the proliferation of CML cell. However, it is gradually found that imatinib is defective in the clinical efficacy. Imatinib is mainly effective for CML patients at early stage, and the complete remission rate of the advanced stage patients treated with imatinib is less than 30%. Some patients may develop imatinib resistance and the resistance proportion increases year by year. The main reason for the imatinib resistance is that BCR-ABL gene mutation or amplification leads to a decrease in sensitivity to imatinib. In response to this phenomenon, it is still necessary to continue to develop a new generation of TKI that can overcome the drug resistance, such as nilotinib, dasatinib and ponatinib. This strategy can not fundamentally cure patients, for it only selectively inhibits the tyrosine kinase activity of the BCR-ABL protein without removing the intracellular BCR-ABL protein. With the accumulation of knowledge of cellular molecular biology, it has become possible to decrease or even eliminate target proteins by activating certain cellular proteases. The BCR-ABL protein degradation strategy is not only a new strategy for molecular target therapy, but also can overcome the problem of to TKI resistance caused by various point mutations of the BCR-ABL gene [3].
In order to provide possible exploration directions and clues, we reviewed the researches on the degradation of the BCR-ABL protein. We searched PubMed database using the terms of “BCR-ABL protein, degradation”, “BCR-ABL protein, down-regulation”, “BCR-ABL protein, inhibitors”, “BCR-ABL protein, proteasome”, “BCR-ABL protein, lysosome” or “BCR-ABL protein, caspase” respectively. 297 papers have been obtained, and 90 of them are related to the BCR-ABL protein degradation. They reflect the situation of exploratory researches on the strategy of targeting the BCR-ABL protein for degradation. These literatures are summarized and refined according to the mechanism of the BCR-ABL protein degradation, which may provide inspiration for future research in this field.
1. Biochemical properties of BCR-ABL
1.1 BCR-ABL's structure, distribution and function
Philadelphia (Ph) chromosome is generated by the chromosome translocation of t(9;22)(q34;q11). The normal c-ABL gene locates in chromosome 9 and consists of 11 exons (1b, 1a, a2-a11). During the chromosome translocation, c-ABL gene can break in any points of these three breakpoints: upstream of 1b, between 1b and 1a, and between 1a and a2. In either case, the first two alternative exons (1b, 1a) of c-ABL gene are always spliced out. The normal BCR gene locates in chromosome 22 and consists of 23 exons (e1-e23). In the generation of the BCR-ABL fusion gene, there are three breakpoint cluster regions in BCR gene: major (M-BCR), minor (m-BCR), and micro (μ-BCR). Break in M-BCR can induce e14a2, e13a2, e14a3 and e13a3 junctions, break in m-BCR can generate e1a2 and e1a3 junctions, and break in μ-BCR can produce e19a2 junction [1]. Although there are such many junctions, e14a2 and e13a2 junctions are the most common junctions in all Ph+ hematological malignancies. The e14a2 and e13a2 junctions generate the same transcript that encodes the p210 BCR-ABL protein, the e19a2 junction creates transcript which encodes the p230 BCR-ABL protein. And the e1a2 junction generates transcript to encode the p190 BCR-ABL protein, which is smaller due to exons' splicing of BCR gene [1, 4].
All domains of the c-ABL protein are contained in three BCR-ABL proteins, and these domains are SRC-homology-2 (SH2) domain, SH3 domain, tyrosine kinase domain, nuclear localization signal (NLS), nuclear export signal (NES), DNA-binding domain (DBD) and actin-binding domain (ABD). However three BCR-ABL proteins have different BCR domains. All three proteins have coiled-coil (CC) oligomerization domain and serine/threonine kinase domain, the p210 and p230 BCR-ABL proteins also have Rho guanine nucleotide exchange factor (Rho-GEF) domain and pleckstrin homology (PH) domain. In addition, the p230 BCR-ABL protein has calcium-dependent lipid-binding (Cal-B) domain and truncated RAC guanosine triphosphatase-activating protein (RAC-GAP) domain [2].
Since the c-ABL protein contains NLS and NES, the c-ABL protein found in both the nucleus and cytoplasm is a protein that shuttles between these two compartments. However the BCR-ABL protein which also have these two domains does not undergo nuclear-cytoplasmic shuttle, it is not imported into the nucleus and remains in cytoplasm [5]. Surprisingly, the BCR-ABL protein with no kinase activity can translocate into the nucleus without affecting cell survival, but artificial nuclear entrapment of the BCR-ABL protein with kinase activity can induce cell apoptosis [6]. The BCR-ABL protein exhibits a polarized distribution in 32D myeloid cells [7]. Further study proved that the p210 BCR-ABL protein is distributed in punctate structures in the cytoplasm, although the greater part of the p210 BCR-ABL protein is diffused rather than in these punctate structures [8].
The c-ABL protein itself is a non-receptor tyrosine kinase, and the BCR protein is a signaling protein containing multiple modular domains. The fusion of the BCR gene to the ABL gene renders the ABL protein the enhanced tyrosine kinase activity and new regulatory domains [9]. The BCR-ABL protein has sustained kinase activity that can phosphorylate itself and signaling proteins thereby activating a series of signaling pathways, resulting cellular processes dysregulation and finally leading to the development of leukemia [4, 9, 10].
1.2 The expression and detection methods of BCR-ABL
The BCR-ABL transcript level may be up to 200-fold greater in the most primitive CML CD34+ progenitors relative to their more differentiated counterparts [4]. Previous study showed the values of the p210 BCR-ABL protein/actin protein in several chronic phase (CP) CML patients were 0.45, 0.33, 0.16, 0.29, 0.66, and those in blastic phase (BP) CML patients were 1.44, 1.13, 1.21, 1.10 [11]. Both the mRNA and protein level of the BCR-ABL in BP CML patients were higher than those in CP CML patients. In CML patients who progress from CP to accelerated phase (AP) or BP, the BCR-ABL mRNA level increased before laboratory or clinical parameters changed [12]. Thus, it is widely considered that the BCR-ABL has dose-dependent effects on the tumorigenicity of CML cell.
Chromosome banding analysis (CBA), fluorescence in situ hybridization (FISH) and Qualitative PCR are recommended in the initial diagnosis of CML, according to the recommendations for the management of CML [13]. During the treatment, Quantitative real-time PCR (RQ-PCR) can be used to determine the transcript level of BCR-ABL for evaluating treatment efficacy and predicting the future response [14]. Mutational analysis by conventional Sanger sequencing is recommended in case of disease progression and treatment failure, for BCR-ABL gene mutants can lead to drug resistance [15].
2. The significance of BCR-ABL-targeted therapy in clinical Ph+ hematologic malignancies
It has already been elucidated that the BCR-ABL was apparently important in both initiation and maintenance of the Ph+ hematological malignancies transformation. The BCR-ABL protein can transform a variety of cell types in vitro including lymphoid and myeloid cell lines, primary bone marrow cells and fibroblast cell lines [7, 9]. Retrovirally transduced BCR-ABL gene in mice bone marrow cells can induce a myeloproliferative disorder in mice resembling CML [4, 7, 9]. However when the mutant BCR-ABL protein without kinase activity expressed in the hematopoietic stem cells (HSCs) of mice, the mice didn't develop leukemia [4].
Before the discovery of BCR-ABL-targeted drugs, the main treatment strategies of CML were chemotherapy and interferon, which have a lot of side effects and can easily lead to drug resistance. Allogeneic stem-cell transplantation may cure CML, but this method is difficult to implement. The development of imatinib which inhibits the kinase activity of the BCR-ABL protein is highly effective for CML treatment, resulting in 98% haematological remission, 87% complete cytogenetic remission, and progression-free survival in 84% of patients [16]. However, mutations in the ABL gene which encoding the kinase domain can reduce the binding of imatinib to the BCR-ABL protein, finally lead to drug resistance and a high recurrence rate in patients [10, 17, 18]. More than 50 kinds of BCR-ABL mutations are currently known to be resistant to imatinib. New tyrosine kinase inhibitors such as nilotinib and dasatinib are generated, but obstacles similar as imatinib also gradually appear [10, 19]. Targeting the degradation pathway of the BCR-ABL protein not only can eliminate the wild-type BCR-ABL protein possessing kinase activity, but also is effective for mutant BCR-ABL proteins.
Proteasome and lysosome pathways are the main pathways of intracellular protein degradation [20, 21]. The ubiquitin proteasome system (UPS) consists of ubiquitin, proteasome, E1 ubiquitin-activating enzymes (E1s), E2 ubiquitin-conjugating enzymes (E2s), E3 ubiquitin ligases (E3s) and deubiquitylating enzymes (DUBs) [22]. The small molecule ubiquitin is transferred to the targeted substrates by the actions of E1s, E2s and E3s, resulting in the translocation of the ubiquitin-modified substrates into the proteasome complex for degradation [23]. Recently there are significant advances in small molecule inhibitors which can target UPS to induce the BCR-ABL protein degradation [23]. Under the external pressures, the cellular BCR-ABL protein degradation mediated by autophagic lysosome pathway can be activated. The BCR-ABL protein to be degraded is initially wrapped by a double membrane to form autophagosomes, next, autophagosomes fuse with lysosomes to form autolysosomes. Finally, the protein within autolysosomes will be degraded by lysosomal hydrolases [24, 25]. Generally, caspases only cleave the substrate protein after specific aspartic acid residue without inducing substrate degradation. The cleavage of the BCR-ABL protein induced by caspases can eliminate the characteristic of the BCR-ABL protein, so this pattern, to some extent, can be regarded as the degradation of the BCR-ABL protein [26, 27].
3. The molecules regulate the BCR-ABL protein degradation via ubiquitin-proteasome pathway
3.1 E3 ligases
The E3 ligases are classified into four families: HECT, RING-finger, U-box, and PHD-finger according to their domain constitution. They are responsible for catalyzing the reaction, by which ubiquitin can be transferred from the cysteine residue of the E2 to the specific substrate's lysine residue or N terminus [28]. Casitas B-lineage lymphoma (c-CBL) and Carboxyl terminus of the Hsc70-interacting protein (CHIP) are two different E3 ligases for the BCR-ABL protein currently known [29, 30].
The RING E3 ligase c-CBL belongs to the small family of CBL E3 ligase which includes c-CBL, CBL-b and CBL-3. All CBL proteins contain an N-terminal tyrosine-kinase-binding (TKB) domain followed by a RING finger (RF) domain and a proline-rich domain [31, 32]. Mao JH et al. firstly reported that c-CBL served as the E3 ligase of the BCR-ABL protein to down-regulate the BCR-ABL protein level by affecting its ubiquitination. c-CBL mediates the ubiquitination of the BCR-ABL protein at the lysine residue (K1517) of the BCR-ABL protein, and the ubiquitin ligation to the BCR-ABL protein occurs mostly at the lysine residue (K48) of ubiquitin. Then the ubiquitinated BCR-ABL protein modified by c-CBL will be transported into proteasome for degradation [29, 33]. Both TKB domain and RF domain of c-CBL are essential for its E3 ligase activity, since mutations in both domains can make c-CBL lose its catalytic activity essential for the BCR-ABL protein degradation.
Zhang QY et al. first discovered that the combination of As4S4 and imatinib had synergistic effects on the treatment of CML; they further confirmed that As4S4 specifically targeted c-CBL to induce the degradation of the BCR-ABL protein. Detailedly, c-CBL is mainly degraded by the proteasome system, and the ubiquitination of c-CBL at the lysine residue (K389) within RF domain can be regulated by itself. As4S4 can inhibit c-CBL's self-ubiquitination process through binding directly to the cysteine residue (C381) of c-CBL, thereby interfering with the self-ubiquitination/degradation of c-CBL. As4S4-induced accumulation of c-CBL promotes the c-CBL-regulated proteasome-mediated BCR-ABL protein degradation, thus inducing BCR-ABL-dependent cell apoptosis [29, 34].
CHIP is a U-box E3 ubiquitin ligase, and is consist of three domains, the N-terminal tetratricopeptide repeat (TPR) domain, the C-terminal U-box domain, and the intermediate helical linker. The U-box domain is responsible for its E3 ligase activity [35]. Tsukahara F et al. considered that both c-CBL and CHIP could induce BCR-ABL-dependent cell growth inhibition by promoting the degradation of the BCR-ABL protein, but the properties of the BCR-ABL proteins recognized by c-CBL and CHIP were different [30]. Both c-CBL and CHIP can recognize the wild-type BCR-ABL protein, the T315I-mutant and E255K-mutant BCR-ABL proteins, and the latter two are imatinib-resistant mutants. In addition, CHIP also can degrade the inactive, immature and unfolded BCR-ABL proteins. Tsukahara F et al. also believed that the CHIP-mediated degradation of the BCR-ABL protein mainly through the proteasome system, while c-CBL caused the degradation of the BCR-ABL protein by lysosome system [30].
The "protein knockout" technique utilizes endogenous ubiquitin-proteasome system to degrade specific protein, proteolysis targeting chimeras (PROTAC) and creating non-endogenous chimeric E3 ligases that can target specific proteins are two approaches of the "protein knockout" technique [36-38]. Non-endogenous chimeric E3 ligases can be constructed by fusing the adaptor domain that can bind to the target protein with a domain that has E3 catalytic activity. SH2-U-box and SH2-RING are two E3 ligases artificially synthesized based on CHIP and c-CBL respectively, and they both can target the BCR-ABL protein for degradation [39]. SH2-U-box consists of SH2 domain and U-box domain, and SH2-RING is composed of SH2 domain and RING domain. The SH2 region derived from growth factor receptor-bound protein 2 (Grb2) can recognize and bind to the phosphorylated tyrosine residue (Y177) of BCR-ABL proteins. U-box domain of CHIP and RING domain of c-CBL has E3 ligase activity, and can induce ubiquitin transferred from E2s to form a covalent bond with the lysine residue of the BCR-ABL protein. Both two E3 ligases can promote the wild-type and mutant BCR-ABL proteins ubiquitination and degradation, and the effect of SH2-U-box is stronger than that of SH2-RING. Furthermore, it has been proved that SH2-U-box could inactivate BCR-ABL-dependent signaling pathways in cell and could induce BCR-ABL-dependent cell growth inhibition [39].
3.2 HSP90
Many newly synthesized proteins in cell are prone to misfolding and aggregation, however, the chaperones allow the client proteins to fold correctly into three-dimensional structures and be functional. According to heat-shock proteins' (HSPs) molecular weight, they are classified into HSP40, HSP60, HSP70, HSP90, HSP100 and the small HSPs. They are several different classes of structurally unrelated chaperones synthesized under environment stress [40]. Heat-shock proteins play significant roles in cell functions, which including modulating protein activity by changing protein conformation, maintaining multiprotein complex assembly or disassembly, and facilitating protein to translocate across organelle membranes. Irreversibly misfolded proteins are degraded by the proteasome and autophagy system [40]. For mature BCR-ABL protein is chaperoned by HSP90, HSP90 is considered to be a potential therapeutic target for CML [41]. The HSP90 consists of four domains: a N-terminal domain, a C-terminal domain, a middle domain and a charged linker region located between N-terminal and middle domain. Both N-terminal domain and C-terminal domain contain ATP/ADP-binding sites. The middle domain can bind with co-chaperones and clients, and it also possesses the ability to discriminate different clients [42].
Multiple HSP90 inhibitors have been reported to be effective in promoting proteasome-mediated BCR-ABL protein degradation by disrupting the interaction between BCR-ABL protein and HSP90. HSP90 ATPase inhibitors and HSP90 histone deacetylase inhibitor are two kinds of inhibitors widely studied by researchers. HSP90 depends on its ATP binding sites to make conformational change for being functional, while HSP90 ATPase inhibitors bind to the ATP/ADP-binding sites of HSP90 resulting in HSP90 function inhibition.
An WG et al. have demonstrated that cellular BCR-ABL protein was stably complexed with HSP90 and HSP90's co-chaperone p23. HSP90 ATPase inhibitor geldanamycin (GA) can induce proteasome-mediated BCR-ABL protein degradation by switching it's chaperone complexes from HSP90-p23-p50 to HSP70-p60, probably because HSP70-p60 complex can mediate the degradation of its client protein [43]. Furthermore, Tsukahara F et al. suggested that both CHIP and c-CBL E3 ligases were involved in the GA-mediated BCR-ABL protein degradation. Because when the expression of CHIP or c-CBL was inhibited, the GA-induced degradation of the BCR-ABL protein was alleviated. The GA-induced BCR-ABL protein degradation was almost completely abrogated when the expression of CHIP and c-CBL were both inhibited [30]. GA's analogue 17-allylamino-17-demethoxygeldanamycin (17-AAG) has also been reported to promote the BCR-ABL protein degradation and the functionary mechanism was similar as GA [44-46]. Another HSP90 ATPase inhibitor novobiocin (NB) also can promote the proteasomal degradation of the BCR-ABL protein. But unlike GA and 17-AGG, NB decreased the binding of the BCR-ABL protein with both HSP90 and HSP70, and disrupted these two BCR-ABL's chaperone complexes [47]. However, the detailed mechanism remains to be explored in further studies. Researchers showed that GA, 17-AGG and NB all had inhibitory effects on BCR-ABL-dependent cell. Tao W et al. also proved that the degradation of the BCR-ABL protein induced by HSP90 ATPase inhibitor AUY922 was a powerful way to inhibit not only imatinib-sensitive but also imatinib-resistant CML cell [48].
Histone deacetylases (HDACs) are classified into four classes. Class I HDACs (1-3, 8) which are predominantly located in nucleus are mainly responsible for mediating the histone deacetylation. Class IIa HDACs (4, 5, 7, 9) and Class IIb HDACs (6 and 10) which are proteins shuttle between nucleus and cytoplasm can deacetylate non-histone proteins. Histone deacetylase inhibitor (HDACi) can inhibit specific HDACs to induce acetylation of HSP90 therefore affecting the function of HSP90 and the stability of HSP90's clients.
Okabe S et al. demonstrated that depsipeptide (FK228) and LAQ824 which are HDACis could inhibit BCR-ABL-dependent cell. They also found that these two HDACis induced HSP90 acetylation and disassociated HSP90 with the BCR-ABL protein, thereby promoting the proteasomal degradation of the BCR-ABL protein [45, 49]. Previous studies showed that HDAC6 was a direct deacetylase for HSP90 and essential for maintaining HSP90 function. Inhibiting the activity of HDAC6 can induce HSP90 hyperacetylation, disrupt HSP90's interaction with its essential co-chaperone p23, and affect the maturation of Glucocorticoid Receptor (GR) which is HSP90's client [50]. According to this, HDAC6 activity inhibition to induce HSP90 hyperacetylation thereby promoting the BCR-ABL protein degradation is the major efficacy mechanism of HDACis.
However, Newbold A et al. found broad HDACi vorinostat and selective HDACi MRLB-223 which can inhibit HADC 1 and 2, both have inhibitory effects on BCR-ABL-dependent cell and can induce the degradation of the BCR-ABL protein [51]. They also declared that decreasing the expression of HDAC6 was sufficient to induce hyperacetylation of HSP90 but to increase the expression of the BCR-ABL protein. Thus, they speculated that inhibiting the activity of HDAC6 to disrupt the BCR-ABL-HSP90 complex was not the major reason of the BCR-ABL protein degradation induced by HDACis. Other effective HSP90 inhibitors which can induce the degradation of the BCR-ABL protein can be seen in Table 1 [48, 49, 52-54].
Compounds confirmed effective in promoting BCR-ABL degradation
| Targeting approach | Compound property | Compound | Target | Proposed working mechanism | Reference |
|---|---|---|---|---|---|
| Targeting ubiquitin-proteasome pathway | Arsenic compound | As4S4 | c-CBL | Enhances c-CBL-mediated ubiquitination and degradation of BCR-ABL | 29 |
| HSP90 ATPase inhibitor | GA, 17-AAG, NB, NVP-AUY922, BIIB021, IPI504 | HSP90 | Destabilizes BCR-ABL by disrupting the complex of BCR-ABL and HSP90 | 43-54 | |
| HSP90 histone deacetylase inhibitor | LAQ824, FK228, MRLB-223, vorinostat | ||||
| Hsp90 inhibitor | EC141, andrographolide | ||||
| Diterpenoid | Oridonin | HSP70 | Actives HSF1 to transcrip- tionally up-regulate HSP70 and ubiquitin | 56 | |
| PP2A activator | Forskolin, butyryl-forskolin, 1,9-dideoxyforskol, FTY720 | PP2A, SHP-1 | Activates PP2A and SHP-1 to dephosphorylate BCR-ABL residues | 65 | |
| Tryptamine derivative | JNJ-26854165 | PP2A, SHP-1 | Promotes the expression of PP2A and SHP-1 to dephosphorylate BCR-ABL residues | 66 | |
| Targeting ubiquitin-lysosome pathway | PTP1B inhibitor | 3-(3,5-dibromo-4-hydroxybenzoyl)-2-ethyl-benzofuran-6-sulfonicacid-(4-(thiazol-2-ylsulfanyl)-phenyl)-amide | PTP1B | Inhibits PTP1B to phosphorylate BCR-ABL residues | 70 |
| Steroidal glycoside | SBF-1 | PTP1B | Blocks the interaction between PTP1B and BCR-ABL | 71 | |
| Tyrphostin derivative | WP1130 | Unknown | Promotes the ubiquitina- tion of BCR-ABL | 72, 73 | |
| Targeting autophagy -lysosome pathway | Arsenic compound | AS2O3 | BCR-ABL | Promotes p62/SQSTM1 -mediated localization of the BCR-ABL to the autolysosome | 76 |
| Tyrosine kinase inhibitor | Imatinib | BCR-ABL | Sequesters BCR-ABL into autolysosome | 77 |
3.3 HSP70 and Hsc70
HSP70 family includes more than 14 proteins (HSP70, HSP70-2, 4, 4L, 5-9, 12a, 14). And HSP70 protein consists of three major functional domains: N-terminal ATPase domain, substrate binding domain and C-terminal domain. HSP70 encoded by HSPA1A, HSPA1B and HSPA1L gene, is the most ubiquitous stress-inducible chaperone. The effects of HSP70 on the BCR-ABL protein' stability are different from those of HSP90, and there are two possible mechanisms to explain it. For one, HSP90 can form chaperone complex with p23, P50, HSP70 and P60. The mature HSP90-p23-p50 chaperone complex can make the client functional, while the immature HSP90-HSP70-p60 chaperone complex cannot. That can help to explain the phenomenon that transforming the BCR-ABL protein's chaperone from HSP90 to HSP70 can induce the BCR-ABL protein degradation. For another, HSP70 can promote the ubiquitination and proteasomal degradation of its interacting protein hypoxia-inducible factor-1α (HIF-1α) by recruiting HSP70's co-chaperone CHIP which is a E3 ligase, indicating that HSP70's cooperation with CHIP can transform HSP70's function from folding protein to degrading protein [55]. It was reported that oridonin can trigger the proteasomal degradation of wild-type and mutant BCR-ABL proteins by targeting HSP70, thereby inducing BCR-ABL-dependent cell growth inhibition and apoptosis. The mechanism behind it was that oridonin bound directly with the cysteine residue of heat shock factor 1 (HSF1) and activated it, which is the major regulator of gene transcription under specific conditions. The activated HSF1 further transcriptionally up-regulated HSP70 and ubiquitin, and finally led to CHIP-mediated ubiquitination and degradation of the BCR-ABL protein [56].
Hsc70 (HSP70-8) chaperone protein is encoded by HSPA8 gene [57]. Tsukahara F et al. found that the over-expression of Hsc70 increased the BCR-ABL protein level by 1.9 times. It was most probably that Hsc70 prevented the immature BCR-ABL protein from misfolding and promoted Hsc70-chaperoned-BCR-ABL protein to be further chaperoned by HSP90 for maturation. Additionally, Hsc70 could associate and stabilize the HSP90-released BCR-ABL protein induced by HSP90 inhibitor, indicating that Hsc70 also can protect the HSP90-unchaperoned immature BCR-ABL protein against CHIP-induced protein degradation [30].
3.4 Co-chaperones
Co-chaperones which associate with the chaperones also play important roles for their clients. For example, the co-chaperones of HSP90 can assist the conformation change of HSP90, regulate the ATPase activity of HSP90 and load HSP90's client proteins [41, 58].
Mucin1 (MUC1) which is a transmembrane oncogenic protein has been reported to interact with diverse effectors and is involved in the pathogenesis of various hematological malignancies [59]. The newly synthesized MUC1 undergoes auto-cleavage and generates two subunits in the endoplasmic reticulum. Extracellular N-terminal subunit (MUC1-N) and transmembrane C-terminal subunit (MUC1-C) form a stable noncovalent heterodimer at the cell surface. MUC1-C contains a 58-amino acid extracellular domain, a 28-amino acid transmembrane domain and a 72-amino acid cytoplasmic domain.
Kawano T et al. demonstrated that inhibiting the expression of MUC1 can decrease BCR-ABL-dependent cell's self-renewal capacity and promote the differentiation of primary CML cell. And these effects were related to the BCR-ABL protein degradation induced by MUC1 silence. MUC1-C cytoplasmic domain (1-45 amino acids) bound directly to the BCR-ABL protein (BCR 197-426 amino acids) thereby stabilizing the BCR-ABL protein. MUC1 also can bind directly to HSP90, but this binding is not intended to maintain the stability of MUC1. Thus it is inferred that MUC1 acts as the HSP90's co-chaperone to maintain the BCR-ABL protein's stability by forming a MUC1-HSP90-BCR-ABL complex [60, 61].
Newly synthesized BCR-ABL protein is stabilized by Hsc70 and then passed on to HSP90 for maturation. In this process, Bcl-2-associated athanogene 1 (BAG1), which is a nucleotide exchange factor of Hsc70, can promote the immature BCR-ABL protein to be unloaded from the Hsc70 for maturation. Accordingly, it can explain why interfering with the expression of BAG1 can cause significant degradation of the BCR-ABL protein [30, 62]. However, BAG1 also can bind to the proteasome and the BCR-ABL protein. After BAG1 recognizing the newly synthesized HSP90-unchaperoned immature BCR-ABL protein via the BCR (1-413 amino acids) domain and the ABL kinase domain, it promotes the BCR-ABL protein to bind with proteasome and to be degraded through the CHIP-mediated degradation pathway. In this process, since Hsc70 and the BCR-ABL protein have competitive binding with BAG1, Hsc70 inhibits the BAG1-directed and CHIP-mediated BCR-ABL protein degradation [30, 62]. Accordingly, BAG1 has two opposite effects on the stability of the BCR-ABL protein.
3.5 Phosphatases
The protein-tyrosine kinases and the protein-tyrosine phosphatases (PTPs) together regulate the phosphorylation of protein, thereby playing critical roles in cell cycle, cell survival and other cellular processes. Src homology region 2 domain-containing phosphatase-2 (SHP-2) is a tyrosine phosphatase comprising the SH2 domain. SHP-2 itself can be phosphorylated by the BCR-ABL protein. Furthermore, SHP-2 can form a complex with the BCR-ABL protein and the complex was crucial for BCR-ABL-dependent hematopoietic cell transformation. Inhibiting the expression of SHP-2 can induce the BCR-ABL protein degradation through the proteasome-mediated pathway. However, when SHP-2's catalytic activity was inhibited, it still can stabilize the BCR-ABL protein, indicating that SHP-2 stabilizing the BCR-ABL protein was independent of its catalytic activity. Since SHP-2 was detected to associate with HSP90, Chen J et al. considered that SHP-2 prevented the proteasome-mediated BCR-ABL protein degradation by the cooperation with HSP90 [63].
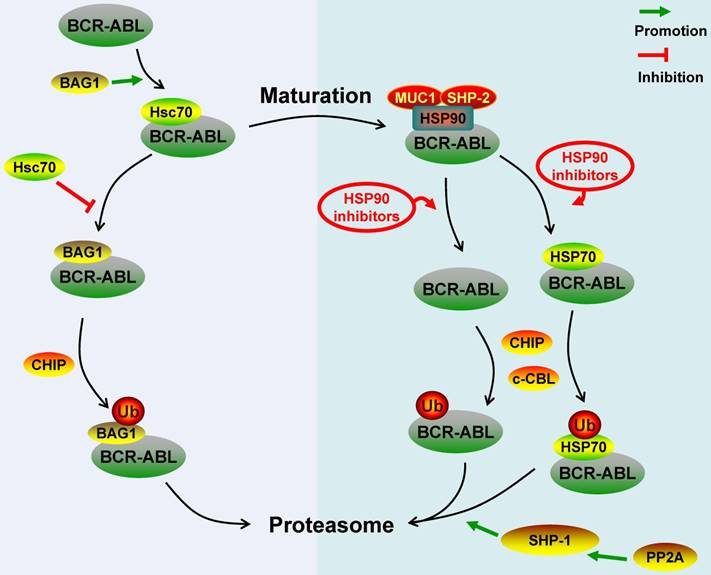
Protein phosphatase 2A (PP2A) is a tumor suppressor. The BCR-ABL protein can induce inactivation of PP2A by promoting the expression of SET, which is a nucleus/cytoplasm-localized phosphoprotein and a potent inhibitor of PP2A. Functional inactivation of PP2A is essential for BCR-ABL-dependent leukemogenesis [64]. Neviani P et al. observed that PP2A activators can inhibit BCR-ABL-dependent leukemogenesis by inducing the wild-type and T315I-mutant BCR-ABL protein degradation via proteasome system. In terms of the mechanism, the activated PP2A can activate Src homology region 2 domain-containing phosphatase-1 (SHP-1), which further dephosphorylated the BCR-ABL protein and made the BCR-ABL protein be prone to be degraded by proteasome [4, 65]. Similarly, You L et al. found that up-regulating the expression of PP2A and SHP-1 by JNJ-26854165 could also promote the proteasomal degradation of the BCR-ABL protein [66]. The BCR-ABL protein dephosphorylated by SHP-1 still has a complete three-dimensional structure, so we proposed that the PP2A- or SHP-1-induced BCR-ABL protein degradation was mediated by c-CBL [30]. Key regulatory molecules involved in the ubiquitin-proteasome degradation pathway of the BCR-ABL protein were summarized in Figure 1.
Schematic represents regulatory molecules involved in the ubiquitin-proteasome degradation pathway of BCR-ABL protein. Both c-CBL and CHIP are E3 ligases of BCR-ABL, except that c-CBL mediates the degradation of mature BCR-ABL protein while CHIP mediates the immature BCR-ABL protein degradation. Newly synthesized BCR-ABL protein is chaperoned by Hsc70 for stabilization, this process is promoted by BAG1. However, BAG1 also can lead immature BCR-ABL protein into proteasome and promote CHIP-mediated BCR-ABL protein degradation via proteasome system. BCR-ABL protein is chaperoned by HSP90 for further maturation and function, MUC-1 and SHP-2 act as HSP90's co-chaperones to stabilize BCR-ABL protein. HSP90 inhibitors disrupt the interaction between HSP90 and BCR-ABL protein or switch the BCR-ABL-associated chaperone complex from HSP90 to HSP70, and all these inhibitors can promote c-CBL- and/or CHIP-mediated BCR-ABL protein proteasome degradation. Additionally, PP2A can activate SHP-1 to dephosphorylate BCR-ABL protein residues, which can make BCR-ABL protein be prone to proteasome-mediated degradation.
Schematic represents regulatory molecules involved in the ubiquitin-lysosome, autophagy-lysosome and caspase-mediated degradation pathway of BCR-ABL protein. PTP1B dephosphorylates BCR-ABL residues to prevent the ubiquitination of BCR-ABL protein, aggregation formation and lysosome-mediated degradation. In the AS2O3-induced BCR-ABL protein autophagy-lysosome degradation, BCR-ABL protein interacts with p62/SQSTM1. In the imatinib-induced BCR-ABL protein autophagy-lysosome degradation, BCR-ABL protein co-locates with autophagy markers LC3 and GABARAP. Caspase 3 and caspase 7 can down-regulate BCR-ABL protein level by cleavage.
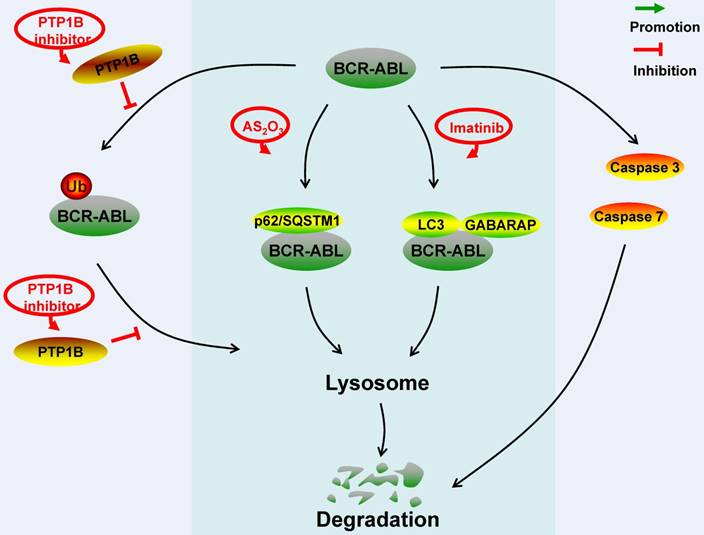
4. The molecules regulate the BCR-ABL protein degradation via ubiquitin-lysosome pathway
In fact, some ubiquitin-modified proteins in cell do not enter the proteasome but enter lysosome for degradation, such as the receptor tyrosine kinase [67]. Here we refer to this pattern of degradation as ubiquitin-lysosome degradation pathway. Protein-tyrosine phosphatase 1B (PTP1B) is considered to be a negative regulator of intracellular signaling pathways and is involved in tumorigenesis [68, 69]. Alvira D et al. revealed that the PTP1B was up-regulated in BCR-ABL-expressing cell and PTP1B expression inhibition could reduce BCR-ABL-dependent cell's viability, indicating that PTP1B plays a role in the BCR-ABL-dependent cell [70]. Besides the PTP1B expression inhibition, inhibiting the activity of PTP1B also can decrease the BCR-ABL protein level, suggesting that the phosphatase activity of PTPT1B is crucial for maintaining the stability of the BCR-ABL protein [70]. Inhibiting the activity of PTP1B increased the ubiquitination of the BCR-ABL protein, and the ubiquitinated BCR-ABL protein further formed aggregation which will be degraded by lysosome. Alvira D et al. considered that the phosphorylation of the BCR-ABL protein induced by PTP1B activity inhibition created a recognition signal for E3 ligases to bind with the BCR-ABL protein, and the binding was significant for the BCR-ABL protein ubiquitination increase [70, 71].
SBF-1, a synthetic steroidal glycoside, had strong anti-tumor effects on the imatinib-sensitive and imatinib-resistant CML cell. The molecular mechanism of the anti-leukemia effects of SBF-1 was probably that SBF-1 promoted the BCR-ABL protein to be ubiquitinated and further be degraded in the lysosome. Specifically, SBF-1 can bind with both the BCR-ABL protein and PTP1B to block the binding of the BCR-ABL protein with PTP1B. That further indicated that PTP1B is crucial for the stabilization of the BCR-ABL protein [71].
Another compound WP1130 was also reported to induce the down-regulation of the BCR-ABL protein; it can increase the ubiquitination of the BCR-ABL protein without affecting the complex of BCR-ABL-HSP90 or BCR-ABL-HSP70. The degradation of the BCR-ABL protein induced by WP1130 can't be attenuated by the proteasome inhibitors, so we speculate that the WP1130-induced BCR-ABL protein degradation is mediated through the ubiquitin-lysosome system. Sun H et al. found that WP1130 also can affect the cellular deubiquitinase activity, thus they considered that WP1130 inhibits the specific deubiquitinases of the BCR-ABL protein and thereby prevents the deubiquitination of the BCR-ABL protein [72, 73]. Other effective inhibitors which can induce the BCR-ABL protein degradation via ubiquitin-lysosome pathway are shown in Table 1.
5. The molecules regulate the BCR-ABL protein degradation via autophagy-lysosome pathway
The BCR-ABL protein can regulate the autophagy via MAPK15 and PI3K/AKT-FOXO4-ATF5-mTOR signaling pathways [74, 75]. Additionally, the BCR-ABL protein itself also can be degraded by the autophagy-lysosome pathway. AS2O3 and imatinib are two reported compounds which can induce the autophagy-lysosome-mediated degradation of the BCR-ABL protein [76, 77].
Both the AS2O3-induced degradation of the BCR-ABL protein and the anti-leukemia effects of AS2O3 can be alleviated by the autophagy inhibition, such as inhibiting the expression of key elements in the autophagic process and reducing the activity of lysosomal hydrolases. In the AS2O3-induced BCR-ABL protein degradation, the BCR-ABL protein was found to interact with autophagosome cargo protein p62/SQSTM1, and they co-located in autophagosome structures and autolysosomes. p62/SQSTM1 has been reported to facilitate the degradation of protein aggregates by binding directly to the autophagy regulator Atg8/LC3 [78]. Moreover, the activity of cathepsin B was essential for the degradation of the BCR-ABL protein induced by AS2O3 [76].
Elzinga BM et al. reported that imatinib exerted anti-leukemia effects not only by inhibiting the tyrosine kinase activity of the BCR-ABL protein but also by promoting the degradation of the BCR-ABL protein through autophagy. In the degradation of the BCR-ABL protein induced by imatinib, the BCR-ABL protein was sequestered into lysosome and co-located with the autophagy markers LC3 and GABARAP [77].
6. The caspase-mediated degradation of the BCR-ABL protein
It was approved that the BCR-ABL protein confers cell the characteristics of differentiation inhibition [79-81]. Furthermore, the BCR-ABL protein was down-regulated during the differentiation of BCR-ABL-dependent cell [82]. In the p53-induced CML cell differentiation process, Di Bacco AM et al. observed that the down-regulation of BCR-ABL protein was dependent on the activation of caspases. They proved that the BCR-ABL protein was cleaved by specific caspases and two ABL-related fragments of 64 and 52 kDa were generated. In further experiment in vitro by constructing several recombinant caspases, they confirmed that caspase 3 and 7 can cleave the BCR-ABL protein, while caspase 6 and 8 don't possess this capability [83]. However, the site of the BCR-ABL protein cleaved by caspase was still unknown.
Survivin is a member of the inhibitor of apoptosis family, which is highly expressed during the blast crisis of Ph+ patients [84]. The expression of survivin can be up-regulated by the BCR-ABL protein. The disruption of survivin also can induce the caspase-dependent down-regulation of the BCR-ABL protein in turn. Survivin disruption activated caspase 3, 7 and 9. One or some of them cleaved the BCR-ABL protein to generate fragments of 85 and 65 kDa, and further made BCR-ABL-dependent cell be more prone to apoptosis induced by tyrosine kinase inhibitor [85, 86].
Conclusion and perspective
Targeting the degradation of the BCR-ABL protein for the treatment of Ph+ hematological malignancies has drawn great attention recently. The key molecules involved in the degradation pathway of the BCR-ABL protein were summarized in Figure 1, 2. Both c-CBL and CHIP are E3 ligases of the BCR-ABL protein, except that c-CBL mediates the degradation of the mature BCR-ABL protein while CHIP mediates the immature BCR-ABL protein degradation. SH2-U-box and SH2-RING which are two E3 ubiquitin ligases specifically targeting the BCR-ABL protein have already been artificially synthesized. HSP90, MUC1, Hsc70 and BAG1 act as chaperones or co-chaperones for the BCR-ABL protein to maintain the stability of the BCR-ABL protein. SHP-2 stabilizes the BCR-ABL protein independent of its catalytic activity; it acts as the co-chaperone of HSP90 to stabilize the BCR-ABL protein. Disrupting the BCR-ABL protein's interaction with HSP90, MUC1, Hsc70, BAG1 and SHP-2 can result in the degradation of the BCR-ABL protein via ubiquitin-proteasome pathway. Although PP2A, SHP-1 and PTP1B all can dephosphorylate the residues of the BCR-ABL protein, they have different effects on the stability of the BCR-ABL protein. The dephosphorylation of the BCR-ABL protein induced by PP2A and SHP-1 can make the BCR-ABL protein undergo the ubiquitin-proteasome-mediated degradation more easily. However, the dephosphorylation of the BCR-ABL protein induced by PTP1B prevents the BCR-ABL protein from being degraded through ubiquitin-lysosome system. Cathepsin B, caspase 3 and 7 are hydrolases that can target the BCR-ABL protein.
Targeting the degradation pathway of the BCR-ABL protein can disrupt the integrity and stability of the BCR-ABL proteins whatever wild type or mutants. Compounds that can induce the degradation of the BCR-ABL protein were shown by Table 1. Compounds targeting the ubiquitin proteasome degradation pathway of the BCR-ABL protein are mainly HSP90 inhibitors. In the aspect of the autophagy lysosome degradation of the BCR-ABL protein, only AS2O3 and imatinib have been found to induce such degradation. Nimmanapalli R et al. reported that LAQ824 enhanced the CML cell's apoptosis induced by imatinib. There were synergistic effects of As4S4 and imatinib in treating BCR-ABL-dependent leukemia. Goussetis DJ et al. raised the possibility that combination of PTP1B inhibitors with AS2O3 may further increase the formation of the p62/SQSTM1-BCR-ABL complex therefore enhancing the anti-leukemic effects. All these indicate that compounds combination may be a therapeutic strategy for the BCR-ABL-dependent leukemia in future.
Since the BCR-ABL protein plays a crucial role in the pathogenesis of BCR-ABL-dependent leukemia, further studies of the molecular mechanisms of the degradation of the BCR-ABL protein may help develop the new target therapy for BCR-ABL-dependent leukemia. A novel therapeutic method-Targeted Protein Degradation (TPD), which selectively degrades abnormal proteins essential for human disorders, has gained widespread attention [87]. A more feasible TPD method called PROTAC has been developed. PROTAC is a molecule that can simultaneously bind E3s and the protein of interest, thus driving the target protein to be ubiquitinated for degradation [88]. Recently, a PROTAC for the BCR-ABL protein degradation has been designed [89]. The compounds-induced proteolysis of selected substrates is undoubtedly a method, for it uses the cell's own degradation system to degrade the pathogenic BCR-ABL protein.
Abbreviations
CML: chronic myeloid leukemia; TKI: tyrosine kinase inhibitor; B-ALL: B-cell acute lymphoblastic leukemia; MPAL: mixed phenotype acute leukemia; AML: acute myeloid leukemia; Ph: Philadelphia; SH2: SRC-homology-2; NLS: nuclear localization signal; NES: nuclear export signal; DBD: DNA-binding domain; ABD: actin-binding domain; CC: coiled-coil; Rho-GEF: Rho guanine nucleotide exchange factor; PH: pleckstrin homology; Cal-B: calcium-dependent lipid-binding; RAC-GAP: RAC guanosine triphosphatase-activating protein; CP: chronic phase; BP: blastic phase; AP: accelerated phase; CBA: chromosome banding analysis; FISH: fluorescence in situ hybridization; RQ-PCR: Quantitative real-time PCR; HSCs: hematopoietic stem cells; UPS: ubiquitin proteasome system; E1s: E1 ubiquitin-activating enzymes; E2s: E2 ubiquitin-conjugating enzymes; E3s: E3 ubiquitin ligases; DUBs: deubiquitylating enzymes; c-CBL: Casitas B-lineage lymphoma; CHIP: Carboxyl terminus of the Hsc70-interacting protein; TKB: tyrosine-kinase-binding; RF: RING finger; TPR: tetratricopeptide repeat; PROTAC: proteolysis targeting chimeras; Grb2: growth factor receptor-bound protein 2; HSPs: heat-shock proteins; GA: geldanamycin; 17-AAG: 17-allylamino-17-demethoxygeldanamycin; NB: novobiocin; HDAC: histone deacetylase; HDACi: Histone deacetylase inhibitor; FK228: depsipeptide; GR: Glucocorticoid Receptor; HIF-1: hypoxia-inducible factor-1; HSF1: heat shock factor 1; MUC1: Mucin1; BAG1: Bcl-2-associated athanogene 1; PTPs: protein-tyrosine phosphatases; SHP-2: Src homology region 2 domain-containing phosphatase-2; PP2A: protein phosphatase 2A; SHP-1: Src homology region 2 domain-containing phosphatase-1; PTP1B: protein-tyrosine phosphatase 1B; TPD: Targeted Protein Degradation.
Supplementary Material
Supplementary figure.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81572796, 81172322 and 81302006), Shanghai Municipal Education Committee (13ZZ089), Science and Technology Committee of Shanghai (14401901500), Science and Technology Committee of Baoshan District (12-E-2), NO.3 People's Hospital affiliated to Shanghai Jiao-Tong University School of Medicine (sy2013-008).
Author Contributions
Feng-Hou Gao and Han-Qing Zhu drafted the outline of the manuscript. Feng-Hou Gao and Han-Qing Zhu conducted the literature review. Han-Qing Zhu assessed the articles and wrote the manuscript. Feng-Hou Gao revised the manuscript. All authors have read and approved the final version of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375-84
2. Zhou T, Medeiros LJ, Hu S. Chronic Myeloid Leukemia: Beyond BCR-ABL1. Curr Hematol Malig Rep. 2018;13:435-45
3. Burslem GM, Smith BE, Lai AC. et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol. 2018;25:67-77 e3
4. Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619-30
5. Melo J. Inviting leukemic cells to waltz with the devil. Nat Med. 2001;7:156-7
6. Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7:228-34
7. Skourides PA, Perera SA, Ren R. Polarized distribution of Bcr-Abl in migrating myeloid cells and co-localization of Bcr-Abl and its target proteins. Oncogene. 1999;18:1165-76
8. Patel H, Marley SB, Greener L. et al. Subcellular distribution of p210(BCR-ABL) in CML cell lines and primary CD34+ CML cells. Leukemia. 2008;22:559-71
9. Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172-83
10. Weisberg E, Manley PW, Cowan-Jacob SW. et al. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345-56
11. Barnes DJ, Schultheis B, Adedeji S. et al. Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene. 2005;24:6432-40
12. Gaiger A, Henn T, Horth E. et al. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood. 1995;86:2371-8
13. Baccarani M, Deininger MW, Rosti G. et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872-84
14. Qin YZ, Huang XJ. Molecular Detection of BCR-ABL in Chronic Myeloid Leukemia. Methods Mol Biol. 2016;1465:1-15
15. Ou J, Vergilio JA, Bagg A. Molecular diagnosis and monitoring in the clinical management of patients with chronic myelogenous leukemia treated with tyrosine kinase inhibitors. Am J Hematol. 2008;83:296-302
16. Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385:1447-59
17. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640-53
18. Helgason GV, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood. 2011;118:2035-43
19. Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834-48
20. Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. 2014;5:5659
21. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780-6
22. Kwon YT, Ciechanover A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem Sci. 2017;42:873-86
23. Bedford L, Lowe J, Dick LR. et al. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29-46
24. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664-73
25. Torgersen ML, Simonsen A. Autophagy: friend or foe in the treatment of fusion protein-associated leukemias. Autophagy. 2013;9:2175-7
26. Yanagawa T, Funasaka T, Tsutsumi S. et al. Regulation of phosphoglucose isomerase/autocrine motility factor activities by the poly(ADP-ribose) polymerase family-14. Cancer Res. 2007;67:8682-9
27. Fan Y, Qu X, Ma Y. et al. Cbl-b promotes cell detachment via ubiquitination of focal adhesion kinase. Oncol Lett. 2016;12:1113-8
28. Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301-7
29. Mao JH, Sun XY, Liu JX. et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2010;107:21683-8
30. Tsukahara F, Maru Y. Bag1 directly routes immature BCR-ABL for proteasomal degradation. Blood. 2010;116:3582-92
31. Davies GC, Ettenberg SA, Coats AO. et al. Cbl-b interacts with ubiquitinated proteins; differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene. 2004;23:7104-15
32. Peschard P, Kozlov G, Lin T. et al. Structural basis for ubiquitin-mediated dimerization and activation of the ubiquitin protein ligase Cbl-b. Mol Cell. 2007;27:474-85
33. Zhang QY, Mao JH, Liu P. et al. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proc Natl Acad Sci U S A. 2009;106:3378-83
34. Yin T, Wu YL, Sun HP. et al. Combined effects of As4S4 and imatinib on chronic myeloid leukemia cells and BCR-ABL oncoprotein. Blood. 2004;104:4219-25
35. VanPelt J, Page RC. Unraveling the CHIP:Hsp70 complex as an information processor for protein quality control. Biochim Biophys Acta Proteins Proteom. 2017;1865:133-41
36. Cromm PM, Crews CM. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol. 2017;24:1181-90
37. Kong F, Zhang J, Li Y. et al. Engineering a single ubiquitin ligase for the selective degradation of all activated ErbB receptor tyrosine kinases. Oncogene. 2014;33:986-95
38. Toure M, Crews CM. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem Int Ed Engl. 2016;55:1966-73
39. Ru Y, Wang Q, Liu X. et al. The chimeric ubiquitin ligase SH2-U-box inhibits the growth of imatinib-sensitive and resistant CML by targeting the native and T315I-mutant BCR-ABL. Sci Rep. 2016;6:28352
40. Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324-32
41. Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213-7
42. Solarova Z, Mojzis J, Solar P. Hsp90 inhibitor as a sensitizer of cancer cells to different therapies. Int J Oncol. 2015;46:907-26
43. An WG, Schulte TW, Neckers LM. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ. 2000;11:355-60
44. Nimmanapalli R, O'Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001;61:1799-804
45. Nimmanapalli R, Fuino L, Bali P. et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126-35
46. Tong WG, Estrov Z, Wang Y. et al. The synthetic heat shock protein 90 (Hsp90) inhibitor EC141 induces degradation of Bcr-Abl p190 protein and apoptosis of Ph-positive acute lymphoblastic leukemia cells. Invest New Drugs. 2011;29:1206-12
47. Wu LX, Xu JH, Zhang KZ. et al. Disruption of the Bcr-Abl/Hsp90 protein complex: a possible mechanism to inhibit Bcr-Abl-positive human leukemic blasts by novobiocin. Leukemia. 2008;22:1402-9
48. Tao W, Chakraborty SN, Leng X. et al. HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex in leukemia cells. Genes Cancer. 2015;6:19-29
49. Okabe S, Tauchi T, Nakajima A. et al. Depsipeptide (FK228) preferentially induces apoptosis in BCR/ABL-expressing cell lines and cells from patients with chronic myelogenous leukemia in blast crisis. Stem Cells Dev. 2007;16:503-14
50. Kovacs JJ, Murphy PJ, Gaillard S. et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601-7
51. Newbold A, Matthews GM, Bots M. et al. Molecular and biologic analysis of histone deacetylase inhibitors with diverse specificities. Mol Cancer Ther. 2013;12:2709-21
52. Liu SH, Lin CH, Liang FP. et al. Andrographolide downregulates the v-Src and Bcr-Abl oncoproteins and induces Hsp90 cleavage in the ROS-dependent suppression of cancer malignancy. Biochem Pharmacol. 2014;87:229-42
53. He W, Ye X, Huang X. et al. Hsp90 inhibitor, BIIB021, induces apoptosis and autophagy by regulating mTOR-Ulk1 pathway in imatinib-sensitive and -resistant chronic myeloid leukemia cells. Int J Oncol. 2016;48:1710-20
54. Peng C, Brain J, Hu Y. et al. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood. 2007;110:678-85
55. Luo W, Zhong J, Chang R. et al. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem. 2010;285:3651-63
56. Huang H, Weng H, Dong B. et al. Oridonin Triggers Chaperon-mediated Proteasomal Degradation of BCR-ABL in Leukemia. Sci Rep. 2017;7:41525
57. Stricher F, Macri C, Ruff M. et al. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9:1937-54
58. Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021-30
59. Stroopinsky D, Kufe D, Avigan D. MUC1 in hematological malignancies. Leuk Lymphoma. 2016;57:2489-98
60. Kawano T, Ito M, Raina D. et al. MUC1 oncoprotein regulates Bcr-Abl stability and pathogenesis in chronic myelogenous leukemia cells. Cancer Res. 2007;67:11576-84
61. Ren J, Bharti A, Raina D. et al. MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene. 2006;25:20-31
62. Alberti S, Esser C, Hohfeld J. BAG-1-a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress Chaperones. 2003;8:225-31
63. Chen J, Yu WM, Daino H. et al. SHP-2 phosphatase is required for hematopoietic cell transformation by Bcr-Abl. Blood. 2007;109:778-85
64. Lucas CM, Harris RJ, Giannoudis A. et al. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood. 2011;117:6660-8
65. Neviani P, Santhanam R, Trotta R. et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355-68
66. You L, Liu H, Huang J. et al. The novel anticancer agent JNJ-26854165 is active in chronic myeloid leukemic cells with unmutated BCR/ABL and T315I mutant BCR/ABL through promoting proteosomal degradation of BCR/ABL proteins. Oncotarget. 2017;8:7777-90
67. de Pril R, Fischer DF, Maat-Schieman ML. et al. Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum Mol Genet. 2004;13:1803-13
68. Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439-46
69. Yip SC, Saha S, Chernoff J. PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci. 2010;35:442-9
70. Alvira D, Naughton R, Bhatt L. et al. Inhibition of protein-tyrosine phosphatase 1B (PTP1B) mediates ubiquitination and degradation of Bcr-Abl protein. J Biol Chem. 2011;286:32313-23
71. Elgehama A, Chen W, Pang J. et al. Blockade of the interaction between Bcr-Abl and PTB1B by small molecule SBF-1 to overcome imatinib-resistance of chronic myeloid leukemia cells. Cancer Lett. 2016;372:82-8
72. Bartholomeusz GA, Talpaz M, Kapuria V. et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109:3470-8
73. Sun H, Kapuria V, Peterson LF. et al. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117:3151-62
74. Colecchia D, Rossi M, Sasdelli F. et al. MAPK15 mediates BCR-ABL1-induced autophagy and regulates oncogene-dependent cell proliferation and tumor formation. Autophagy. 2015;11:1790-802
75. Sheng Z, Ma L, Sun JE. et al. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118:2840-8
76. Goussetis DJ, Gounaris E, Wu EJ. et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120:3555-62
77. Elzinga BM, Nyhan MJ, Crowley LC. et al. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol. 2013;88:455-62
78. Pankiv S, Clausen TH, Lamark T. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
79. Ray S, Bullock G, Nunez G. et al. Enforced expression of Bcl-XS induces differentiation and sensitizes chronic myelogenous leukemia-blast crisis K562 cells to 1-beta-D-arabinofuranosylcytosine-mediated differentiation and apoptosis. Cell Growth Differ. 1996;7:1617-23
80. Ghaffari S, Daley GQ, Lodish HF. Growth factor independence and BCR/ABL transformation: promise and pitfalls of murine model systems and assays. Leukemia. 1999;13:1200-6
81. Fang G, Kim CN, Perkins CL. et al. CGP57148B (STI-571) induces differentiation and apoptosis and sensitizes Bcr-Abl-positive human leukemia cells to apoptosis due to antileukemic drugs. Blood. 2000;96:2246-53
82. Eisbruch A, Blick M, Evinger-Hodges MJ. et al. Effect of differentiation-inducing agents on oncogene expression in a chronic myelogenous leukemia cell line. Cancer. 1988;62:1171-8
83. Di Bacco AM, Cotter TG. p53 expression in K562 cells is associated with caspase-mediated cleavage of c-ABL and BCR-ABL protein kinases. Br J Haematol. 2002;117:588-97
84. Badran A, Yoshida A, Wano Y. et al. Expression of the antiapoptotic gene survivin in chronic myeloid leukemia. Anticancer Res. 2003;23:589-92
85. Wang Z, Sampath J, Fukuda S. et al. Disruption of the inhibitor of apoptosis protein survivin sensitizes Bcr-abl-positive cells to STI571-induced apoptosis. Cancer Res. 2005;65:8224-32
86. Wang Z, Pelus LM. Disruption of Survivin in K562 cells elevates telomerase activity and protects cells against apoptosis induced by the Bcr-abl kinase inhibitor STI571. Cancer Ther. 2008;6:603-10
87. Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138-44
88. Raina K, Crews CM. Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol. 2017;39:46-53
89. Demizu Y, Shibata N, Hattori T. et al. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett. 2016;26:4865-9
Author contact
![]() Corresponding author: Feng-Hou Gao, e-mail: fenghougaocom
Corresponding author: Feng-Hou Gao, e-mail: fenghougaocom