Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(10):2176-2184. doi:10.7150/jca.32731 This issue Cite
Research Paper
UBE2C overexpression in melanoma and its essential role in G2/M transition
Guolong Liu1*, Jun Zhao2*, Boyu Pan1, Gang Ma1, Liren Liu1 ![]()
1. Department of Gastrointestinal Cancer Biology, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin 300060, P.R. China
2. Department of Bone & Soft Tissue Tumor, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin 300060, P.R. China
*The authors contributed equally to this work.
Received 2019-1-1; Accepted 2019-4-7; Published 2019-5-21
Abstract
Ubiquitin‑conjugating enzyme E2C (UBE2C) is a key regulator of cell cycle progression, and its aberrant expression has been implicated in various malignancies. However, its clinical and biological roles in malignant melanoma is still unclear. In this study, we found a significant high expression level of UBE2C in melanoma by an in silico analysis of The Cancer Genome Atlas (TCGA) database, which was further validated using fresh melanoma samples. The KM plotter showed that UBE2C level was statistically related to the overall survival (OS) of melanoma patients (p<0.01). RNA interference of UBE2C inhibited the growth of melanoma cells via deactivating ERK/Akt signaling pathways, and blocked the G2/M transition through downregulation of both the level and the activity of mitosis promoting factor (MPF), triggering the apoptosis of melanoma cells. Further, silencing of UBE2C significantly inhibited the xenografted tumor growth on nude mice, indicating an important role of UBE2C in melanoma growth in vivo. Together, our results show that UBE2C may serve as a novel prognostic biomarker as well as a potential therapeutic target for melanoma.
Keywords: UBE2C, melanoma, G2/M arrest, cell cycle, apoptosis
Introduction
Melanoma is the most aggressive form of skin malignancy, accounting for more than 80% of all skin tumor-related deaths [1]. Its incidence has been increasing faster than that of any other tumor type during the last decades, though representing only 4% of all skin malignancy cases [2]. In spite of recent advancement in the therapeutic approach for melanoma, such as immunotherapy, photothermal and targeted molecular therapy, the long term survival of melanoma patients remains poor [3, 4]. Therefore, it is an urgent need to elucidate the detailed mechanisms underlying melanoma tumorigenesis, so as to develop effective therapies for this deadly disease.
UBE2C belongs to Ubiquitin-conjugating enzyme (E2) family, which mediate the transfer of activated ubiquitin molecule either directly or from the E3 ligase to the preferred lysine residue(s) on a substrate [5]. The physiological role of UBE2C or its homologs in mitosis has been well characterized in various eukaryotes, including human cells. Acting in concert with the anaphase-promoting complex/cyclosome (APC/C) E3 ligase, UBE2C promotes cell cycle through mitosis by destructing mitotic cyclin B1[6-8]. UBE2C has also been reported to be required to dissociate the Mad2-Cdc20 complex by ubiquitylating Cdc20, thus abrogating the spindle-assembly checkpoint [9]. Furthermore, UBE2C is involved in the dissociation of sister chromatids in late mitosis by inducing APC/C catalyzed degradation of securin, an enzyme that prevents the chromatids segregation [10]. Notably, UBE2C has been proposed to be rate-limiting in the late G1 phase, when it is required to degrade cyclin A by APC/C to prevent premature DNA replication[11]. However, the role of UBE2C in interphase beyond this finding remains largely elusive.
Given UBE2C plays a critical role in proper cell cycle progression, its aberrant expression may lead to pathological consequences. Indeed, cells over-expressing UBE2C failed to maintain spindle checkpoint activity after entering mitosis, resulting in chromosome missegregation and aneuploidy [9]. Intriguingly, ube2c transgenic mice were prone to a broad spectrum of spontaneous tumors, demonstrating a driving role of UBE2C overexpression in tumorigenesis [12]. In human, amplification and/or overexpression of ube2c gene have been reported in various malignancies, and high expression of UBE2C was associated with poor clinical outcome [5, 13-22]. Furthermore, loss-of-function assays revealed that silencing of UBE2C expression could reduce the proliferation rate of both normal and cancerous cells [16, 18, 23]. Thus, UBE2C has emerged as a biological marker for cancer as well as a potential target for anti-cancer therapy.
In this study, we first observed the overexpression of UBE2C in malignant melanoma and showed that high UBE2C level was associated with poor survival in these patients. The following loss-of-function assays demonstrated that UBE2C played an essential role in G2/M transition rather than functioning as a mitotic regulator in melanoma cells. Further mechanism exploration revealed that down-regulation of both the level and the activity of MPF led to the G2/M arrest upon UBE2C suppression. These findings suggest UBE2C may represent a novel diagnostic and prognostic marker as well as a promising therapeutic target for malignant melanoma.
Materials and Methods
In silico analysis of UBE2C expression in melanoma
In order to determine the expression level of UBE2C in melanoma, we performed an in silico analysis using The Cancer Genome Atlas database (TCGA). The association between UBE2C expression level and the overall survival of melanoma patients was assessed using Kaplan Meier Plotter based on TCGA data.
Patients and melanoma samples
Fresh melanoma tissues and normal control tissues were collected from 9 melanoma patients who received radical resection at the Department of Bone & Soft Surgery in Tianjin Medical University Cancer Institute and Hospital (Tianjin, China). Upon resection, these samples were stored in a liquid nitrogen container until extracting RNAs. None of these patients received neo-adjuvant chemo-radiation therapy and the relevant clinical data were available. The resected melanoma samples from these patients were confirmed histologically. The experimental procedure involving melanoma patients was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (Protocol# 20150304) and was performed according to the relevant regulations. Written informed consents were signed and obtained from all participants, and the study conformed to the ethical principles set forth by the Declaration of Helsinki.
Cell lines and reagent
Melanoma cell lines A375 and SK-MEL-28 were purchased from ATCC (Manassas, VA, USA). A375 cells were cultured in DMDM with 20% FBS, whereas SK-MEL-28 cells were cultured in MEM with 10% FBS. All cells were maintained under humanized condition (37°C, 5% CO2) and the continual culture did not exceed two months. MG132 was purchased from Cell Signaling Technology (cat#PD98059, Danvers, MA, USA).
Lentiviral shUBE2C construction
The shRNA oligos of UBE2C were designed according to its sequence in the NCBI database as following: 5'-GTTCCTCACGCCCTGCTAT-3' and 5'-CTCCCGTCATGTGCTTCAC-3'. The fragments of shRNA were inserted into the lentivirus vector and transfected into 293T cells with packaging vectors using Lipofectamine 3000. After 48 hours, the recombinant lentivirus was collected from the media for further infection.
qRT-PCR analysis
RNA was extracted using TRIzol (Thermo Fisher Scientific) and cDNAs were synthesized using PrimeScript™ RT reagent Kit (TaKaRa). The qPCR assays were performed using SYBR® Premix Ex Taq™ II (TaKaRa) on QuantStudio™ 5 Real-Time PCR System (Thermo Fisher Scientific). GAPDH was used as the internal control simultaneously. One representative result of two independent assays was shown.
Western blot analysis
Cell lysis buffer (100 mM NaCl, 10 mM EDTA (pH 8.0), 50 mM Tris-Cl (pH 8.0) and 0.5% (v/v) Triton X-100) with EDTA-free complete protease and phosphatase inhibitors (Roche) was used to extract proteins. Lysates were separated on 10% SDS-PAGE gel and transferred to PVDF membranes. The targets were detected using Amersham Imager 600 (GE). GAPDH was used as the loading control.
Cell functional assays
Cell viability was measured by WST-1 (4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) assay according to the manufacturer's instruction (Roche). Briefly, after lentivirus infection, A375 and SK-MEL-28 cells were reseeded in a 96‑well plate, respectively, at a density of 1x104 cell/well. This assay entails the addition of 10 mL WST-1 reagents per 100 mL cell cultures in a 96-well plate. These cultures were incubated for 30 minutes and the absorbance at 450 nm was determined by an ELISA Microplate Reader (Bio-Rad).
In cell cycle assay, cells were collected and washed with pre‑cooling PBS and fixed with cooling 70% ethanol overnight at 4˚C. After washing twice with pre‑cooling PBS, the cells were resuspended and incubated in 200 μl PBS containing 50 μg/ml propidium iodide (PI) solution and 100 μg/ml RNase for 30 min at room temperature.
In apoptosis assay, cells were assessed with Annexin V‑FITC/PI apoptosis detection Kit II (BD) on a flow cytometer (BD Biosciences, San Diego, CA, USA) following the manufacturer's instruction. Data were analyzed using CellQuest software.
Nude mice study
BALB/c Nude mice purchased from Beijing Weitong Lihua Experimental Animal Technology Co., Ltd. In subcutaneous tumor experiments, all experiments were performed in accordance with the official recommendations of the Chinese animal community. A375/shNC or A375/shUBE2C cells (5 x 106 in 0.15 ml sterilized PBS buffer) were implanted subcutaneously into 5-week-old male nude mice. The right flank of mouse was injected with shUBE2C cells and the left flank of mouse was injected with shNC cells. Tumor volume was calculated every 2 days using the following formula: V = (length) x (width) x (width)/2, and the subcutaneous tumors were removed and fixed with 10 % buffered formalin for immunohistochemistry analysis.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6 (La Jolla, USA) software. Statistical comparisons were performed by two-tailed t-test or one-way analysis of variance for parameter test, and quantitative data are expressed as the means ± SD of three independent experiments. Comparisons of tumor growth curves in vivo were completed by repeated-measures variance analysis. A value of P<0.05 was considered a statistically significant difference.
Results
Overexpression of UBE2C was associated with poor OS in the patients with melanoma
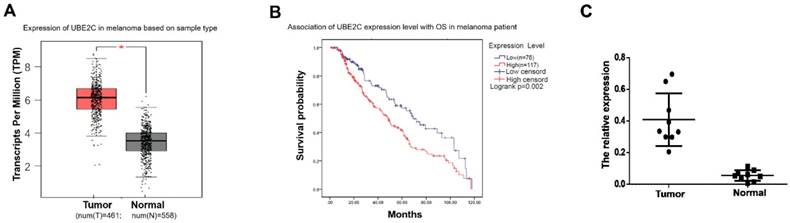
To determine the expression status of UBE2C in melanoma, we first analyzed the mRNA expression profiles and the associated clinical characteristics in 461 malignant melanomas compared to 558 normal samples from The Cancer Genome Atlas (TCGA) dataset. As shown in Fig. 1A, significant overexpressions of UBE2C were observed in melanoma tissues, which implied an oncogenic role it may play in melanoma. Kaplan-Meier analysis demonstrated that patients with high UBE2C expression had worse OS compared to patients with low UBE2C expression, suggesting that the expression level of UBE2C may serve as a potential prognostic marker for melanoma patients (Fig. 1B). Next, we determined the expression level of UBE2C using the clinical samples collected from our hospital to valid the above in silico result. Indeed, our qRT‑PCR analysis showed significant higher UBE2C mRNA levels in melanoma tissues compared to those in control tissues (Fig. 1C).
The clinical significance of UBE2C expression level in melanoma. (A) TCGA's datasets were used for bioinformatics analysis to determine UBE2C mRNA level in melanoma. A higher UBE2C mRNA expression level was found in melanoma than that in normal tissue (p<0.05). (B) According to the data from KM plotter, high UBE2C mRNA level was associated with poor overall survival of the patients with melanoma (p<0.01). HR, hazard ratio. (C) qRT-PCR assay validated UBE2C overexpression in melanoma using fresh tissues (p<0.01).
Downregulation of UBE2C suppressed melanoma cell growth via inactivation of ERK/AKT signaling pathways
Having demonstrated the overexpression of UBE2C in malignant melanoma, we then investigated the potential biological role of UBE2C in melanoma cells using loss-of-function assays. Lentivirus‑mediated shRNA (shUBE2C) was constructed to knockdown the expression of UBE2C in A375 and SK-MEL-28 melanoma cells. After 48 hours post-infection, the knockdown efficiency of shUBE2C was evaluated by qRT-PCR and western blot. As shown in Fig. 2A-B, the expression of UBE2C was significantly suppressed by shUBE2C at both mRNA and protein levels in those melanoma cells.
Knockdown of UBE2C suppressed cell growth via inhibiting ERK/AKT signaling pathways. (A) qRT‑PCR analysis of UBE2C mRNA levels in A375 and SK-MEL-28 cells following lentiviral shNC or shUBE2C infection. Data are expressed as mean ± SD of 3 independent experiments. ***P<0.001. (B) Western blot analysis of UBE2C protein levels in A375 and SK-MEL-28 cells following lentiviral shNC or shUBE2C infection. GAPDH was used as the loading control. (C) WST-1 assays were performed to determine cell viability in A375 and SK-MEL-28 cells after lentiviral infection of shNC or shUBE2C. **P<0.01, ***P<0.001. (D-E) Western blot analysis of key protein levels in ERK/AKT signaling pathways after lentiviral infection of shNC and shUBE2C in A375 and SK-MEL-28 cells, respectively. GAPDH was used as the loading control.
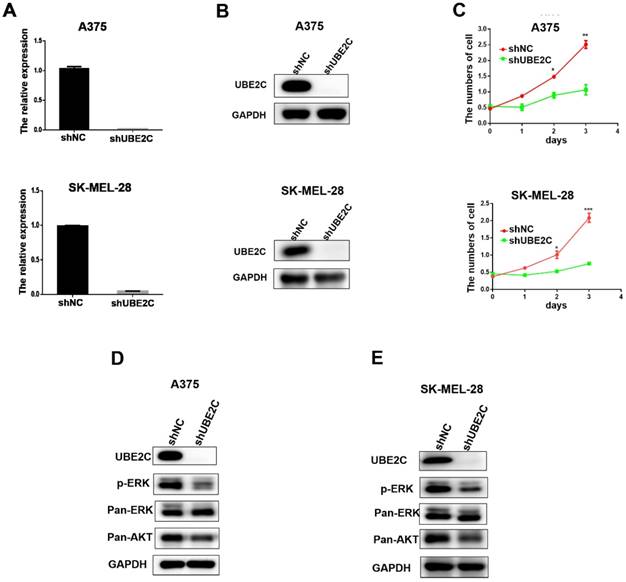
Next, the growths of A375 and SK-MEL-28 melanoma cells were respectively determined by WST-1 assay, following lentiviral shUBE2C infection for 24, 48 and 72 hours (Fig. 2C). The result showed that silencing of UBE2C significantly inhibited the growth rates of both A375 and SK-MEL-28 melanoma cells, demonstrating an important role of UBE2C in the regulation of cell growth.
To explore the potential mechanism involved in the growth inhibitory phenotype upon knockdown of UBE2C, we determined the status of critical proteins in survival-related signaling pathways in melanoma cells. Among those, the levels of phosphorylated ERK (p-ERK) and total Akt (pan-Akt) were dramatically reduced in the UBE2C knockdown cells (Fig. 2D-2E). These results indicated that knockdown of UBE2C expression inhibited the melanoma cell growth via inactivating ERK/AKT signaling pathways.
Silencing of UBE2C arrested melanoma cells at G2/M phase through downregulation of both the level and the activity of MPF
To determine whether above growth inhibitory phenotype was due to the disturbed cell cycle progression, we analyzed the cell cycle profile upon UBE2C silencing by flow cytometry with PI staining in A375 and SK-MEL-28 cells (Fig. 3A-3D). The results showed that the percentage of cells in G2/M phase doubled after knockdown of UBE2C (increased from 13.04% to 26.96% in A375 cells and from 23.35% to 40.85% in SK-MEL-28 cells, respectively), which indicated that silencing of UBE2C arrested these melanoma cells at G2/M phase.
Knockdown of UBE2C led to G2/M arrest via inhibiting MPF in melanoma cells. (A-B) Flow cytometry with PI staining was used to analyze the cell cycle distribution of A375 and SK-MEL-28 cells following shNC or shUBE2C infection. (C-D) Statistical analysis of cell percentage in G0/G1, S, G2/M in A375 and SK-MEL-28 cells with shNC or shUBE2C infection, Data are expressed as mean ± SD of 3 independent experiments. **P<0.01, ***P<0.001. (E-F) Expression levels of MPF complex, composed of cyclin B1 and CDK1, as well as the potential regulators of MPF activity were determined by western blot in A375 and SK-MEL-28. GAPDH was used as the loading control. (G) CDK2 and Cyclin E were detected in A375 cells after infected with shNC, and mRNA level of cyclinB1 was determined by qRT-PCR. (I) The protein level of cyclinB1 was determined after UBE2C-depletion with and without 10uM MG132 treatment for 4 hours by western blot.
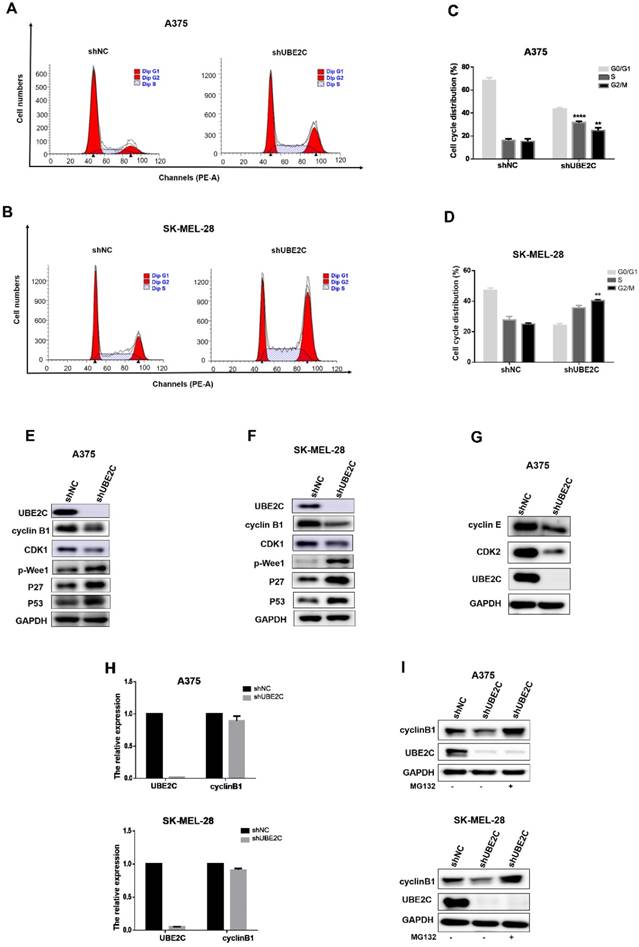
The cell-cycle transition at G2/M is controlled by MPF (M-phase-promoting factor), a complex consisting of CDK1 and cyclin B1. Thus, to further elucidate the mechanism underlying the shUBE2C-induced G2/M arrest, we determined the levels of CDK1 and cyclin B1 after knockdown of UBE2C in A375 and SK-MEL-28 cells, respectively. As shown in Fig. 3E-3F, the levels of both cyclinB1 and CDK1 were decreased in UBE2C-knockdown cells compared to those in control cells. Notably, without affecting cyclinB1 expression at the transcriptional level, silencing of UBE2C lowered the protein level of cyclinB1 by promoting its proteasome-mediated degradation, as the shUBE2C-induced down-regulation of cyclinB1 could be rescued by the addition of MG132 (Fig. 3H-3I). These results indicated that knockdown of UBE2C could down-regulate cyclinB1 and CDK1 levels, and thus reduced MPF complex formation, resulting in cell cycle arrest at G2/M phase.
It is well established that phosphorylation at Y15 by inhibitory kinase Wee1 interferes with full activation of CDK1 [24]. We then determined the phosphorylation status of Wee1 and found that the levels of phosphorylated Wee1 (p-Wee1, an active form of Wee1 kinase) were significantly increased upon UBE2C silencing in melanoma cells (Fig. 3E-3F). Moreover, the kinase activity of MPF is strictly regulated by Cdk inhibitors (CKIs), such as p21, p27 and p57. Among those, we observed a dramatic accumulation of p27, as well as its upstream regulator p53, resulted from the knockdown of UBE2C (Fig. 3E-3F). Thus, p53-dependent p27 accumulation may play an important role in blocking the activity of MPF. Together, these results indicated that silencing of UBE2C led to cell cycle blockage at G2/M phase in melanoma cells, which could be attributed to down-regulation of MPF level as well as inhibition of MPF activity.
Additionally, we observed a significant increase of A375 cell population in S phase (from 14.87% to 32.86% after shUBE2C infection, Fig. 3A and 3C), accompanied with decreased CDK2 and Cyclin E levels, indicating silencing of UBE2C resulted in an S phase arrest as well in A375 cells (Fig. 3G).
Knockdown of UBE2C induced apoptosis in melanoma cells
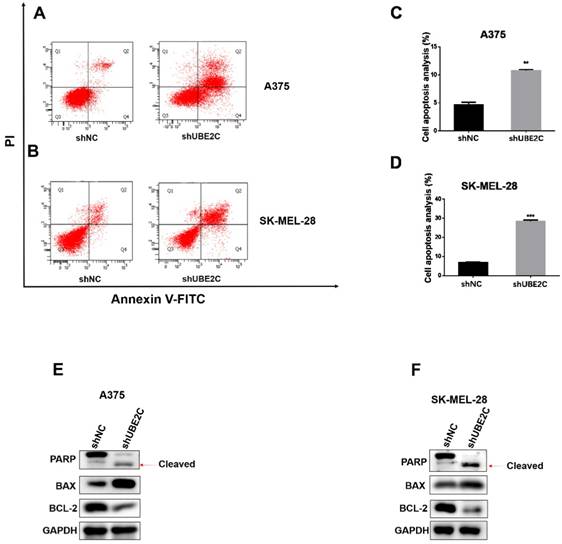
As prolonged cell cycle blockage would eventually lead to apoptosis of the arrested cells, we next determined whether UBE2C knockdown could enhance apoptosis in melanoma cells. The apoptotic events were detected by flow cytometry with Annexin V-FITC/PI double staining of A375 and SK-MEL-28 cells. As expected, knockdown of UBE2C elicited a remarkable increase of apoptotic population, which was further confirmed by the promoted PARP cleavage and increased BAX/Bcl-2 ratio using western blot (Fig. 4E-4F). These results showed that silencing of UBE2C promoted cell apoptosis in both A375 and SK-MEL-28 cells, contributing to the shUBE2C-induced growth inhibitory phenotype of melanoma cells.
Knockdown of UBE2C induced apoptosis of melanoma cells. (A-B) Flow cytometry with Annexin V‑FITC/PI double staining was employed to analyze cell apoptosis in A375 and SK-MEL-28 cells with shNC or shUBE2C infection. (C) Statistical analysis of apoptotic population in A375 and SK-MEL-28 cells after shNC or shUBE2C infection. Data are expressed as mean ± SD of 3 independent experiments. **P<0.01, ***P<0.001. (E-F) Expression levels of apoptotic marker BAX, Bcl‑2 and cleaved PARP were analyzed by western blot in A375 and SK-MEL-28 cells after infected with shNC or shUBE2C. GAPDH was used as the loading control.
Silencing of UBE2C inhibited xenografted melanoma growth in vivo
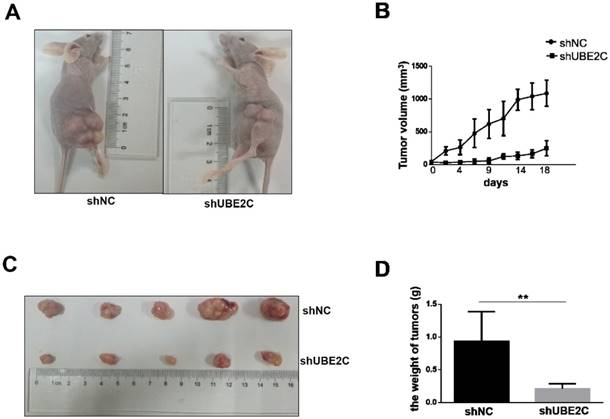
The inhibitory effect of UBE2C knockdown on melanoma cell growth was further investigated in vivo. Xenograft melanomas were established by subcutaneously injecting A375 cells into the dorsal flank of BALB/c nude mice. Seven BALB/c nude mice were included in the experiment, of which 5 developed tumors on either side of the flank. As shown in Fig. 5A-5B, the development of tumor was notably suppressed by silencing of UBE2C. By day 18, the tumor volume of shUBE2C-group was approximately 6-fold smaller than that of the control group. Consistently, the tumor weights showed a striking difference between the two groups (Fig. 5C-5D). These results provided convincing evidence showing that knockdown of UBE2C could suppress xenografted melanoma growth in vivo.
Knockdown of UBE2C inhibits the xenografted tumor growth in mice. (A) BALB/c nude mice were injected with A375 cells treated by shNC or shUBE2C to establish the experimental model. At day 18, the tumor masses on shUBE2C group mice were much smaller than that of shNC group mice. (B) The tumor volumes were measured and calculated once every two or three days. (C-D) The tumors were resected and weighted on day 16. Statistical comparisons of tumor size and weight were performed by repeated-measures variance analysis (**P<0.01).
Discussion
The role of UBE2C in proper mitotic progression has been well characterized in various eukaryotes [25]. In fission yeast, genomic disruption of Ube2C homolog UbcP4 gene resulted in a failure of anaphase onset with condensed chromosomes [26]. Mutants of Vihar, Ube2C homolog in Drosophila, arrested cells in metaphase with defective spindles [27]. Also, dominant-negative UBE2C caused clam and mammalian (COS) cells to accumulate in mitosis before anaphase [28]. Of note, an early study showed that depletion of UbcP4 protein in Schizosaccharomyces pombe led to defective cell cycle progression at both G2/M and metaphase/anaphase transitions [29]. However, several lines of evidence showed that RNAi-mediated depletion of UBE2C from human cells gave rise to only partial or little mitotic defects [6, 11, 30]. This is presumably due to the complementary function of UBCH5, another E2 partner of APC/C E3 ligase existing in human cells. UBCH5 and UBE2C are both responsible for adding the first ubiquitin molecules onto APC/C targets, after which APC/C ligase switches its E2 partner to E2-25K for extending K48-linked chains on those ubiquitin molecules[26, 30].
In this study, we observed little if any defect related to spindle checkpoint, but rather a G2 arrest in interphase upon silencing of UBE2C in human melanoma cells. Consistently, recent studies showed that both pharmacological and biological downregulation of UBE2C blocked the cells at G2 phase without overt mitotic defect in various human tumor cell lines [15, 31-33]. However, the involved mechanism underlying defective G2/M transition caused by UBE2C depletion remains unclear.
The MPF, a complex composed of Cyclin B1 and CDK1, is the key regulator of mitosis [34]. Proper formation, activation and cellular translocation of the complex are the absolute requirements for a successful entry into mitosis in most eukaryotes [35-37]. During the cell cycle, while CDK1 is constitutionally expressed through all the phases, the protein level of cyclin B1 begins to accumulate in late S-phase and peaks at early M phase to reach the required level for MPF assembly, which represents a crucial step in the G2/M transition [5, 38, 39]. Here, our results show that silencing of UBE2C down-regulated both CDK1 and cyclinB1 levels, thus interfered with the formation of Cyclin B1-CDK1 complex, leading to the blockage of G2/M transition.
As an ubiquitin-conjugating enzyme, UBE2C has been proposed to participate in ubiquitination and subsequent proteasome-mediated degradation of cyclin B1 by APC/C ligase in late metaphase, and UBE2C depletion leads to accumulation of cyclin B1 protein in several species [28, 29, 40]. Surprisingly, we observed a decreased protein level, rather than an accumulation of cyclin B1 upon silencing of UBE2C in melanoma cells. As the down-regulation of cyclin B1 level could be rescued by inhibition of proteasome activity in our study, we speculate that another “pre-mitotic” ubiquitin machinery, instead of the “mitotic mode” (UBE2C-APC/C), is involved in the “premature degradation” of cyclin B1 prior to mitotic entry, and that silencing of UBE2C facilitates the activity of this “pre-mitotic” machinery through an unknown mechanism.
It is well-established that the shift of phosphorylation-dephosphorylation of CDK1 kinase determines the active status of MPF [41]. In late G2 phase, upon binding with cyclin B1, the triggered conformational change of CDK1 facilitates its self-dephosphorylation at Tyt15, resulting in activation of MPF, which in turn promotes the G2/M transition [24]. On the other hand, Tyrosine kinase Wee1 plays an important role in hampering the activity of MPF by catalyzing phosphorylation of CDK1 at Tyt15 [42]. Our result showed an increased kinase activity of Wee1, evidenced by an increased level of phosphorylated Wee1 upon silencing of UBE2C, which may contribute to the G2/M arrest in those melanoma cells. Moreover, MPF activity could also be modulated by Cip/Kip family member, such as p21, p27 and p57 [24]. Among those, we observed a dramatic accumulation of p27 in UBE2C-depleted melanoma cells, in line with the previous findings showing that p27 could either directly inactivating MPF or inhibit the activity of MPF by sequestering it in the nucleus, causing failure of cells to complete the transition from G2 to M phase [43, 44].
Cell cycle checkpoints provide an important protective mechanism for eukaryotic cells to ensure proper operation of the cycle and maintain genomic integrity. In spite of multiple cycle checkpoints exist, cancer cells are inclined to rely on G2/M checkpoint to avoid cell death caused by extensive DNA damage, as G1 checkpoint defects is a common feature of malignant cells [45]. G2/M arrest reduces the proliferation rate of cancer cells and prolonged arrest would eventually lead to apoptosis, which renders it an attractive strategy for cancer therapy. Indeed, we observed a significant enhanced apoptotic population in UBE2C-depleted melanoma cells by flow cytometry. Thus, silencing of UBE2C leads to G2/M arrest and induces apoptosis of melanoma cells, suggesting that UBE2C could be a potential therapeutic target for melanoma. Moreover, these UBE2C-suppression approach further supplements the G2/M-blockage based strategy for cancer therapy.
Abbreviations
UBE2C: Ubiquitin‑conjugating enzyme E2C; TCGA: The Cancer Genome Atlas; OS: overall survival; APC/C: anaphase-promoting complex/cyclosome; MPF: M phase promoting factor; MAPK: Mitogen-activated protein kinase.
Acknowledgements
This work was jointly funded by National Natural Science Foundation of China (81572416) and National Key R&D Program of China (Grant No.2016YFC1303200).
Authors' contributions
Liren Liu designed and supervised the study and finalized the manuscript. Guolong Liu performed the experiments, analyzed data and participated in manuscript writing. Jun Zhao and Boyu Pan engaged in study design and data analysis. Boyu Pan and Gang Ma participated in preparation of manuscript and data analysis. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. AlQathama A, Prieto JM. Natural products with therapeutic potential in melanoma metastasis. Natural product reports. 2015;32:1170-82
2. Kuphal S, Bosserhoff A. Recent progress in understanding the pathology of malignant melanoma. The Journal of pathology. 2009;219:400-9
3. Pavri SN, Clune J, Ariyan S, Narayan D. Malignant Melanoma: Beyond the Basics. Plastic and reconstructive surgery. 2016;138:330e-40e
4. de Carvalho Lima EN, Piqueira JRC, Maria DA. Advances in Carbon Nanotubes for Malignant Melanoma: A Chance for Treatment. Molecular diagnosis & therapy. 2018
5. Shen Z, Jiang X, Zeng C, Zheng S, Luo B, Zeng Y. et al. High expression of ubiquitin-conjugating enzyme 2C (UBE2C) correlates with nasopharyngeal carcinoma progression. BMC Cancer. 2013;13:192
6. Rape M, Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004;432:588-95
7. Rape M, Reddy SK, Kirschner MW. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 2006;124:89-103
8. Yamanaka A, Hatakeyama S, Kominami K, Kitagawa M, Matsumoto M, Nakayama K. Cell cycle-dependent expression of mammalian E2-C regulated by the anaphase-promoting complex/cyclosome. Molecular biology of the cell. 2000;11:2821-31
9. Reddy SK, Rape M, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921-5
10. Xie C, Powell C, Yao M, Wu J, Dong Q. Ubiquitin-conjugating enzyme E2C: a potential cancer biomarker. The international journal of biochemistry & cell biology. 2014;47:113-7
11. Walker A, Pines J. UbcH10 has a rate-limiting role in G1 phase but might not act in the spindle checkpoint or as part of an autonomous oscillator. Journal of cell science. 2008;121:2319-26
12. van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. The Journal of cell biology. 2010;188:83-100
13. Parris TZ, Danielsson A, Nemes S, Kovacs A, Delle U, Fallenius G. et al. Clinical implications of gene dosage and gene expression patterns in diploid breast carcinoma. Clin Cancer Res. 2010;16:3860-74
14. Lin J, Raoof DA, Wang Z, Lin MY, Thomas DG, Greenson JK. et al. Expression and effect of inhibition of the ubiquitin-conjugating enzyme E2C on esophageal adenocarcinoma. Neoplasia. 2006;8:1062-71
15. Bavi P, Uddin S, Ahmed M, Jehan Z, Bu R, Abubaker J. et al. Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am J Pathol. 2011;178:2109-20
16. Wagner KW, Sapinoso LM, El-Rifai W, Frierson HF, Butz N, Mestan J. et al. Overexpression, genomic amplification and therapeutic potential of inhibiting the UbcH10 ubiquitin conjugase in human carcinomas of diverse anatomic origin. Oncogene. 2004;23:6621-9
17. Ieta K, Ojima E, Tanaka F, Nakamura Y, Haraguchi N, Mimori K. et al. Identification of overexpressed genes in hepatocellular carcinoma, with special reference to ubiquitin-conjugating enzyme E2C gene expression. International journal of cancer Journal international du cancer. 2007;121:33-8
18. Berlingieri MT, Pallante P, Guida M, Nappi C, Masciullo V, Scambia G. et al. UbcH10 expression may be a useful tool in the prognosis of ovarian carcinomas. Oncogene. 2007;26:2136-40
19. Wang H, Zhang C, Rorick A, Wu D, Chiu M, Thomas-Ahner J. et al. CCI-779 inhibits cell-cycle G2-M progression and invasion of castration-resistant prostate cancer via attenuation of UBE2C transcription and mRNA stability. Cancer research. 2011;71:4866-76
20. Cunha IW, Carvalho KC, Martins WK, Marques SM, Muto NH, Falzoni R. et al. Identification of genes associated with local aggressiveness and metastatic behavior in soft tissue tumors. Translational oncology. 2010;3:23-32
21. Lu J, Wen M, Huang Y, He X, Wang Y, Wu Q. et al. C2ORF40 suppresses breast cancer cell proliferation and invasion through modulating expression of M phase cell cycle genes. Epigenetics. 2013;8:571-83
22. Fujita T, Ikeda H, Taira N, Hatoh S, Naito M, Doihara H. Overexpression of UbcH10 alternates the cell cycle profile and accelerate the tumor proliferation in colon cancer. BMC Cancer. 2009;9:87
23. Pallante P, Berlingieri MT, Troncone G, Kruhoffer M, Orntoft TF, Viglietto G. et al. UbcH10 overexpression may represent a marker of anaplastic thyroid carcinomas. British journal of cancer. 2005;93:464-71
24. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079-93
25. Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nature reviews Molecular cell biology. 2006;7:644-56
26. Seino H, Yamao F. Two ubiquitin-conjugating enzymes, UbcP1/Ubc4 and UbcP4/Ubc11, have distinct functions for ubiquitination of mitotic cyclin. Molecular and cellular biology. 2003;23:3497-505
27. Mathe E, Kraft C, Giet R, Deak P, Peters JM, Glover DM. The E2-C vihar is required for the correct spatiotemporal proteolysis of cyclin B and itself undergoes cyclical degradation. Current biology: CB. 2004;14:1723-33
28. Townsley FM, Ruderman JV. Dominant-negative cyclin-selective ubiquitin carrier protein E2-C/UbcH10 blocks cells in metaphase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:2362-7
29. Osaka F, Yamao F. A ubiquitin-conjugating enzyme in fission yeast that is essential for the onset of anaphase in mitosis. Molecular and cellular biology. 1997;17:3388-97
30. Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007;130:127-39
31. Jiang L, Bao Y, Luo C, Hu G, Huang C, Ding X. et al. Knockdown of ubiquitin-conjugating enzyme E2C/UbcH10 expression by RNA interference inhibits glioma cell proliferation and enhances cell apoptosis in vitro. Journal of cancer research and clinical oncology. 2010;136:211-7
32. Wang H, Wang Q. CCI-779 inhibits cell-cycle G2-M progression and invasion of castration-resistant prostate cancer via attenuation of UBE2C transcription and mRNA stability. Cancer research. 2011;71:4866-76
33. Palumbo A Jr, Da Costa NM, De Martino M, Sepe R, Pellecchia S, de Sousa VP. et al. UBE2C is overexpressed in ESCC tissues and its abrogation attenuates the malignant phenotype of ESCC cell lines. Oncotarget. 2016;7:65876-87
34. Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503-8
35. Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends in cell biology. 2006;16:55-63
36. Lindqvist A, van Zon W, Karlsson Rosenthal C, Wolthuis RM. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS biology. 2007;5:e123
37. Porter LA, Donoghue DJ. Cyclin B1 and CDK1: nuclear localization and upstream regulators. Progress in cell cycle research. 2003;5:335-47
38. Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell. 1993;73:1393-402
39. Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82-7
40. Kobirumaki F, Miyauchi Y, Fukami K, Tanaka H. A novel UbcH10-binding protein facilitates the ubiquitinylation of cyclin B in vitro. Journal of biochemistry. 2005;137:133-9
41. Zachariae W, Nasmyth K. Whose end is destruction: cell division and the anaphase-promoting complex. Genes & development. 1999;13:2039-58
42. O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. The EMBO journal. 1997;16:545-54
43. Pagano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Molecular cell. 2004;14:414-6
44. Foijer F, Wolthuis RM, Doodeman V, Medema RH, te Riele H. Mitogen requirement for cell cycle progression in the absence of pocket protein activity. Cancer cell. 2005;8:455-66
45. Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug discovery today. 2012;17:194-202
Author contact
![]() Corresponding author: Professor Liren Liu, MD/PhD, Department of Gastrointestinal Cancer Biology, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin 300060, P.R. China. E-mail: liulirenedu.cn; Tel: +86-22-23340123-6512.
Corresponding author: Professor Liren Liu, MD/PhD, Department of Gastrointestinal Cancer Biology, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin 300060, P.R. China. E-mail: liulirenedu.cn; Tel: +86-22-23340123-6512.